

Abstract

Sulfur copolymers with high sulfur content find a broad range of applications from Li–S batteries to catalytic processes, self-healing materials, and the synthesis of nanoparticles. Synthesis of sulfur-containing polymers via the inverse vulcanization technique gained a lot of attention due to the feasibility of the reaction to produce copolymers with high sulfur content (up to 90 wt %). However, the interplay between the cross-linker and the structure of the copolymers has not yet been fully explored. In the present work, the effect of the amount of 1,3-diisopropenyl benzene (DIB) cross-linker on the structural stability of the copolymer was thoroughly investigated. Combining X-ray diffraction and differential scanning calorimetry, we demonstrated the partial depolymerization of sulfur in the copolymer containing low amount of cross-linker (<30 wt % DIB). On the other hand, by applying NMR and electron paramagnetic resonance techniques, we have shown that increasing the cross-linker content above 50 wt % leads to the formation of radicals, which may severely degrade the structural stability of the copolymer. Thus, an optimum amount of cross-linker is essential to obtain a stable copolymer. Moreover, we were able to detect the release of H2S gas during the cross-linking reaction as predicted based on the abstraction of hydrogen by the sulfur radicals and therefore we emphasize the need to take appropriate precautions while implementing the inverse vulcanization reaction.
1. Introduction
Abundant natural availability and over 70 million ton excess annual production of elemental sulfur by petroleum refineries creates the opportunity to develop new chemistry and applications to utilize sulfur.1−3 Processing and reaction of sulfur is difficult because of its limited solubility in organic solvents and incompatibility with the majority of chemicals and reagents; thus, the synthesis of a high-sulfur-content material is a challenging effort. Elemental sulfur occurs in its most stable eight-membered ring form (S8), which melts around 119 °C and forms sulfur rings of 8–35 sulfur atoms. Further temperature increase to 159 °C leads to the formation of a high-molecular-weight polysulfane via ring-opening polymerization. The polysulfane formed is unstable at room temperature and reverts back to its stable S8 form due to the presence of sulfur radicals at the polymer chain end.4
To overcome the depolymerization issue, Pyun and co-workers developed a new synthetic method called inverse vulcanization technique.5 In this method, the polymeric sulfur radicals are stabilized by reacting with an aromatic divinyl molecule to produce a highly cross-linked network that prevents depolymerization. They also demonstrated that this method could be scaled up to the kilogram scale.6 Initially, 1,3-diisopropenyl benzene (DIB) was utilized as the co-monomer because of its high boiling point (ca. 230 °C) and highly reactive divinyl group, which can form a cross-linked structure with sulfur at high temperature. Later, many researchers followed similar chemistry to prepare sulfur-rich copolymers using various vinyl, allyl, and alkylnyl monomers.7−13 The versatility of this synthetic approach further extends it to many naturally occurring monomers to produce polymers with a high sulphur content.14−17 The high sulfur content in these materials gives them unique properties that can be used in many applications such as Li–S batteries,10,18−20 as photoelectron catalysts for water splitting,21 as IR transparent high-refractive-index optical materials,22−24 in mercury detection and mercury removal from wastewater,15,16,25,26 self-healing materials,13,27 and synthesis of nanoparticles.2,28
In recent literature,29−31 reports of copolymer synthesis via inverse vulcanization method focus primarily on the preparation of materials with high sulfur content (from 50 to 90 wt %) and direct use for applications. However, the structural stability and the effect of cross-linker on the copolymer are not yet fully explored.
In this work, we employed advanced NMR and electron paramagnetic resonance (EPR) spectroscopic techniques to elucidate the role of DIB on the copolymer structure. The S-DIB copolymers with variable DIB content ranging from 10 to 70 wt % were synthesized via the inverse vulcanization technique. The solid-state NMR spectral editing and relaxation techniques and solution NMR were applied to clarify the changes induced in the copolymer structure by varying the DIB content. With the aid of EPR and electron nuclear double resonance (ENDOR) spectroscopy, we assessed the formation of radicals with increasing weight percentage of DIB. Additionally, we were able to detect the release of H2S gas during the reaction of DIB with sulfur, both in the NMR and Fourier transform infrared (FTIR) spectra, and by using a particular experimental setup that was specifically designed to enable the identification of H2S gas release.
2. Results and Discussion
The reaction of sulfur with DIB at 180 °C results in the formation of highly cross-linked sulfur copolymers. The chemical and physical properties of the sulfur copolymers depend mainly on the amount of cross-linker; hence, sulfur copolymers with the DIB ratio varying from 10 to 70 wt % were prepared. The schematic diagram for the copolymer preparation is presented in Scheme S1 (Supporting Information). All samples were characterized by X-ray diffraction (XRD), differential scanning calorimetry (DSC), and FTIR spectroscopy. Details of sample characterization are provided in the Supporting Information.
2.1. Solid-State CP MAS NMR Studies and Characterization of Sulfur Copolymers
2.1.1. Cross-Polarization/Total Sideband Suppression Technique (CP/TOSS)
The structure of the prepared sulfur copolymers was characterized by 1H and 13C solid-state NMR techniques.
The 1H NMR spectra of the investigated samples consist of two broad peaks, corresponding to aromatic and aliphatic groups (see Figure S1 in the Supporting Information) and do not reveal much information on the structure of the copolymers. Therefore, we applied 1H–13C CP MAS NMR, employing in particular the total sideband suppression technique (CP/TOSS) to suppress the sidebands.
The 1H–13C CP/TOSS MAS NMR spectra of S-DIB copolymers for three distinct DIB concentrations are depicted in Figure 1a. The two peaks at δ = 130–150 ppm are assigned to the aromatic carbons and the peak at δ = 58.6 ppm corresponds to the tertiary C–S bond. The broad multiple peaks between δ = 20–50 ppm are assigned to the merging of peaks originating from methylene (CH2), C–S, and CH3 groups. To further analyze those multiple peaks, we conducted cross-polarization–polarization inversion (CPPI) experiments.
Figure 1.

