

Abstract
The all-trans-retinoic acid (atRA) hydroxylase Cyp26a1 is essential for embryonic development and may play a key role in regulating atRA clearance also in adults. We hypothesized that loss of Cyp26a1 activity via inducible knockout in juvenile or adult mice would result in decreased atRA clearance and increased tissue atRA concentrations and atRA-related adverse effects. To test these hypotheses, Cyp26a1 was knocked out in juvenile and adult male and female Cyp26a1 floxed mice using standard Cre–Lox technology and tamoxifen injections. Biochemical and histological methods were used to study the effects of global Cyp26a1 knockout. The Cyp26a1 knockout did not result in consistent histopathological changes in any major organs. Cyp26a1−/− mice gained weight normally and exhibited no adverse phenotypes for up to 1 year after loss of Cyp26a1 expression. Similarly, atRA concentrations were not increased in the liver, testes, spleen, or serum of these mice, and the Cyp26a1 knockout did not cause compensatory induction of lecithin:retinol acetyltransferase (Lrat) or retinol dehydrogenase 11 (Rdh11) mRNA or a decrease in aldehyde dehydrogenase 1a1 (Aldh1a1) mRNA in the liver compared with tamoxifen-treated controls. However, the Cyp26a1−/− mice showed increased bone marrow cellularity and decreased frequency of erythroid progenitor cells in the bone marrow consistent with a retinoid-induced myeloid skewing of hematopoiesis. In addition, the Cyp26a1 knockout decreased clearance of exogenous atRA by 70% and increased atRA half-life 6-fold. These findings demonstrate that despite lacking a major impact on endogenous atRA signaling, Cyp26a1 critically contributes as a barrier for exogenous atRA exposure.
Keywords: retinoic acid, retinoid, retinol, cytochrome P450, vitamin A, cytochrome, hematopoiesis
Introduction
Vitamin A and its active metabolite all-trans-retinoic acid (atRA)2 are necessary for normal fetal development, regulation of cell division, reproduction, and controlling myeloproliferative processes in all mammals (1–3). All of these processes rely on spatiotemporal regulation of the concentration gradients of atRA, and too high (4) or too low atRA concentrations can be detrimental to life (5–7). This is well-demonstrated by the male infertility resulting from vitamin A deficiency and depletion of atRA in the testis after dosing with Aldh1a inhibitors (8), by the teratogenic effects of retinoids (9, 10), and by the development of acute promyelocytic leukemia when retinoic acid receptor is mutated impairing retinoid signaling (11). Despite these well-characterized phenotypes of vitamin A and retinoid imbalance and the extensive characterization of generation of atRA gradients during embryonic and fetal development (1, 5), the detailed enzymological processes that regulate tissue-specific concentrations of atRA and hence retinoid signaling in adult mammals are still not completely understood.
It is well-established that vitamin A is stored in the liver stellate cells in the form of retinyl esters (RE, primarily retinyl palmitate) and that the liver stores serve to provide vitamin A to the various tissues in the body (12, 13). The storage of vitamin A within the stellate cells is mediated by esterification of retinol by lecithin:retinol acetyltransferase (Lrat) (14) and hydrolysis of the esters by retinyl ester hydrolases. Vitamin A is supplied to the circulation from the hepatocytes in the form of retinol bound to retinol-binding protein RBP4. Retinol is taken up to target tissues from the circulation, and it undergoes oxidation to atRA in the tissues via a two-step process mediated by Rdh and Aldh1a enzymes (12, 13, 15). The clearance of atRA from tissues is believed to be mediated by the cytochrome P450 (Cyp) family 26 enzymes (7, 16). It has been shown that inhibition of the Aldh1a enzymes decreases tissue atRA concentrations and signaling (17, 18) and that inhibition of Cyp26 enzymes increases tissue atRA concentrations and retinoid signaling (19–21). However, the relative tissue-specific roles of individual Aldh1a and Cyp26 enzymes in regulating atRA signaling and concentrations in adult animals are not known. The understanding of the role of the individual enzymes is largely derived from knockout mice, in which the enzymes are knocked out already during fetal development (1). Because of the significance of retinoid signaling in embryogenesis, many such knockout mice die during development (15) and therefore yield very little information of the role of the enzymes of interest in vitamin A homeostasis in adult animals. Inducible and tissue-specific knockout models have provided important new tools to address this issue by selectively eliminating the activity of key retinoid-metabolizing enzymes in adulthood. These models have clearly demonstrated the dichotomy of the need of specific enzymes in fetal development versus adulthood. For example, knockout of Aldh1a2 leads to lethality of the embryos early on in gestation (1, 23), but when Aldh1a2 is knocked-out via the inducible Cre–Lox system postnatally, the animals show no obvious adverse phenotype (24). The goal of this study was to determine whether the previously shown critical need for Cyp26a1 activity during fetal development (25, 26) extends to a necessity of this enzyme in regulating atRA homeostasis in adult animals.
The family of Cyp26 enzymes includes three enzymes that are very efficient in oxidizing atRA and are expressed in all mammalian species (27–29). In mouse models, knockout of Cyp26a1 or Cyp26b1 during embryonic development is lethal (30), and therefore, it has been hypothesized that Cyp26a1 and Cyp26b1 are the critical atRA hydroxylases also in adult mammals (7, 16). The function of Cyp26c1 in clearing retinoids is less clear (29, 31). Cyp26a1 protein and mRNA were detected in human livers (32), and Cyp26a1 was predicted to be the main human liver atRA hydroxylase (33). In contrast, Cyp26b1 appears to be the main atRA hydroxylase in extrahepatic tissues in adults (28, 32). Cyp26a1 seems to also be the enzyme that is readily induced upon atRA treatment (32, 34) and thus controls exogenous retinoid clearance. Based on the existing data, we hypothesized that post-natal global conditional knockout of Cyp26a1 in adult or juvenile mice would result in a significant increase in tissue atRA concentrations, specifically in the liver and testis, as these tissues would have decreased clearance of atRA. We also predicted that increased atRA concentrations in plasma would result in activation of retinoid signaling and subsequent retinoid-related toxicity. These hypotheses were tested using a ROSA26CreERT to induce global knockout of the Cyp26a1 floxed allele upon treatment with tamoxifen. We aimed to determine the role of Cyp26a1 in atRA clearance and in regulating atRA signaling in juvenile and adult mice.
Results
Phenotypic characterization of the Cyp26a1−/− mice
The transgenic strains of mice were generated successfully, and Cyp26a1 was knocked out in these mice in a genotype-dependent manner by tamoxifen injections as confirmed by RT-PCR analysis (Fig. 1). Heterozygous Cyp26a1+/− mice were included in the analysis, as it was assumed these mice should have a 50% decrease in the expression of Cyp26a1 compared with the Cyp26a1+/+ (control) mice. The RT-PCR analysis suggested that the Cyp26a1+/− animals indeed had reduced Cyp26a1 mRNA expression compared with Cyp26a1+/+ controls (Fig. 1), although the expression levels of Cyp26a1 mRNA were highly variable. The Cyp26a1 knockout was induced in either juvenile (21 days old) or adult mice (60 days old) to explore possible differences in the role of Cyp26a1 in the different ages of mice. Following the tamoxifen injections, the Cyp26a1−/− mice gained weight at similar pace as their Cyp26a1+/+ controls (Fig. 2), and the mice showed no gross physical or behavior abnormalities and showed normal grooming. When Cyp26a1 knockout was induced in juvenile animals, the male Cyp26a1−/− mice gained weight faster than the controls (p < 0.0001), but this effect was not reproduced in the female mice or in the male mice in which Cyp26a1 was knocked out in adulthood (Fig. 2), suggesting this was a transient effect. Overall, selective loss of Cyp26a1 activity appeared to have no obvious adverse effects on the mice, and the mice survived up to a year after tamoxifen injections without physical or behavioral abnormalities.
Figure 1.
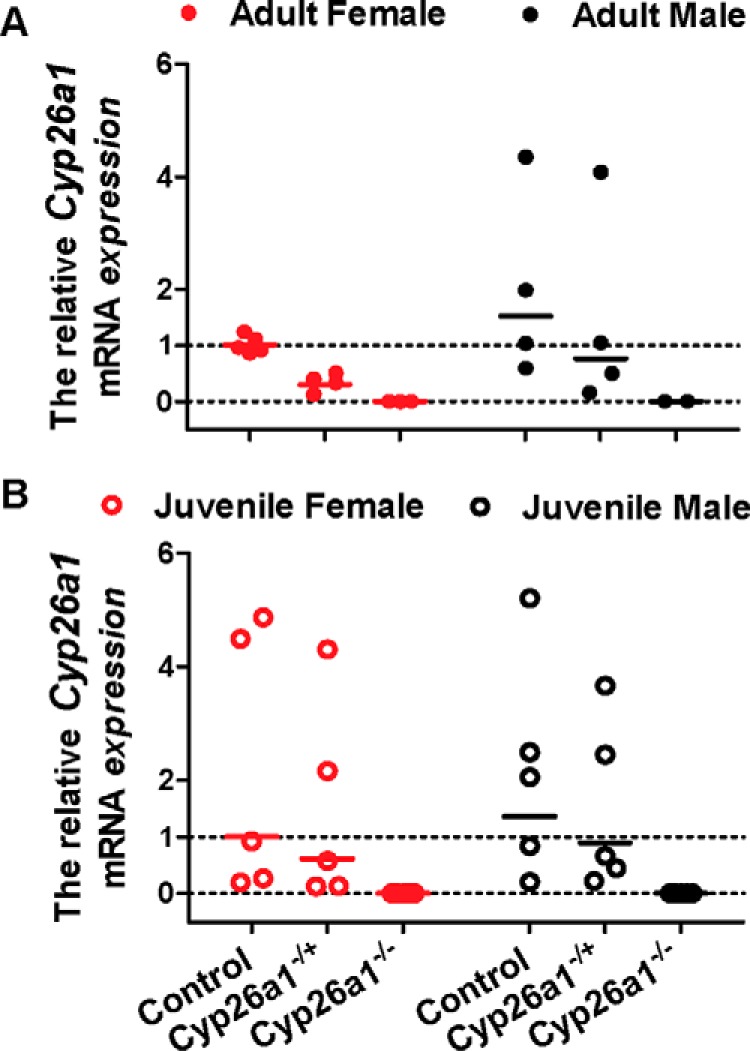
Analysis of Cyp26a1 mRNA expression in the Cyp26a1−/− mice compared with Cyp26a1+/− and Cyp26a1+/+ (control) mice. The Cyp26a1 deletion was induced either in adulthood (A) or in juvenile animals (B), and liver was collected 2 months after completion of tamoxifen treatment. A value of 0 was assigned for relative mRNA expression to mice in the Cyp26a1−/− group that had no detectable Cyp26a1 mRNA in the liver. One female Cyp26a1−/− mouse was excluded from all the analyses due to incomplete knockout of Cyp26a1 based on RT-PCR analysis. Each data point represents an individual mouse and is the average value of two RT-PCR analyses performed on separate days. Each analysis was performed in duplicate. The solid lines show the group geometric mean.
Figure 2.

Body weight gain in female (A and B) and male (C and D) Cyp26a1+/+, Cyp26a1+/−, and Cyp26a1−/− mice. The notation “Adult” (left panels) indicates that tamoxifen injections were done in 2-month old mice, and “Juvenile” (right panels) indicates the mice in which tamoxifen injections were done at 21 days of age. The days on the x axis refer to the number of days elapsed after the tamoxifen injections were completed. Data points indicate mean values, and error bars show S.D.
Standard histopathological analysis was done in all three genotype groups in mice in which Cyp26a1 knockout was induced as juveniles or adults. No significant histological abnormalities were noted in Cyp26a1−/− mice versus Cyp26a1+/− or Cyp26a1+/+ mice in any of the tissues analyzed (Fig. 3). All mice, including controls, had minimal to mild chronic to chronic-active inflammation of the mesenteric fat with low numbers of histiocytes with vacuolated cytoplasm, most likely related to intraperitoneal tamoxifen injections. Sporadic minimal to mild lymphocytic salivary gland adenitis, minimal hepatic necrosis, and minimal to mild gastritis were observed in mice from all genotypes, and these observations were considered background lesions.
Figure 3.

Hematoxylin and eosin (H&E)-stained representative images of the liver (top row, ×200 original magnification; bar, 100 μm); kidney (2nd row, ×400 original magnification; bar, 10 μm); spleen (3rd row, ×100 original magnification; bar, 100 μm); and testis (bottom row, ×200 original magnification; bar, 50 μm) from Cyp26a1+/+ (left), Cyp26a1+/− (middle), and Cyp26a1−/− (right) male mice.
atRA has been shown to play a critical role in hematopoiesis, and Cyp26 enzymes are known to degrade atRA. Hematopoiesis in Cyp26a1−/− mice compared with Cyp26a1+/+ mice was analyzed by complete blood counts and peripheral blood flow cytometry in 9-week-old mice, 5 weeks after tamoxifen injections (Fig. 4 and Figs. S1 and S2). Peripheral blood analysis via flow cytometry showed a significant decrease in the peripheral blood CD3+ T-cells and a significant increase in the peripheral blood myeloid cells in the Cyp26a1−/− mice when compared with Cyp26a1+/+ mice. No significant differences in the B220+ B-cells were observed between the two groups (Fig. 4).
Figure 4.

Flow cytometry analysis of peripheral blood white blood cells (WBC). A shows total white blood cell counts in peripheral blood obtained from Cyp26a1+/+ mice (circles) and Cyp26a1−/− mice (squares) at different time points after tamoxifen injections. B–D show the flow cytometry analysis of peripheral blood in Cyp26a1+/+ (control) and Cyp26a1−/− mice 5 weeks after tamoxifen injections. The horizontal lines indicate mean values of the frequencies of CD3+ T cells (B), B220+ B cells (C), and myeloid cells (Gr-1+ cells) (D), and error bars indicate standard deviation (S.D.). Each data point represents an individual mouse. The filled and open symbols represent male and female mice, respectively. Significant differences were analyzed by unpaired Student's t test; *, p < 0.05. Representative flow cytometry plots are shown in Figs. S1 and S2.