(a) 1H–13C CP/TOSS MAS NMR spectra of S-DIB copolymers with different weight percentage of DIB. (b) 1H–13C CP MAS spectra of S-DIB-50-50 copolymer with (A) polarization inversion (PI = 38 μs) and without (B, C) polarization inversion (PI = 1 μs), for long (contact time (CT) = 2 ms) and short (CT = 50 μs) contact times, respectively. (c) 1H–13C CP/TOSS spectra of the S-DIB-50-50 copolymer for different values of the CT ranging from 1500 to 50 μs. (d) Evolution of 13C spin magnetization as a function of the experimental contact time in the cross-polarization experiment of S-DIB-50-50 copolymer.
2.1.2. Cross-Polarization–Polarization Inversion (CPPI)
The technique of cross-polarization–polarization inversion (CPPI) is similar to heteronuclear single quantum correlation (HSQC)–distorsionless enhancement by polarization transfer (DEPT) method commonly applied for MAS spectral editing. In this method, it is possible to distinguish between 13CH and 13CH2 as well as 13CH3 and nonprotonated carbon signals depending on the polarization inversion time (PIT). The CPPI experiment at low MAS speed of 5 kHz, with a polarization inversion time (PIT) of 38 μs, results in zero signal for 13CH (peak b), inverted signal for 13CH2, whereas the quaternary 13C (peak c) and CH3 (peak e) peaks are unaffected.
The pulse sequence was initially applied to the known polypropylene (PP) polymer containing CH, CH2, and CH3 carbon as a model sample (see Figure S2, Supporting Information). The experiment was carried at low MAS speed of 5 kHz with PI = 38 μs. For comparison, another experiment without polarization (PIT = 1 μs) was also carried. CPPI spectra of PP without polarization inversion (PIT = 1 μs) showed all three major peaks at δ = 27.1, 31.5, and 49.3 ppm, respectively, corresponding to 13C peaks of 13CH3, 13CH, and 13CH2 groups, respectively. However, by implementing polarization inversion with PI = 38 μs, the 13CH peak disappeared completely; the 13CH2 peak was inverted, whereas the 13CH3 peak was unaffected. Thus, a CPPI pulse sequence with similar parameters was applied to S-DIB samples to distinguish between carbon nuclei belonging to different groups.
Spectra of S-DIB-50-50 copolymer obtained by using CP combined with polarization inversion (CPPI) are displayed in Figure 1b. The CPPI spectra of S-DIB copolymer acquired without polarization inversion (PIT = 1 μs) are similar to the corresponding CP MAS spectra comprising 13C peaks from all C sites along with aromatic sidebands (Figure 1b,A). However, by applying inversion of the polarization with a PI time equal to 38 μs (Figure 1b,B), the peak at 131.6 ppm (peak b) disappeared completely, confirming that it belongs to the aromatic CH group. The other peaks at 150 ppm (peak a), 58.8 ppm (peak c), and 27 ppm (peak e) are unaffected, indicating that they belong to the aromatic 13C, quaternary 13C, and methyl 13CH3 groups, respectively. Interestingly, the intensity of the peak at 33.2 ppm (peak d) is reduced significantly, suggesting that the peaks of methyl carbon from different carbon environments overlap. When the CPPI was applied without polarization inversion (PIT = 1 μs) and with the contact time (CT) reduced to 50 μs (Figure 1b,C), the aromatic CH carbon peak appeared again at 131.6 ppm as expected, whereas the quaternary aromatic and quaternary aliphatic carbon peaks at 150 and 58.8 ppm, respectively, were nullified. Methyl CH3 peaks also disappeared completely because the 1H–13C dipolar coupling is diminished due to faster free rotation of the CH3 groups. However, the intensity of the peak at ∼33.2 ppm (peak d) is reduced to more than half of its original intensity. The reduction in the intensity of peak (d) either under polarization inversion (Figure 1b,B), or by reducing the contact time without polarization inversion (Figure 1b,C) could originate from the overlapping of CH peaks. It is also worth noting that the broad peak observed between 33 and 52 ppm for low contact time without polarization inversion (Figure 1b,C) disappears completely under polarization inversion (Figure 1b,B). This could be due to the presence of the CH2 peak. These results provide further evidence that the CH2 groups react with sulfur radicals and partially convert to CH under cross-linking reactions.
2.1.3. Cross-Polarization Kinetics
In 1H–13C CP NMR experiments, the relative intensities of various carbon peaks depend on the relative effectiveness of the dipolar coupling between each individual carbon spin and the surrounding protons. Because the strength of the 13C–1H dipolar coupling is determined by the local environment of the carbon nucleus, nuclei in different groups will be characterized by different rates for maximal magnetization transfer (relative cross-polarization rate: CH3 (static) > CH2 > CH ≥ CH3 (rotating) > C (quaternary)). It is thus possible in the cross-polarization experiments to distinguish between different types of carbon nuclei by varying the contact time (CT) between the C and H spins and monitoring the corresponding change in the relative signal intensities.
Decreasing CT decreases the intensities of nonprotonated 13C due to the absence of polarization transfer from proton. At short CT, the peak from methyl 13C disappears completely due to the weak coupling, whereas the rigid 13CH and 13CH2 intensities are less affected. Figure 1c shows the 1H–13C CP/TOSS spectra of the S-DIB-50-50 copolymer for different values of the CT ranging from 1500 to 50 μs. The observed signals reveal a wide distribution of carbon kinetics in the S-DIB system. For long contact times (CT = 1500 μs), protonated and nonprotonated carbon peaks are clearly observed. However, with decreasing CT time, the intensities of nonprotonated and 13CH3 peaks decreased.
The spectrum obtained for 50 μs contact time, i.e., for the rapidly cross-polarizing, least-mobile components, showed mainly signals from the copolymer. The aromatic 13CH peak appears at 130 ppm—without any change—whereas the aromatic 13C and the tertiary 13C peaks disappear completely. It is also worth noting that the presence of a peak at 33.2 ppm can be assigned to the CH group. This also further supports the presence of CH in the copolymer.