To further investigate the potential impact of loss of Cyp26a1 activity on hematopoiesis, bone marrow cellularity and differentiation status were analyzed. The bone marrow cellularity of Cyp26a1−/− mice was significantly increased, whereas there was no difference in the frequency and number of hematopoietic progenitor cells in the bone marrow of the Cyp26a1−/− mice when compared with Cyp26a1+/+ controls as measured by CFU cells (Fig. S3). However, when the bone marrow progenitor cell colonies were analyzed, the Cyp26a1−/− mice had significantly lower frequency and absolute number of erythroid CFUs when compared with Cyp26a1+/+ mice (p < 0.05) (Fig. 5). In addition, the frequency of myeloid CFUs was significantly greater in Cyp26a1−/− mice when compared with Cyp26a1+/+ mice (p < 0.05), but the absolute number of myeloid CFU was not altered in Cyp26a1−/− mice (Fig. 5). The frequency or number of early progenitor cells (CFU-Mix; common myeloid progenitors, CFU mix of both erythroid and myeloid) was not different (p > 0.05) between the two groups of mice (Fig. 5).
Figure 5.

Analysis of bone marrow hematopoiesis in mice 5 weeks after tamoxifen treatment as measured by CFU cells (CFU-C). A–C show the percentage of total CFU-C as specific types of CFU-C: myeloid (A), erythroid (B), and mixed (C). D–F show the absolute number of individual types of CFU per femur for myeloid (CFU-GM; D), erythroid (BFU-E; E) and CFU mix (F) within the progenitor populations found in the bone marrow. In all panels, the horizontal lines indicate mean values, and error bars indicate S.D. Each data point represents an individual mouse. The filled and open symbols represent male and female mice, respectively. Significant differences were analyzed by unpaired Student's t test; *, p < 0.05; **, p < 0.01; ***, p < 0.001.
Impact of Cyp26a1−/− on vitamin A homeostasis
To determine whether the global elimination of Cyp26a1 in adult or juvenile mice impacts vitamin A homeostasis in select tissues or serum, atRA, retinol, and retinyl ester concentrations were measured in serum, liver, spleen, and testis of the mice (Table 1). No differences in atRA, retinol, or retinyl ester concentrations were observed between Cyp26a1−/− mice and Cyp26a1+/+ mice in serum (Fig. 6), liver (Fig. 7), testis (Fig. S4), or spleen (Fig. S5) regardless of whether Cyp26a1 was deleted in adult or in juvenile animals. Of the tissues analyzed, the liver had the highest and most variable atRA concentrations, 10–20 pmol/g, with testis concentrations being very consistent, 11–13 pmol/g. The spleen and serum atRA concentrations were over 70% lower than liver and testis, 2–4 pmol/g and 2–4 nm, respectively. Testis and spleen retinol and retinyl ester concentrations were negligible (0.3–1 nmol/g retinol and 0.8–1.6 nmol/g retinyl esters) compared with the serum concentrations of 1 μm (retinol) and liver stores of 100–200 nmol/g retinol and 200–1400 nmol/g retinyl esters (Table 1). Although this study was not designed to compare the retinoid concentrations in the mice of different ages, the data collected show that the retinyl ester and possibly retinol concentrations are lower in the liver of younger mice (Fig. 7 and Table 1).
Table 1.
Retinoid concentrations in mouse serum and tissues
Data are shown as mean ± S.D.; numbers in parentheses indicate the number of mice in each group. F, female; M, male.
| Retinyl ester |
Retinol |
atRA |
||||
|---|---|---|---|---|---|---|
| Female | Male | Female | Male | Female | Male | |
| μm | μm | nm | ||||
| Serum | ||||||
| Adult | ||||||
| Cyp26a1+/+ | 0.06 ± 0.02 (5) | 0.20 ± 0.21 (4) | 1.0 ± 0.2 (5) | 1.9 ± 0.1 (4) | 2.8 ± 0.6 (5) | 3.4 ± 1.0 (4) |
| Cyp26a1+/− | 0.06 ± 0.04 (5) | 0.10 ± 0.06 (4) | 1.1 ± 0.2 (5) | 1.5 ± 0.1 (4) | 2.9 ± 0.4 (5) | 3.4 ± 0.7 (4) |
| Cyp26a1−/− | 0.08 ± 0.02 (4) | 0.07 ± 0.02 (4) | 1.0 ± 0.1 (4) | 1.7 ± 0.1 (4) | 3.2 ± 0.7 (4) | 3.6 ± 0.8 (4) |
| Juvenile | ||||||
| Cyp26a1+/+ | 0.10 ± 0.07 (5) | 0.08 ± 0.04 (5) | 1.1 ± 0.1 (5) | 1.6 ± 0.1 (5) | 2.1 ± 0.7 (5) | 1.9 ± 0.3 (5) |
| Cyp26a1+/− | 0.08a (2) | 0.11 ± 0.15 (6) | 0.8a (2) | 1.5 ± 0.2 (6) | 1.6a (2) | 2.4 ± 0.6 (6) |
| Cyp26a1−/− | 0.06 ± 0.02 (4) | 0.05 ± 0.02 (5) | 0.9 ± 0.2 (5) | 1.5 ± 0.1 (5) | 2.1 ± 0.6 (5) | 2.4 ± 0.6 (5) |
| nmol/g liver | nmol/g liver | pmol/g liver | ||||
| Liver | ||||||
| Adult | ||||||
| Cyp26a1+/+ | 1261 ± 567 (5) | 1098 ± 344 (4) | 154 ± 42 (5) | 116 ± 48 (4) | 11.2 ± 2.6 (5) | 19.1 ± 11.1 (4) |
| Cyp26a1+/− | 1365 ± 478 (4) | 942 ± 378 (4) | 226 ± 143 (4) | 150 ± 31 (4) | 11.6 ± 3.8 (4) | 21.4 ± 10.5 (4) |
| Cyp26a1−/− | 1243 ± 401 (4) | 906 ± 260 (4) | 192 ± 58 (4) | 137 ± 41 (4) | 14.5 ± 3.0 (4) | 18.5 ± 2.2 (4) |
| Juvenile | ||||||
| Cyp26a1+/+ | 415 ± 103 (4) | 315 ± 76 (4) | 82 ± 41 (4) | 141 ± 80 (4) | 8.1 ± 2.2 (4) | 22.3 ± 1.5 (4) |
| Cyp26a1+/− | 440 ± 153 (4) | 267 ± 110 (4) | 182 ± 92 (4) | 113 ± 61 (3) | 8.2 ± 1.2 (4) | 26.7 ± 3.4 (4) |
| Cyp26a1−/− | 424 ± 105 (4) | 318 ± 87 (4) | 102 ± 31 (4) | 80 ± 23 (4) | 9.4 ± 2.8 (4) | 18.1 ± 2.5 (4) |
| nmol/g testis | nmol/g testis | pmol/g testis | ||||
| Testis | ||||||
| Adult | ||||||
| Cyp26a1+/+ | 0.8 ± 0.1 (3) | 0.3 ± 0.1 (3) | 11.1 ± 1.3 (3) | |||
| Cyp26a1+/− | 0.8 ± 0.0 (3) | 0.3 ± 0.1 (3) | 12.6 ± 1.3 (3) | |||
| Cyp26a1−/− | 0.9 ± 0.2 (3) | 0.3 ± 0.0 (3) | 11.6 ± 1.2 (3) | |||
| Juvenile | ||||||
| Cyp26a1+/+ | 1.1 ± 0.1 (3) | 0.3 ± 0.0 (3) | 12.9 ± 2.8 (3) | |||
| Cyp26a1+/− | 1.0 ± 0.3 (3) | 0.3 ± 0.0 (3) | 11.0 ± 1.0 (3) | |||
| Cyp26a1−/− | 0.8 ± 0.2 (3) | 0.4 ± 0.1 (3) | 11.0 ± 1.3 (3) | |||
| nmol/g spleen | nmol/g spleen | pmol/g spleen | ||||
| Spleenb | ||||||
| Adult | ||||||
| Cyp26a1+/+ | 1.5 ± 0.5 (8) | 0.8 ± 0.1 (8) | 2.8 ± 0.5 (5; 4F,1 M) | |||
| Cyp26a1+/− | 1.3 ± 0.2 (9) | 0.9 ± 0.4 (9) | 2.6 ± 0.4 (4; 3F,1 M) | |||
| Cyp26a1−/− | 1.6 ± 0.8 (8) | 1.0 ± 0.4 (8) | 3.3 ± 1.1 (5; 3F,2 M) | |||
a Data are mean values of two samples.
b atRA concentrations in several spleen samples (three in Cyp26a1+/+ group, five in Cyp26a1+/− group, and three in Cyp26a1−/− group) were excluded due to poor LC separation of the atRA peak from an interfering peak in those samples. Data of retinoid concentrations in spleen were not separated based on sex because in each experimental group atRA concentrations were quantified in less than three spleen samples for male mice.
Figure 6.

Serum retinyl esters (RE) (A and B), retinol (ROL) (C and D), RBP4 (E and F), and atRA (G and H) concentrations in Cyp26a1−/− mice compared with Cyp26a1+/− and Cyp26a1+/+ (control) mice. The notation Adult indicates that tamoxifen injections were done in 2-month old mice, and Juvenile indicates the mice in which tamoxifen injections were done at 21 days of age. In all panels, the horizontal lines indicate mean values, and error bars indicate S.D. Each data point represents an individual mouse. Significant differences were analyzed by two-way ANOVA; ***, p < 0.001; €, p < 0.05, €€€, p < 0.001.
Figure 7.

Liver concentrations of retinyl esters (RE) (A and B), retinol (ROL) (C and D), and atRA (E and F) in Cyp26a1−/− mice compared with Cyp26a1+/− and Cyp26a1+/+ (control) mice. In all panels, the horizontal lines indicate mean values, and error bars indicate S.D. Each data point represents an individual mouse. Significance of differences was analyzed by two-way ANOVA; *, p < 0.05; €€€, p < 0.001.
The conditional knockout of Cyp26a1 had no impact on tissue retinoid concentrations, but a sex difference was observed in the vitamin A metabolome. In serum, retinol and RBP4 concentrations were ∼1.4–1.7-fold higher (p < 0.05) in male mice when compared with age-matched females both in the cohort of mice in which Cyp26a1 was deleted as juveniles and in the cohort of mice in which Cyp26a1 was deleted in adulthood (Fig. 6). This sex difference was independent of the genotype of the mice. No sex difference in retinyl ester or atRA serum concentrations was observed. Interestingly, the higher serum retinol concentrations in male mice translated to significantly (p < 0.05) higher concentrations of atRA in the liver of male mice when compared with female mice regardless of the genotype and age of the mice at time of sacrifice. The number of spleen samples available for analysis was not sufficient to assess sex differences in spleen retinoid concentrations.
As no differences were observed in atRA concentrations and vitamin A metabolome between the genotypes, despite the expected critical role of Cyp26a1 in regulating retinoid homeostasis, we hypothesized that other enzymes such as Cyp26b1, Lrat, and Aldh1a1 may be transcriptionally regulated to compensate for the lack of Cyp26a1 activity. The mRNA expression of these enzymes in the mouse liver was measured together with the marker of atRA signaling (Rarβ) (Fig. 8). Surprisingly, no differences were observed in Aldh1a1, Lrat, Rarβ, or Rdh11 mRNA in any of the mouse groups (Fig. 10). In the mouse liver, Cyp26c1 and Rdh1 mRNAs were not detected (Ct >40), whereas Cyp26b1, Aldh1a2, and Rarγ mRNA expression was either undetermined (Ct >40) or not quantifiable (38 < Ct <40) for all groups of mice, demonstrating lack of compensatory induction of any of these genes.
Figure 8.

Analysis of the mRNA expression of Lrat (A and B), Aldh1a1 (C and D), Rdh11 (E and F), and Rarβ (G and H) in the livers of female (red symbols) and male (black symbols) Cyp26a1−/−, Cyp26a1+/−, and Cyp26a1+/+ (control) mice. Each data point represents an individual mouse and is the average value of duplicate measurements performed on two separate days. The solid lines show the group geometric mean.
Figure 10.