The dependence of the intensities of the peaks a–d with the contact time CT for the S-DIB-50-50 copolymer is shown in Figure 1d. This change in the peak intensities as a function of CT depicts the rate of build-up of the carbon cross-polarized magnetization. The experimental results in Figure 1d were analyzed using the following expression31
| 1 |
where
which, in a cross-polarization
transfer experiment
between two spin systems I and S, describes the time evolution of
the magnetization mS of the S spins (13C in our case) as a function of the contact time CT with
the I spins (1H in our case). The above equation is valid
in the limiting case of 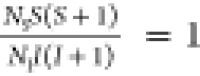 , where N is the number
of spins. This condition is fulfilled for 13C at the natural
isotopic abundance in organic solids, where NI/NS ≅ 150.
, where N is the number
of spins. This condition is fulfilled for 13C at the natural
isotopic abundance in organic solids, where NI/NS ≅ 150.
This time evolution is characterized by two different relaxation times. The TIS term, which is a cross-polarization transfer time constant, characterizes the rate of the energy transfer between the I and S systems, whereas the T1ρ term is the rotating frame spin lattice relaxation time of the I spins. The rate of spin lattice relaxation in the rotating frame is most sensitive to motions occurring at the precession frequency ω1 = γH1 of the magnetization about the spin-locking field H1 in the rotating frame. Fast magnetization decay rates with low T1ρ values correspond to motions with a correlation time τc ≈ 2π/ω1, whereas slow magnetization decay rates with long T1ρ values indicate motions that are very fast or very slow on the time scale of the rotating frame Larmor frequency ω1.
In this context, eq 1 has been fitted to the experimental data of Figure 1d, and the results are shown as lines in this figure.
The fit of eq 1 to the peak d (CH) experimental data gives TIS = 0.6 ms and T1ρ = 10 ms, whereas the other groups—peak a: quaternary C, peak b: aromatic CH, and peak e: methyl CH3—give similar TIS of about 1 ms and T1ρ > 10 ms. For these groups, therefore, the T1ρ term has negligible contribution to the fitting curves of Figure 1d. This is the reason why there is no decay of the fitting curves above 2000 μs. The results of the fits are shown in Table 1.
Table 1. Chemical Shift Assignment of the Experimental NMR Spectra Together with the Obtained Characteristic Relaxation Times from the CP and 13C T1 Experiments.
| peaks | δ (13C) (ppm) | assignment | TIS (ms) | T1ρ (H) (ms) | T1 (s) |
|---|---|---|---|---|---|
| e | 27 | methyl CH3 | 1 | 20 | 1.7 and 0.2 |
| d | 33.2 | CH | 0.6 | 10 | 0.17 |
| c | 58.8 | quaternary aliphatic C | 1 | 30 | 6.8 |
| 54.6 (from solution NMR) | |||||
| b | 131.6 | aromatic CH | 1 | 30 | 14.7 |
| a | 150 | quaternary aromatic C | 1 | 30 | 11 |
2.1.4. 13C Spin-Lattice Relaxation Behavior
In the semi-logarithmic plot of Figure 2, the peak intensities for aromatic 13C (peak a), aromatic 13CH (peak b), aliphatic quaternary 13C (peak c), aliphatic 13CH (peak d), and methyl 13C (peak e) carbons in the S-DIB-50-50 copolymer are plotted against the decay time τ for the 13C T1 spin-lattice relaxation.
Figure 2.

13C T1 relaxation decays of the carbon peaks (aromatic 13C, aromatic 13CH, aliphatic quaternary 13C, aliphatic 13CH, and methyl 13C) in S-DIB-50-50 copolymer.
The experimental data were analyzed using the familiar exponential decay function, and the results are shown in Table 1 along with the chemical shifts assignment of the observed peaks according to the attained experimental spectra. Also shown are the TIS, T1ρ (H), and T1 values for each peak obtained from the dynamic CP and 13C T1 experiments.
It is observed that aromatic carbons and tertiary carbon showed longest T1 relaxation. The peak at 33.2 ppm (peak d) showed the shortest T1 relaxation. From the CPPI spectra, it has already been proven that the broad peak at 33.2 ppm results from the merging of the peaks originating from CH2 to CH3 groups and the CH attached to the sulfur.
Therefore, there could be multiple contributions to the faster relaxation of the peak at 33.2 ppm: during the reaction, the sulfur radicals attach on the vinyl groups, resulting in the formation of a new C–S bond; moreover, the detachment of additional hydrogen by sulfur radicals could result in unstable SCSH, which could equally well contribute to the observed faster relaxation.
It is also observed that the peak d at 33.2 ppm has all its dynamic parameters reduced in comparison with the corresponding ones of the other groups. This is an interesting finding obtained from two different dynamic experiments (CP kinetics and T1 spin-lattice relaxation) signaling the significance of this specific NMR peak in the analysis of the experimental results in relation to the corresponding EPR observations.
2.2. Structural Analysis of Sulfur Copolymers via Solution NMR Spectroscopy
The prepared sulfur copolymers were also characterized by solution NMR. 1H and 13C NMR spectra, taken by dissolving the samples in CDCl3, are shown in Figure 3b,c, respectively. As can be clearly observed in Figure 3b, the alkene =CH2 proton peaks at δ = 5.13 and 5.40 ppm are completely absent from the spectra of the samples with a DIB content lower than 50 wt %. This indicates that in these samples, DIB has completely reacted with sulfur. However, the spectra of the samples with a DIB content higher than >50 wt % show two low intensity peaks corresponding to alkene protons, suggesting the presence of unreacted DIB monomer in the final product. It seems thus reasonable to conclude that the DIB percentages >50 wt % lead to an incomplete cross-linking reaction between DIB and sulfur. Moreover, all samples show a new peak around δ = 1.61 ppm, which can be assigned to thiol groups. Most probably, the highly reactive sulfur radicals abstract hydrogen from the methyl carbon during the cross-linking reaction, generating thiol moieties. The enlarged area from 1 to 2.5 ppm of the aliphatic region of the 1H NMR spectra for three different DIB mass percentages (30, 50, and 70%) is presented in the Supporting Information (see Figure S3). The abstraction of hydrogens results in the appearance of multiple complex peaks between δ = 3.5–1.00 ppm. It is well known that the thiol group is a functional group containing an active hydrogen atom; therefore, the chemical shift of the 1H NMR spectrum can be quite large, extending from 0.9 to 2.5 ppm, but with a most probable value around 1.5 ppm.32,33
Figure 3.

Solution NMR spectra in CDCl3. (a) 1H NMR spectrum of DIB monomer. (b) Comparison of 1H NMR spectra of S-DIB copolymers with different weight ratio of components. (c) 13C, DEPT-135, and DEPT-90 spectra of S-DIB-50-50 and (d) HSQC–DEPT overlapped with 1H NMR spectra for S-DIB-50-50.
The copolymers were further characterized with phase-edited HSQC and DEPT 90, and 135 experiments. The spectra of the 50–50 sample were identical to previously reported5 and the rest of the samples showed similar spectral characteristics, with the 30–70 sample showing unreacted DIB present. A more detailed analysis of the 1H spectra based on the interpretations of the chemical shifts indicates that the methyl groups are connected to a saturated bond. Moreover, the signals around 8.5 ppm could be due to close in space and nearly coplanar aromatic rings of the DIB units.
In Figure 3c, the 13C NMR spectrum is compared with the DEPT-135 and DEPT-90 spectra. In 13C NMR spectrum, the disappearance of alkene carbon peaks and the appearance of new C–S peaks between δ = 20–60 ppm supports the cross-linking reaction between DIB and sulfur. The disappearance of peaks in DEPT-135 between δ = 133.2–149.0 ppm and peak at δ = 54.6 ppm indicates the nonprotonated aromatic and aliphatic quaternary carbon, respectively. Inversion of the peak at δ = 48.2 ppm in DEPT-135 suggests the presence of the CH2 group. The two distinct peaks at δ = 34.3 and 39.3 ppm in DEPT-90 confirms the presence of the CH peaks. The peaks between δ = 20.1–29.3 are from methyl group in DIB. The DEPT-135 spectrum of S-DIB-30-70 with 70 wt % DIB (see Figure S4) shows series of inverted peaks between δ = 39.5–49.8 ppm for CH2 group. This suggests the presence of different environment of the CH2 group compared to the samples with lower amount of DIB. This could be due to the self-cross-linking of excess amount of DIB in S-DIB-30-70. This result also confirms the higher reactivity of sulfur toward the vinyl group and the sulfur-DIB reaction is dominated over DIB–DIB self-cross-linking reaction.
HSQC–DEPT NMR experiments were also utilized to obtain further evidence on the presence of thiol proton and for a complete structural analysis of the S-DIB copolymers. The HSQC–DEPT-135 spectrum overlapped with 1H NMR of S-DIB-50-50 copolymer is presented in Figure 3d. The HSQC–DEPT results are in good agreement with the 1H, 13C, and 13C DEPT NMR experiments. The negative peak (green) in HSQC–DEPT at δ = 3.2 ppm with secondary carbon at δ = 48 ppm in 13C NMR confirms the CH2 moieties. The peaks at δ = 2.91 and 3.19 ppm further support the presence of methine protons. However, the peaks at δ =1.61 ppm observed in 1H NMR give no signal in HSQC–DEPT confirming the generation of thiol group during the reaction. Further support to the above results stems from FTIR spectroscopy. The FTIR spectra of S-DIB-50-50 show a weak and rather broad band at 2594 cm–1 (see Figure S5), not present in neat DIB, that can be attributed to the −SH groups. Once −SH groups are formed, hydrogen sulfide (H2S) may be produced by the disproportionation of R–S–SH (to sulfane components (oxidation) and H2S (reduction)). Therefore, it is important to know if there is any release of H2S gas during the cross-linking reaction. Thus, an experiment was designed to identify the release of H2S during the reaction.
2.3. Experimental Identification of H2S Gas Release
The reaction setup is shown in Figure 4. In this setup, the reaction tube containing equal weight ratio of sulfur and DIB was kept in an oil bath and connected with the glass connector and the other end of the connector was dipped in a glass vial containing 5 mL of 1 M solution of silver nitrate (AgNO3). Any gas released from the reaction tube was bubbled through the silver nitrate solution. The reaction tube was purged with nitrogen and sealed. The temperature of the oil bath was slowly increased to 180 °C, where it was maintained for 1 h to ensure complete reaction. Initially, the reaction mixture melts to form a yellow solution, whereas the color of the silver nitrate solution remains transparent. As the reaction proceeds, the reaction mixture turns dark brown and the silver nitrate solution slowly turns dark gray and a dense black powder starts to settle at the bottom of the vial (see the enlarged digital image in Figure 4c). The formed black precipitate was filtered and analyzed with XRD, which confirmed the formation of Ag2S (see Figure 4).
Figure 4.

XRD comparison of AgNO3 and Ag2S, inset picture (a, b) shows the digital image of experimental setup to detect the release gas before and after the reaction, respectively. (c) Enlarged image of AgNO3 solution showing change in color from clear solution to black precipitate.
2.4. EPR Study
Room-temperature EPR signals in solid state were observed for S-DIB copolymers containing more than 50 wt % DIB, whereas samples with a low DIB content showed very weak or negligible signals. Although the signal intensity correlates strongly with the DIB content, it was found that there are other factors that can contribute to it as well. For instance, the thermal treatment of the previously prepared sample S-DIB-30-70 at 180 °C for 15 min resulted in enhancement of the EPR signal by a factor of 2. Figure 5B shows the relevant EPR spectra together with the recordings of signals 2 and 5 days after annealing.
Figure 5.

(A) Continuous-wave (CW) X-band EPR spectra of S-DIB-30-70 (blue trace) and S-DIB-50-50 (red trace) copolymers. (B) CW X-band EPR spectra of S-DIB-30-70 copolymer before and after annealing at 180 °C (equal amounts). (C) CW X-band EPR spectra of elemental sulfur before and after thermal annealing at 180 °C.