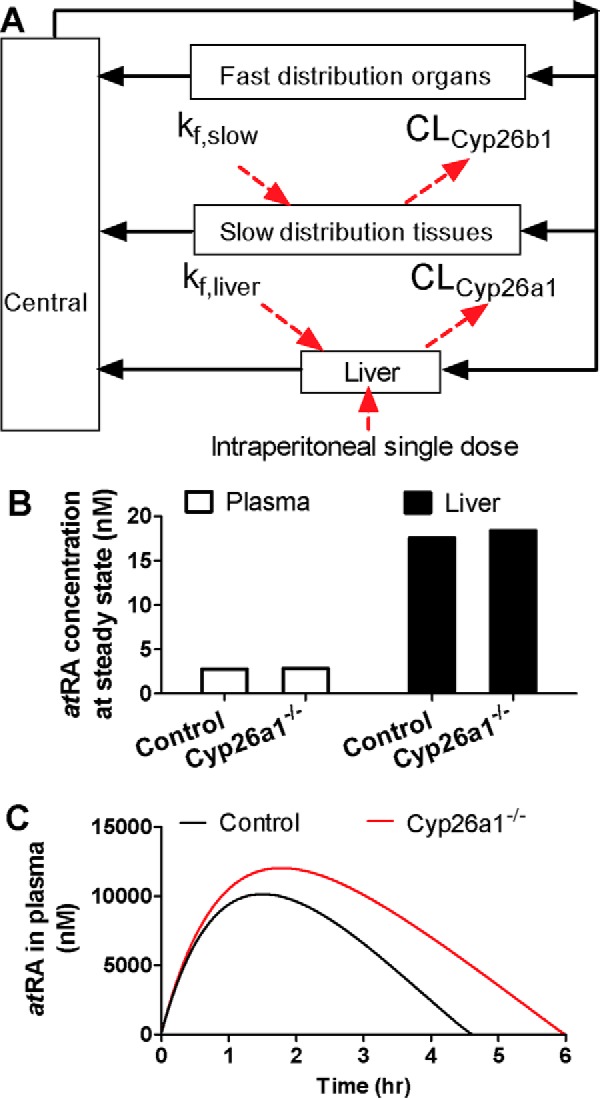
Simulation of the changes in endogenous and exogenous atRA concentrations in Cyp26a1−/− mice compared to Cyp26a1+/+ mice. The structure of the semi-physiologically–based model used to simulate atRA disposition is shown in A. Central compartment represents the central circulation system; fast-distributing organ compartment represents the visceral organs; and slow-distributing tissue compartment represents skin, adipose, bone, and muscle. kf, slow and kf, liver represent the endogenous formation rate of atRA in slow-distribution tissues and liver, respectively. CLCyp26b1 and CLCyp26a1 represent the atRA clearances by Cyp26a1 and Cyp26b1, respectively, modeled based on Michaelis-Menten kinetics. The intraperitoneal single dose of atRA was introduced to the liver compartment by a first-order absorption process. B shows the simulated steady-state (SS) atRA concentrations in plasma in the Cyp26a1+/+ (control) and Cyp26a1−/− mice. The simulated plasma concentrations are 2.77 and 2.85 nm in Cyp26a1+/+ and Cyp26a1−/− mice, respectively, and simulated hepatic atRA concentrations are 17.57 and 18.38 nm, in Cyp26a1+/+ and Cyp26a1−/− mice, respectively. The simulated plasma concentration versus time curve of atRA following i.p. dosing to Cyp26a1+/+ and Cyp26a1−/− mice is shown in C. The black curves show simulations for Cyp26a1+/+ (control) mice and red curves for Cyp26a1−/− mice.
Impact of Cyp26a1−/− on exogenous atRA pharmacokinetics
Cyp26a1 is highly inducible by atRA and appears to respond rapidly to changing atRA exposures (32, 34). Based on this inducibility, a likely role of Cyp26a1 in adults is to protect the organism from excess atRA exposure from dietary sources. It has been hypothesized that Cyp26a1, especially in the liver, plays an important role in clearance of exogenously administered atRA (33). To test whether Cyp26a1 is indeed important for the clearance of exogenously administered atRA in mice, the pharmacokinetics of atRA given i.p. was evaluated in Cyp26a1−/− and Cyp26a1+/+ mice. The exposure to atRA was significantly greater in Cyp26a1−/− than in Cyp26a1+/+ mice (Fig. 9). The AUC of atRA was increased over 3-fold from 17,400 ± 5,380 to 63,288 ± 24,524 h·nmol/liter (p = 0.011), and the systemic clearance was decreased from 2.0 ± 0.5 to 0.6 ± 0.2 liters/h/kg (p = 0.002) in the Cyp26a1−/− mice compared with the Cyp26a1+/+ mice. The decrease in the systemic clearance was accompanied by an almost 6-fold increase in atRA half-life from 0.9 ± 0.4 to 5.5 ± 1.8 h (p = 0.003) in the Cyp26a1−/− mice.
Figure 9.
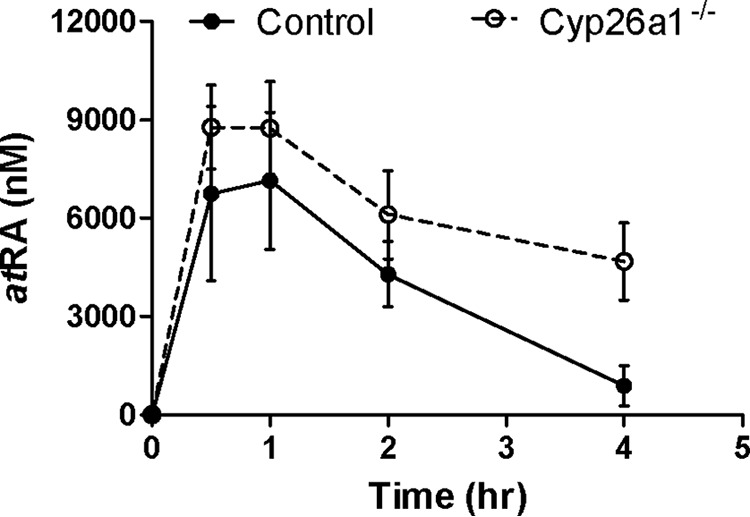
Pharmacokinetics of atRA following a 10 mg/kg i.p. injection to Cyp26a1+/+ (control) (closed symbols and solid line) and Cyp26a1−/− mice (open symbols and dashed line). Each data point indicates the mean value of plasma atRA concentrations measured in mice (n = 4), and error bars show S.D.
Pharmacokinetic modeling
To explore the mechanisms that contribute to the differences between the impact of deletion of Cyp26a1 on endogenous and exogenous atRA, a semi-physiologically-based pharmacokinetic model was developed to simulate the relative roles of hepatic and extrahepatic Cyp26a1 and Cyp26b1 in atRA clearance (Fig 10 and Table S1). The model was used to test the hypothesis that endogenous atRA concentrations are predominantly regulated by extrahepatic Cyp26b1, whereas exogenous administration of atRA results in saturation of Cyp26b1 (low Km), and the clearance of exogenous atRA is mainly regulated by activity of Cyp26a1 (high Km). This hypothesis is supported by the lower apparent Km value of atRA for human CYP26B1 compared with CYP26A1 (28). The simulation predicted a <5% increase in steady-state atRA concentrations in plasma and liver in Cyp26a1−/− mice compared with Cyp26a1+/+ mice, an insignificant difference in atRA concentrations. However, because of the saturation of Cyp26b1 after exogenous atRA administration, the model predicted a 1.5-fold increase in atRA AUC and an increase in atRA Cmax (Fig. 10).
Discussion
The CYP26 enzymes are believed to be the main mammalian atRA hydroxylases responsible for the clearance of atRA from variety of retinoid target tissues (7, 15, 16). Indeed, recombinant CYP26 enzymes efficiently hydroxylate atRA to 4-OH-atRA and other direct oxidation products (27–29, 36). It has also been shown that CYP26B1 and CYP26C1 interact with the cellular retinoic acid binding proteins (CRABPs), with CRABPs likely directly delivering atRA for metabolism by these CYPs (29, 37). Based on their biochemical characterization, CYP26A1 and CYP26B1 appear to be functionally redundant, although CYP26B1 has a higher affinity to atRA and a lower capacity as atRA hydroxylase (28), potentially pointing to a different biological significance. A key difference between the two main CYP26 enzymes is their apparent distinct tissue expression that may point to different roles of these enzymes. CYP26A1 is the main liver CYP26 enzyme, and CYP26B1 appears to be expressed in extrahepatic tissues (28, 33). Overall, the in vivo role of each of these CYP26 enzymes in atRA clearance will depend on the expression levels, the presence of binding proteins in the specific cells, and the concentrations of atRA at the site of metabolism. Therefore, the relative role of the enzymes in atRA homeostasis is difficult to predict.