After annealing, the spectrum becomes stronger and shows a very modest decay over time. Moreover, for elemental sulfur, no EPR signal was detected either before or after thermal treatment (Figure 5C).
The typical intensity of the observed signals corresponds to about 3 × 1013 spins/mg, and the spectrum consists of a structureless derivative with g = 2.0044 ± 0.0005 and a linewidth of ΔBpp = 0.63 mT (Figure 5A). A careful analysis of the line shape shows that the spectrum can be fitted with at least two Gaussian derivatives, which implies inhomogeneously broadened lines. This is further supported by progressive saturation measurements shown in Figure 6.
Figure 6.

Progressive microwave saturation for the sample S-DIB-30-70 at room temperature. The solid line is the fit using the equation I(P) = (1 + P/P1/2)−b/2, where I is the normalized EPR amplitude divided by the square root of the incident microwave power P, P1/2 is the power at which the signal attains half of its unsaturated value, and b is the inhomogeneity parameter. Simulation parameters: P1/2 = 80.6 μW, b = 1.96.
The obtained saturation parameter b = 1.96 implies a modest homogeneous broadening character for the CW EPR spectra of copolymers (b = 1 indicates a completely inhomogeneously broadened character, whereas b = 3 is for a homogeneously broadened line).
Although the observed spectral parameters are typical for radical EPR signals, they do not provide unambiguous information about their origin. Possible candidates include organic radicals related to DIB, or sulfur-centered radicals in the polymer network.
To get further insight into the hyperfine couplings, we employed ENDOR spectroscopy. Figure 7 shows the Mims ENDOR spectrum of the sample S-DIB-30-70 measured with the medium wave (mw) pulse sequence π/2-τ-π/2-T-π/2-τ-echo, where the frequency of an additional rf pulse, placed between the second and third mw pulses, is swept. Apart from the intense peak at the proton Larmor frequency, νH = 14.7 MHz (dashed line), originating from weakly coupled matrix protons, the spectrum contains a doublet that corresponds to the hyperfine coupling of about 3 MHz. This signal is assigned to the proximal proton nuclei, which are close to the paramagnetic center, i.e., methyl group hydrogens close to the tertiary carbon of DIB.
Figure 7.

Mims ENDOR spectrum of sample S-DIB-30-70. Experimental conditions: temperature, 90 K; mw frequency, 9.722 GHz; magnetic field, 346.5 mT; τ = 96 ns; T = 10 μs; length of rf π-pulse, 9 μs; length of mw π/2-pulse, 16 ns.
On the other hand, the S-centered radicals (e.g., thiyl radicals, RS·) are characterized by strongly anisotropic g values with a rhombic symmetry and cannot account for the observed signals.34 To the best of our knowledge, the only compatible S-related radicals with our observations are sulfonyl radicals of the type RSO2,35 which, however, if present, would be masked by strong EPR signal from the radicals on the tertiary carbon on DIB. According to the NMR results (Figure 3b), the samples containing more than 50 wt % of DIB showed the presence of unreacted double bonds, which through a self-cross-linking reaction form DIB polymers, which are unstable and depolymerize via radical mechanism at room temperature due to low ceiling temperature of DIB.36 These results are also supported by the absence of radicals in copolymers with a DIB content lower than 50 wt %. Conclusively, the observed EPR signals arise mainly from the radicals formed on the tertiary carbon of the DIB polymer.
2.5. XRD and DSC Study
Detailed characterization and the spectra of XRD and DSC of pure elemental sulfur and S-DIB copolymers with different weight ratio are presented in the Supporting Information (see Figures S6 and S7). Both XRD and DSC of sulfur gave evidence for the reformation of elemental sulfur in the copolymer with the lower amount of cross-linker (i.e., DIB < 30 wt % with respect to the amount sulfur). It is well known that the long chains of polysulfur are unstable at room temperature and slowly revert to the more stable eight-membered sulfur ring. However, the mechanism of unzipping is still unclear. When low amount of DIB is used, the cross-linking density is lower and the copolymers formed contain long chains of sulfur and, hence, the sulfur chains are not completely terminated with the cross-linker. As a result, the long chains of sulfur could partially undergo unzipping and results in the formation of elemental sulfur in the copolymer. This is also evident by the partial decrease in the color intensity of the freshly prepared samples and aged samples (see Figure S7a,b). However, the samples containing higher amount of cross-linkers (DIB > 30–50 wt %) results in the formation of shorter chains of sulfur with a high cross-linking density and leads to a more stable copolymer. The results are also supported by the XRD, where the complete absence of crystalline sulfur peaks indicates the complete cross-linking of sulfur and the formation of amorphous copolymer. Similar results are also observed in DSC, where the sulfur-melting peaks are completely absent in the copolymer with a higher amount of cross-linkers.
The overall effect of the cross-linker on the sulfur is demonstrated in Scheme 1.
Scheme 1. Effect of Cross-Linker on the Sulfur Copolymer with Different Weight Ratio of DIB in the Copolymer.

3. Conclusions
This work presents a detailed analysis and characterization of sulfur-DIB polymers prepared by the inverse vulcanization technique. The presence of crystalline sulfur observed in the samples with a low content of cross-linkers may be attributed to the reduced cross-linking density, which leads to the formation of long sulfur chains. Similar to polysulfur, these long sulfur chains are unstable at room temperature and revert to stable monomeric sulfur. In the samples containing a higher amount of DIB, the NMR spectra confirmed the presence of unreacted alkene group, which leads to the formation of radicals, as evident from the EPR and ENDOR spectra. The presence of radicals can significantly degrade the stability of the copolymer. The above results are considered to be of great importance for optimizing the sulfur–DIB ratio to get a stable polymer. Moreover, this survey revealed the release of H2S gas during the inverse vulcanization reaction, thereby contributing to improving the protocol of the required precautions in the application of this synthetic method.
4. Materials and Methods
4.1. Materials
Elemental sulfur (S8, 99.9% pure, Sigma-Aldrich) and 1,3-diisopropenylbenzene (DIB, 97%, TCI America) were used without further purification.
4.2. Synthesis of Sulfur-(1,3-diisopropenylbenzene) (S-DIB) Copolymer
The sulfur-1,3-diisopropenylbenzene (S-DIB) copolymers were prepared via the inverse vulcanization technique according to a previously reported method.5 A representative example of the synthetic procedure is given by the synthesis of S-DIB-50-50 as follows: sulfur (S8, 5 g) was added to a 50 mL glass beaker equipped with a magnetic stir bar and heated to T = 180 °C in a thermostated oil bath until a clear orange molten phase was formed. 1,3-Diisopropenylbenzene (DIB, 5 mL) was then slowly added to the molten sulfur via syringe. The resulting mixture was stirred at T = 180 °C until the liquid mixture underwent vitrification and formed a solid mass. The vitrified mass was further heated for around 30 min to ensure complete reaction. The solidified mass was recovered by shattering the glass beaker and carefully separating the product and glass fragments. A similar procedure was followed for the synthesis of all S-DIB samples with the DIB mass percentage varying from 10 to 70% by weight. The prepared samples were denoted as S-DIB-x-y, where x and y indicate the percentage of sulfur and DIB, respectively.
4.3. Sample Characterization
The synthesized sulfur copolymers were characterized by X-ray diffraction (XRD), differential scanning calorimetry (DSC), and Fourier transform infrared (FTIR) spectra.
The diffraction (XRD) patterns were collected using an analytical X’Pert PRO Powder Diffractometer (Cu Kα radiation 1.5406 Å, 40 kV, 40 mA) in the range of 5–80° 2θ scale, with a step size of 0.02°. The DSC analysis was carried out on a Discovery series (TA instruments) for temperatures ranging from −30 to 200 °C at a ramp rate of 10 °C/min under nitrogen atmosphere.
4.4. NMR Measurements
The solution NMR experiments were carried out at 298 K on a Bruker Avance NMR spectrometer operating at 500.13 MHz for 1H and 125.77 MHz for 13C and at 296 K on a Bruker Avance III NMR spectrometer operating at 250.13 MHz for 1H and 62.90 MHz for 13C. The samples were prepared by dissolving DIB monomer and S-DIB copolymers in CDCl3. DEPT and 2D-HSQC-edited experiments were performed using the instrument’s library of pulse sequences.
The solid-state NMR measurements were run on a Bruker AVANCE 400 (B0 = 9.4 T) spectrometer operating at 400.23 MHz for 1H and 100.65 MHz for 13C. The spectra were recorded at room temperature using cylindrical 4 mm o.d. zirconia (ZrO2) rotors. 1H NMR experiments were carried out at the spinning speed of 12 kHz. 1H–13C cross-polarization/total sideband suppression (CP/TOSS) experiment was performed at a spinning speed of 12 kHz to get a sideband-free spectrum. A contact time of 2000 ms and a 1H 90° pulse length of 5 μs were used. Typically, 1024 scans were acquired with a relaxation delay of 5 s. A cross-polarization combined with polarization inversion (CPPISPI) was used to provide spectral editing of the 13C spectrum. Here, a short, polarization-inverting spin-lock period of 38 μs is applied on the 13C and 1H channels following the initial contact time of 2000 μs to achieve spectral editing. The CPPISPI experiment was carried out at a low MAS speed of 5 kHz and the number of scans was fixed to 1024 with a relaxation delay of 5 s. This experiment nulls methine, inverts methylene, and leaves methyl and quaternary carbon signals without much change, thus leading to spectral discrimination.
4.5. EPR Measurements
Continuous-wave (CW) EPR measurements at the X-band were performed on a Bruker ESP 380E spectrometer equipped with an EN 4118X-MD4 Bruker resonator. The EPR experiments were performed at room temperature (293 K) and at a microwave frequency of 9.646 GHz. The microwave power was 6.4 μW. The modulation amplitude used was 0.25 mT and the modulation frequency was 100 kHz, whereas the number of accumulated scans was fixed to 20. The measurements at cryogenic temperatures were performed using a helium cryostat from Oxford Inc. The microwave frequency was measured using a HP 5350B microwave frequency counter and the temperature was stabilized using an Oxford ITC4 temperature controller. Pulse EPR measurements at X-band (mw frequency 9.722 GHz) were performed on a Bruker ESP 380E spectrometer equipped with an EN 4118X-MD4 Bruker resonator. Mims-type electron nuclear double resonance (ENDOR) experiments were carried out at 90 K with the pulse sequence of π/2-τ-π/2-T-π/2-τ-echo, with a π/2 pulse of length 16 ns, a radio frequency pulse of length 9 μs, and a waiting time τ of 96 ns between the pulses.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsomega.8b00031.
Schematic representation of the S-DIB copolymer synthesis; solid-state 1H MAS NMR spectra of the DIB monomer and sulfur copolymers with different weight percentage of DIB; 1H–13C CP MAS spectra of the model polymer polypropylene-isotactic (PPI); 1H NMR spectra of the DIB monomer and of sulfur copolymers dissolved in CDCl3 and 13C DEPT 135 spectra of S-DIB-30-70; FTIR spectra of the DIB monomer and S-DIB copolymers; DSC thermograms and XRD patterns of pure elemental sulfur and S-DIB copolymers (PDF)
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
This work was supported by Abu Dhabi National Oil Company (ADNOC), Abu Dhabi, United Arab Emirates.
The authors declare no competing financial interest.