The importance of Cyp26a1 and Cyp26b1 during embryonic development and body patterning is unequivocally established, and both of these enzymes are critical for correct embryonic development in a plethora of developing organs (1). However, the consequences of specific loss of activity of the CYP26 enzymes in adult animals and humans are less well-understood. At present, no homozygous complete loss-of-function mutations of CYP26A1 or CYP26B1 enzymes in humans have been characterized, presumably as such mutations would be lethal during embryonic or fetal development. However, predicted diminished function mutations and heterozygous microdeletions of CYP26 enzymes in humans have been shown to result in various phenotypes (38–41). Although it is likely the phenotypes observed in these cases are largely due to defects in fetal development, some phenotypic differences were also observed in these individuals in post-natal life. Haploinsufficiency of CYP26B1 led to a lack of induction of this CYP upon atRA treatment in cultured lymphoblastoid cells when compared with control (41). The microdeletion of CYP26A1, in contrast, resulted in accelerated bone maturation and increased circulating 13-cis-RA concentrations, but did not affect circulating atRA concentrations compared with unaffected family members or age-matched controls (40). In mice, simultaneous inhibition of both Cyp26a1 and Cyp26b1 has been shown to result in tissue-specific but temporary increase in atRA concentrations and retinoid signaling, accompanied by a significant induction of Cyp26a1 in the liver (19). The temporal changes were considered to be due to the short half-life of talarozole, the inhibitor used. Based on these findings, we hypothesized that global deletion of Cyp26a1 in mice after birth would result in increased atRA concentrations in tissues and atRA related adverse effects.
The findings from this study show that the phenotype of global knockout mice relating to atRA elimination during embryonic and fetal development does not translate to adult animals, as no apparent retinoid toxicity or lethal phenotype was observed in the Cyp26a1−/− mice. Surprisingly, the liver atRA concentrations and mRNA expression of retinoid-associated genes were unaltered in the Cyp26a1−/− mice. These findings suggest that in fact Cyp26b1 is the key enzyme for endogenous atRA clearance in adult mice. This hypothesis is supported by our simulations of endogenous atRA homeostasis in which Cyp26b1 was assigned as the main contributor to atRA clearance and as the high-affinity, low-capacity enzyme in extrahepatic tissues. Cyp26a1 was assigned as the low-affinity, high-capacity enzyme in the liver, consistent with the biochemical characterization of the human CYP26 enzymes. Our kinetic simulations show that despite Cyp26a1 being the sole Cyp26 enzyme in the liver, the predicted change in endogenous atRA concentrations in the liver is too small to detect experimentally (<25%). The findings of this study are also consistent with our previous analysis that the pharmacological effects of talarozole, a pan-CYP26 inhibitor, are mainly due to the more potent inhibition of Cyp26b1 by talarozole (19). Previous cell-type–specific knockout models of Cyp26 enzymes also support our conclusion that extrahepatic Cyp26b1 is quantitatively more important in regulating endogenous atRA homeostasis than Cyp26a1. T-cell–specific knockout of Cyp26b1 showed that Cyp26b1 is required for T-cell–mediated colitis but that T-cell development was unaffected by Cyp26b1 knockout (42). However, Cyp26b1 knockout T-cells were more sensitive to retinoid-initiated differentiation in vitro (42). In the testis, deletion of Cyp26a1 in germ cells or Sertoli cells had no effect on spermatogenesis, whereas deletion of Cyp26b1 in Sertoli cells resulted in mild spermatogenic defects (43). Our findings of lack of effect of global Cyp26a1 deletion on testis morphology are consistent with these previous findings.
The observed minor phenotypes of the specific effects of Cyp26a1 knockout on myeloid skewing of hematopoiesis are in agreement with the expected role of atRA in the bone marrow and the presumed role of Cyp26 enzymes in maintaining the niche in the bone marrow. Current knowledge suggests that knockout of Cyp26a1 will increase endogenous atRA concentrations in the bone marrow niche and result in skewed hematopoiesis toward myelopoiesis and away from erythropoiesis (44, 45). Indeed, our studies clearly demonstrate a decrease in erythroid lineage and an increase in the frequency of myeloid cells. The mild increase in bone marrow cellularity and peripheral blood myeloid cells with decreased peripheral blood T-cells is also expected given the proliferative role that retinoids have in mouse hematopoiesis.
Although we conclude that Cyp26a1 plays only a minor role in regulating endogenous atRA homeostasis, our results demonstrate that Cyp26a1 is the key enzyme in clearing exogenous atRA. This is evident from the 3-fold increase in atRA AUC in the Cyp26a1−/− mice compared with controls after i.p. administration of atRA. The different role of Cyp26a1 in endogenous and xenobiotic atRA clearance was unexpected but does shed light to the functional differences between Cyp26a1 and Cyp26b1, and the requirement of multiple atRA clearing Cyp enzymes in all chordates. Our simulations suggest that the high affinity of atRA to Cyp26b1, amplified by the substrate delivery of atRA to Cyp26b1 by CRABPs, result in saturation of this enzyme upon administration of exogenous atRA and therefore diminished contribution of this enzyme to atRA clearance. However, the higher Km value of atRA toward Cyp26a1 allows Cyp26a1 to clear exogenous atRA. This role in clearing exogenous atRA may extend to a role of Cyp26a1 in protecting the liver from toxic atRA levels under conditions of excessive vitamin A intake and subsequent atRA formation from the diet. We can speculate that the high affinity of atRA to Cyp26b1 is necessary for the appropriate function of this enzyme in homeostatic processes. If the saturation of Cyp26b1 following exogenous atRA administration translates to humans, this may be a critical component and prerequisite for explaining the activity of therapeutically administered atRA.
This study clearly demonstrated a sex difference in circulating retinol, RBP4, and liver atRA concentrations in mice. It is possible that the sex difference is a mouse-specific phenomenon, and further studies are needed to determine whether sex differences also exist in humans. The biological consequences of the sex difference are not clear, but these findings suggest that attention should be devoted to understanding sex differences in retinoid concentrations. Based on the observed nearly 2-fold difference in liver atRA concentrations between male and female mice, the data here suggest that the sexes should be analyzed separately for liver and serum retinoid homeostasis or that studies should be conducted only in one sex. In addition, the fact that the higher liver atRA concentrations in male mice corresponded with higher circulating retinol and RBP4 concentrations in male mice may indicate that liver atRA concentrations regulate RBP4-retinol excretion from the liver.