Supplementary Material
References
- Meyer B.; Kharasch N.; Sulphur I.. Elemental Sulfur: Chemistry and Physics; Interscience: New York, London, 1965. [Google Scholar]
- Chung W. J.; Simmonds A. G.; Griebel J. J.; Kim E. T.; Suh H. S.; Shim I.-B.; Glass R. S.; Loy D. A.; Theato P.; Sung Y.-E.; Char K.; Pyun J. Elemental Sulfur as a Reactive Medium for Gold Nanoparticles and Nanocomposite Materials. Angew. Chem., Int. Ed. 2011, 50, 11409–11412. 10.1002/anie.201104237. [DOI] [PubMed] [Google Scholar]
- Gerson F.; Huber W.. Paramagnetic Organic Species and Their Generation. In Electron Spin Resonance Spectroscopy of Organic Radicals. Gerson F.; Huber W., Eds.; Wiley-VCH Verlag GmbH & Co: Germany, 2004; pp 10−36. [Google Scholar]
- Meyer B. Elemental sulfur. Chem. Rev. 1976, 76, 367–388. 10.1021/cr60301a003. [DOI] [Google Scholar]
- Chung W. J.; Griebel J. J.; Kim E. T.; Yoon H.; Simmonds A. G.; Ji H. J.; Dirlam P. T.; Glass R. S.; Wie J. J.; Nguyen N. A.; Guralnick B. W.; Park J.; Somogyi Á.; Theato P.; Mackay M. E.; Sung Y.-E.; Char K.; Pyun J. The use of elemental sulfur as an alternative feedstock for polymeric materials. Nat. Chem. 2013, 5, 518–524. 10.1038/nchem.1624. [DOI] [PubMed] [Google Scholar]
- Griebel J. J.; Li G.; Glass R. S.; Char K.; Pyun J. Kilogram scale inverse vulcanization of elemental sulfur to prepare high capacity polymer electrodes for Li-S batteries. J. Polym. Sci., Part A: Polym. Chem. 2015, 53, 173–177. 10.1002/pola.27314. [DOI] [Google Scholar]
- Arslan M.; Kiskan B.; Yagci Y. Combining Elemental Sulfur with Polybenzoxazines via Inverse Vulcanization. Macromolecules 2016, 49, 767–773. 10.1021/acs.macromol.5b02791. [DOI] [Google Scholar]
- Salman M. K.; Karabay B.; Karabay L. C.; Cihaner A. Elemental sulfur-based polymeric materials: Synthesis and characterization. J. Appl. Polym. Sci. 2016, 133, 43655 10.1002/app.43655. [DOI] [Google Scholar]
- Dirlam P. T.; Simmonds A. G.; Kleine T. S.; Nguyen N. A.; Anderson L. E.; Klever A. O.; Florian A.; Costanzo P. J.; Theato P.; Mackay M. E.; Glass R. S.; Char K.; Pyun J. Inverse vulcanization of elemental sulfur with 1,4-diphenylbutadiyne for cathode materials in Li-S batteries. RSC Adv. 2015, 5, 24718–24722. 10.1039/C5RA01188D. [DOI] [Google Scholar]
- Gomez I.; Mecerreyes D.; Blazquez J. A.; Leonet O.; Ben Youcef H.; Li C.; Gómez-Cámer J. L.; Bondarchuk O.; Rodriguez-Martinez L. Inverse vulcanization of sulfur with divinylbenzene: Stable and easy processable cathode material for lithium-sulfur batteries. J. Power Sources 2016, 329, 72–78. 10.1016/j.jpowsour.2016.08.046. [DOI] [Google Scholar]
- Sun Z.; Xiao M.; Wang S.; Han D.; Song S.; Chen G.; Meng Y. Sulfur-rich polymeric materials with semi-interpenetrating network structure as a novel lithium-sulfur cathode. J. Mater. Chem. A 2014, 2, 9280–9286. 10.1039/c4ta00779d. [DOI] [Google Scholar]
- Lin H.-K.; Liu Y.-L. Reactive Hybrid of Polyhedral Oligomeric Silsesquioxane (POSS) and Sulfur as a Building Block for Self-Healing Materials. Macromol. Rapid Commun. 2017, 38, 1700051 10.1002/marc.201700051. [DOI] [PubMed] [Google Scholar]
- Arslan M.; Kiskan B.; Yagci Y. Recycling and Self-Healing of Polybenzoxazines with Dynamic Sulfide Linkages. Sci. Rep. 2017, 7, 5207 10.1038/s41598-017-05608-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez I.; Leonet O.; Blazquez J. A.; Mecerreyes D. Inverse Vulcanization of Sulfur using Natural Dienes as Sustainable Materials for Lithium–Sulfur Batteries. ChemSusChem 2016, 9, 3419–3425. 10.1002/cssc.201601474. [DOI] [PubMed] [Google Scholar]
- Parker D. J.; Jones H. A.; Petcher S.; Cervini L.; Griffin J. M.; Akhtar R.; Hasell T. Low cost and renewable sulfur-polymers by inverse vulcanisation, and their potential for mercury capture. J. Mater. Chem. A 2017, 5, 11682–11692. 10.1039/C6TA09862B. [DOI] [Google Scholar]
- Crockett M. P.; Evans A. M.; Worthington M. J. H.; Albuquerque I. S.; Slattery A. D.; Gibson C. T.; Campbell J. A.; Lewis D. A.; Bernardes G. J. L.; Chalker J. M. Sulfur-Limonene Polysulfide: A Material Synthesized Entirely from Industrial By-Products and Its Use in Removing Toxic Metals from Water and Soil. Angew. Chem., Int. Ed. 2016, 55, 1714–1718. 10.1002/anie.201508708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khawaja S. Z.; Vijay Kumar S.; Jena K. K.; Alhassan S. M. Flexible sulfur film from inverse vulcanization technique. Mater. Lett. 2017, 203, 58–61. 10.1016/j.matlet.2017.05.133. [DOI] [Google Scholar]
- Hu G.; Sun Z.; Shi C.; Fang R.; Chen J.; Hou P.; Liu C.; Cheng H.-M.; Li F. A Sulfur-Rich Copolymer@CNT Hybrid Cathode with Dual-Confinement of Polysulfides for High-Performance Lithium–Sulfur Batteries. Adv. Mater. 2017, 29, 1603835 10.1002/adma.201603835. [DOI] [PubMed] [Google Scholar]