In conclusion, this study shows that Cyp26a1 is the critical enzyme in clearing exogenous atRA, but it has a minor role in modulating endogenous retinoid homeostasis in postnatal life. The results show that deletion of Cyp26a1 in the bone marrow niche, likely due to decreased atRA depletion, results in decreased erythroid lineage and decreased peripheral T-cells, hallmarks of retinoid-induced myeloid bias. Finally, the data shown here provide an explanation to the need of multiple Cyp26 enzymes as the analysis suggests Cyp26a1 is the high-capacity low-affinity Cyp26 enzyme responsible for exogenous atRA clearance, whereas Cyp26b1 is the high-affinity, low-capacity enzyme regulating endogenous atRA concentrations.
Experimental procedures
Ethics statement, animal care and breeding
All the procedures involving mice prior to the commencement of these studies were approved by either Washington State University Committee on the Use and Care of Animals or the Institutional Animal Care and Use Committee at Johns Hopkins Animal Facility. The mouse colonies were maintained in a temperature- and humidity-controlled environment with food and water provided ad libitum. The following mouse strains were used in these studies: tamoxifen-inducible Cre recombinase line B6.129-Gt(ROSA)26Sortm1(cre/ERT2)TyjlJ (stock number 008463), purchased from JAX Mice (Bar Harbor, ME), and the Cyp26a1 floxed line (25) kindly donated by Dr. Martin Petkovitch (43). Cyp26a1 floxed mice were bred to ROSA26CreERT mice using a standard Cre–Lox breeding scheme, and the following three genotypes were analyzed: 1) Cyp26a1 WT (controls, called Cyp26a1+/+ after tamoxifen injections); 2) Cyp26a1 heterozygous for flox (heterozygous, called Cyp26a1+/− after tamoxifen injections); and 3) Cyp26a1 homozygous for flox (knockouts, called Cyp26a1−/− after tamoxifen injections). All three groups of mice were homozygous for ROSA26CreRT and were treated with tamoxifen to induce expression of Cre recombinase as described below.
Animals were genotyped and knockouts assessed by PCR using the following primer sets: for Tam-Cre forward primer AAAGTCGCTCTGAGTTGTTAT and reverse primer GGAGCGGGAGAAATGGATATG; for Cyp26a1 Flox forward primer ACATTGCAGATGGTGCTTCA and reverse primer CGTATTTCCTGCGCTTCATC. Tamoxifen (Sigma) was dissolved in 10% ethanol and 90% sesame oil at a concentration of 20 mg/ml, and the solution was wrapped in foil to protect it from light. Mice from all three genotype groups were i.p. injected with 80 mg/kg tamoxifen once a day for 5 consecutive days beginning at 21 days post-partum (dpp) for the juvenile induction studies or 60 dpp for the adult induction studies. The mice were fed LabDiet 5K67 containing 1.5 ppm of carotene and 20 IU/g of vitamin A. The animals were then left to recover for 8 weeks after the final tamoxifen injection before being euthanized for tissue collection, except for the mice used for blood and bone marrow flow cytometry. These mice were injected with tamoxifen at 4 weeks of age and then allowed to recover for 5 weeks after tamoxifen injections before euthanasia for blood and bone marrow collection. These mice and the mice used for long-term follow-up studies were fed a Teklad Global 18% Protein-extruded rodent diet (Teklad Diets, Madison WI) containing 30 IU/g of vitamin A. An additional cohort of mice were injected with tamoxifen at 4 weeks of age and followed for 1 year for development of any adverse effects. Animal health was monitored via assessment of weight and body condition twice a week after tamoxifen injection. For assessment of hematopoiesis, peripheral blood counts were performed regularly. Animals were euthanized by CO2 asphyxiation followed by cervical dislocation. Bone marrow (BM) mononuclear cells were obtained upon flushing femurs with 1 ml of PBS. Blood was collected by cardiac puncture immediately following CO2 asphyxiation and cervical dislocation.
Histological analyses
For all histological analyses, mice were euthanized and subjected to routine necropsy (46). For tissues other than the testes, mouse tissues were immersed in 10% neutral-buffered formalin for 48 h and then stored in 70% ethanol until routine processing in which they were embedded in paraffin, cut into 4–5 μm sections, and hematoxylin and eosin (H&E)–stained. Testes were immersed in Bouins fixative overnight and then washed in a graded series of ethanol before being embedded and sectioned and stained routinely with H&E. To characterize the impact of Cyp26a1 knockout in juvenile animals, three Cyp26a1−/− mice (1 male and 2 females), three Cyp26a1+/− mice (2 males and 1 female), and three Cyp26a1+/+ mice (1 male and 2 females) at 12 weeks of age (about 3 months) were examined histologically following tamoxifen administration at 21 dpp of age (juveniles). To study the impact of induction of Cyp26a1 knockout in adulthood, five Cyp26a1−/− mice (3 males and 2 females), four Cyp26a1+/− (2 males and 2 females), and four Cyp26a1+/+ mice (2 males and 2 females) at 4 months of age were examined histologically following tamoxifen administration at 60 dpp of age (adults). Tissues examined included the following: decalcified cross-section of the skull, including brain; lung; thymus; heart; kidney; liver; pancreas and mesentery; spleen; reproductive tract; bladder and gastrointestinal tract. Slides were read by a board-certified veterinary pathologist who was blinded to the genotype and treatment status of the adult-induction mice and the Cyp26a1+/− juvenile-induction mice. Cyp26a1+/+ and Cyp26a1−/− juvenile-induction mice were read in an unblinded fashion. Representative images were taken using NIS-Elements BR 3.2 64-bit and plated in Adobe Photoshop Elements. Image white balance, brightness, and contrast were adjusted using auto-corrections applied to the entire image. Original magnification is stated.
Flow cytometry and CFU cell (CFU-C) assays
Peripheral blood was obtained from the tail vein, and red blood cells were lysed using red blood cell lysing buffer (Sigma). The cells were then centrifuged at 400 × g for 5 min, and supernatant was removed. The remaining white blood cells were stained using phycoerythrin-conjugated anti-mouse–CD3, allophycocyanin, anti-mouse–CD45R/B220, Alexa488–anti-mouse–Ly-6G/Gr-1, and fluorescein (FITC)–anti-mouse–CD45 (BioLegend, San Diego). Upon staining, cells were resuspended in running buffer containing 4 μg/ml 7-aminoactinomycin D (Invitrogen) and analyzed by flow cytometry using BD LSRII flow cytometer and BDFACSDiva software (BD Biosciences).
BM progenitor assays in methylcellulose-based medium with recombinant cytokines were performed as described previously (45). Briefly, 25,000 BM mononuclear cells were resuspended in MethoCultTM GFM3434 (StemCell Technologies, Vancouver, Canada). Cells were plated in triplicate, incubated at 37 °C, and scored for the presence of hematopoietic colonies 14 days later. CFUs were classified as CFU-erythroid (CFU-E), CFU granulocyte and macrophage (CFU-GM), and common myeloid progenitors, CFU mix of both erythroid and myeloid (CFU-Mix), according to microscopic characteristics.