- Fu C.; Li G.; Zhang J.; Cornejo B.; Piao S. S.; Bozhilov K. N.; Haddon R. C.; Guo J. Electrochemical Lithiation of Covalently Bonded Sulfur in Vulcanized Polyisoprene. ACS Energy Lett. 2016, 1, 115–120. 10.1021/acsenergylett.6b00073. [DOI] [Google Scholar]
- Li B.; Li S.; Xu J.; Yang S. A new configured lithiated silicon-sulfur battery built on 3D graphene with superior electrochemical performances. Energy Environ. Sci. 2016, 9, 2025–2030. 10.1039/C6EE01019A. [DOI] [Google Scholar]
- Zhuo S.; Huang Y.; Liu C.; Wang H.; Zhang B. Sulfur copolymer nanowires with enhanced visible-light photoresponse. Chem. Commun. 2014, 50, 11208–11210. 10.1039/C4CC05574H. [DOI] [PubMed] [Google Scholar]
- Kleine T. S.; Nguyen N. A.; Anderson L. E.; Namnabat S.; LaVilla E. A.; Showghi S. A.; Dirlam P. T.; Arrington C. B.; Manchester M. S.; Schwiegerling J.; Glass R. S.; Char K.; Norwood R. A.; Mackay M. E.; Pyun J. High Refractive Index Copolymers with Improved Thermomechanical Properties via the Inverse Vulcanization of Sulfur and 1,3,5-Triisopropenylbenzene. ACS Macro Lett. 2016, 5, 1152–1156. 10.1021/acsmacrolett.6b00602. [DOI] [PubMed] [Google Scholar]
- Anderson L. E.; Kleine T. S.; Zhang Y.; Phan D. D.; Namnabat S.; LaVilla E. A.; Konopka K. M.; Ruiz Diaz L.; Manchester M. S.; Schwiegerling J.; Glass R. S.; Mackay M. E.; Char K.; Norwood R. A.; Pyun J. Chalcogenide Hybrid Inorganic/Organic Polymers: Ultrahigh Refractive Index Polymers for Infrared Imaging. ACS Macro Lett. 2017, 6, 500–504. 10.1021/acsmacrolett.7b00225. [DOI] [PubMed] [Google Scholar]
- Griebel J. J.; Nguyen N. A.; Namnabat S.; Anderson L. E.; Glass R. S.; Norwood R. A.; Mackay M. E.; Char K.; Pyun J. Dynamic Covalent Polymers via Inverse Vulcanization of Elemental Sulfur for Healable Infrared Optical Materials. ACS Macro Lett. 2015, 4, 862–866. 10.1021/acsmacrolett.5b00502. [DOI] [PubMed] [Google Scholar]
- Hasell T.; Parker D. J.; Jones H. A.; McAllister T.; Howdle S. M. Porous inverse vulcanised polymers for mercury capture. Chem. Commun. 2016, 52, 5383–5386. 10.1039/C6CC00938G. [DOI] [PubMed] [Google Scholar]
- Abraham A. M.; Kumar S. V.; Alhassan S. M. Porous sulphur copolymer for gas-phase mercury removal and thermal insulation. Chem. Eng. J. 2018, 332, 1–7. 10.1016/j.cej.2017.09.069. [DOI] [Google Scholar]
- Xiang H. P.; Qian H. J.; Lu Z. Y.; Rong M. Z.; Zhang M. Q. Crack healing and reclaiming of vulcanized rubber by triggering the rearrangement of inherent sulfur crosslinked networks. Green Chem. 2015, 17, 4315–4325. 10.1039/C5GC00754B. [DOI] [Google Scholar]
- Bear J. C.; Peveler W. J.; McNaughter P. D.; Parkin I. P.; O’Brien P.; Dunnill C. W. Nanoparticle-sulphur “inverse vulcanisation” polymer composites. Chem. Commun. 2015, 51, 10467–10470. 10.1039/C5CC03419A. [DOI] [PubMed] [Google Scholar]
- Griebel J. J.; Glass R. S.; Char K.; Pyun J. Polymerizations with elemental sulfur: A novel route to high sulfur content polymers for sustainability, energy and defense. Prog. Polym. Sci. 2016, 58, 90–125. 10.1016/j.progpolymsci.2016.04.003. [DOI] [Google Scholar]
- Lim J.; Pyun J.; Char K. Recent Approaches for the Direct Use of Elemental Sulfur in the Synthesis and Processing of Advanced Materials. Angew. Chem., Int. Ed. 2015, 54, 3249–3258. 10.1002/anie.201409468. [DOI] [PubMed] [Google Scholar]
- Boyd D. A. Sulfur and Its Role In Modern Materials Science. Angew. Chem., Int. Ed. 2016, 55, 15486–15502. 10.1002/anie.201604615. [DOI] [PubMed] [Google Scholar]
- Zhao B.; Zheng Y.; Weng Z.; Cai S.; Gao C. The electrophilic effect of thiol groups on thiol-yne thermal click polymerization for hyperbranched polythioether. Polym. Chem. 2015, 6, 3747–3753. 10.1039/C5PY00307E. [DOI] [Google Scholar]
- Lian Q.; Li Y.; Yang T.; Li K.; Xu Y.; Liu L.; Zhao J.; Zhang J.; Cheng J. Study on the dual-curing mechanism of epoxy/allyl compound/sulfur system. J. Mater. Sci. 2016, 51, 7887–7898. 10.1007/s10853-016-0044-z. [DOI] [Google Scholar]
- Kutney G.The Sulfur Entrepreneur. In Sulfur, 2nd ed.; Kutney G., Ed. ChemTec Publishing: Oxford, 2013; pp 187−208. [Google Scholar]
- Bassindale A. R.; Iley J. N.. NMR and ESR of Organosulphur Compounds. In Sulphur-Containing Functional Groups (1993); John Wiley & Sons, Inc., 2010; pp 245–291. [Google Scholar]
- Lutz P.; Beinert G.; Rempp P. Anionic polymerization and copolymerization of 1,3- and 1,4-diisopropenylbenzene. Die Makromol. Chem. 1982, 183, 2787–2797. 10.1002/macp.1982.021831114. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.