Analysis of mRNA expression
Total RNA was extracted from mouse livers using TRI Reagent (Invitrogen). The concentration and purity of RNA were determined by Nanodrop 2000c spectrophotometer (Thermo Fisher Scientific, Waltham, MA). Total mRNA was reverse-transcribed into cDNA using TaqMan reverse transcription reagent kit (Applied Biosystems, Foster City, CA). To quantify mRNA, real-time PCR was performed using a StepOnePlus real-time PCR instrument (Applied Biosystems) with TaqMan gene expression master mix, probes, and primers as described previously (32). The mRNAs of Cyp26a1, Cyp26b1, Cyp26c1, Aldh1a1, Aldh1a2, Lrat, Rdh1, Rdh11, Rarβ, and Rarγ were measured. β-Actin was used as the housekeeping gene. The following primers were purchased from Applied Biosystems (Foster City, CA): Cyp26a1 (Hs00175627_m1); Cyp26b1 (Mm00558507_m1); Cyp26c1 (Mm03412454_m1); Aldh1a1 (Mm00657317_m1); Aldh1a2 (Mm00501306_m1); Rdh1 (Mm00650636); Rdh11 (Mm00458129_m1); Lrat (Mm00469972_m1); Rarβ (Mm01319677_m1); Rarγ (Mm00441091); and β-actin (Mm00607939_s1). The relative mRNA expression level normalized to the housekeeping gene was calculated using 2−ΔΔCt method (ΔΔCt = ΔCt, individual − AvgΔCt, control, female) (47).
Quantification of atRA, retinol, retinyl esters, and RBP4 in mouse serum and tissues
ROL, RE, and atRA concentrations were measured in the serum, liver, spleen, and testes of the mice from the three different experimental groups. 13-cis-RA was also monitored in all samples but was not detected in any of the mouse tissues and was detectable but below quantifiable levels in mouse serum. Oxidized metabolites of RA were not measured in any tissues due to the recent finding that they are either not quantifiable or not detectable in mouse liver (22). atRA and 13-cis-RA were purchased from MilliporeSigma. atRA-d5, 13-cis-RA-d5, all-trans-retinyl palmitate, and retinol were purchased from Toronto Research Chemicals (North York, Ontario, Canada) or Sigma. All-trans-retinyl palmitate-d4 (RP-d4) and all-trans-retinol–d8 (ROL-d8) were purchased from Cambridge Isotopes (Andover, MA). The sample preparation was done under a yellow light to prevent retinoid isomerization and degradation. Mouse tissues (liver 100–120 mg, testis 50–80 mg, or the whole spleen) were first homogenized on ice with 0.9% saline in a 5 to 1 ratio of saline to tissue weight. Mouse serum (100 μl) was diluted with 5× volume of saline. To quantify RE and ROL in mouse liver, liver homogenates (15 μl) were transferred into 1.7-ml Eppendorf tubes and were further diluted with 185 μl of 0.9% saline. After a 20-μl internal standard (IS) mixture (10 μm ROL-d8 and 100 μm RP-d4) was added, samples were kept on ice for 10 min, and then tissue proteins were precipitated by the addition of ice-cold acetonitrile (780 μl) followed by 30-min centrifugation at 18,000 × g at 4 °C. The supernatant was transferred to amber vials for ROL measurement, and 100 μl of supernatant was further diluted with 900 μl of acetonitrile for RE measurement.
A liquid–liquid extraction, adapted from published procedures, was used to measure atRA in mouse livers (17, 22). Briefly, liver homogenates were transferred into a glass tube and IS (5 μl of 2 μm atRA-d5) was added. Acetonitrile with 1% formic acid (2 ml) was added followed by vortexing and the addition of 10 ml of hexanes. The sample was centrifuged at 300 × g for 2 min to facilitate phase separation. The top hexane layer containing atRA was transferred to another glass tube and evaporated under N2 flow. The dry residues were resuspended in 100 μl of 60% acetonitrile.
To measure serum and spleen RE, ROL, and atRA, diluted mouse serum or spleen homogenates were added to 10 μl of IS mixture containing 0.5 μm atRA-d5, 4 μm ROL-d8, and 4 μm RP-d4. Then, 2 ml of 25 mm KOH dissolved in ethanol was added to each sample; the sample vortexed, and 10 ml of hexanes was added. The top organic layer, which contained ROL and RE, was transferred to a new glass tube, and 120 μl of 4 m HCl was added to the aqueous layer followed by another 10 ml of hexanes. The top organic layer containing atRA was transferred to a new glass tube. Glass tubes with a hexane layer were dried down under N2 flow. Residues containing ROL and RP were reconstituted in 200 μl of ACN, whereas those containing atRA were resuspended in 100 μl 60% ACN.
To measure RE, ROL, and atRA in mouse testis, testis homogenates (120 μl) were transferred into a 1.7-ml Eppendorf tube followed by the addition of 8 μl of IS mixture containing 0.5 μm atRA-d5, 4 μm ROL-d8, and 4 μm RP-d4. After gentle vortexing, 240 μl of ice-cold acetonitrile was added, and the mixture was vortexed and left on ice for 10 min. Then, the sample was centrifuged at 18,000 × g at 4 °C for 30 min. The supernatant containing atRA, ROL, and RE was transferred into an amber MS vial for further LC-MS/MS analysis.
All retinoid samples, except spleen samples, were analyzed using AB Sciex 6500 QTRAP mass spectrometer (AB Sciex LLC; Framingham, MA) coupled with Shimadzu UFLC XR DGU-20A5 (Shimadzu Corp.; Kyoto, Japan). Spleen atRA samples were analyzed using AB Sciex 5500 QTRAP mass spectrometer coupled with an Agilent 1290 UHPLC (Agilent Technologies; Santa Clara, CA). The LC-MS/MS method used for measuring atRA in spleen was the same as described previously for atRA measurement in the small intestine (35). Methods used for measuring ROL and RP in all tissues and atRA in serum, liver, and testis were based on published LC-MS/MS methods (22, 35) with minor modifications. The chromatographic conditions, mobile phase, and gradient were as described previously for all retinoids (22), except that to quantify atRA in serum, liver, and testis, the column temperature was set at 33 °C. Multiple reaction monitoring (MRM) transitions selected to monitor analytes included m/z 269 > 93, 95 (ROL and RE); m/z 277 > 98, 102 (ROL-d8); and m/z 273 > 94, 98 (RP-d4). MS parameters were set as the following: CUR 50 p.s.i., transmission EM 350 °C, GS1 and GS2 70; NC 3 μA; CAD −2 p.s.i., DP 35 V; EP 4 V; CE 30 V; and CXP 10 V. MRM transitions of m/z 301 > 205 and m/z 306 > 208 were used to monitor and quantify atRA, 13-cis-RA and atRA-d5, respectively. MS/MS/MS (MS3) scans were collected in parallel for qualitative confirmation of analyte identity. MS parameters were set as listed above except GS1 50 p.s.i. and CE 17 V. In MS3 mode, product ions [M + H]+ derived from the second precursor ion of the MRM transition (m/z 301 > 205 > 109–220 Da) were collected as described previously (22). All solvents used in LC-MS/MS analysis were Optima LC-MS grade purchased from Thermo Fisher Scientific (Pittsburgh, PA). Standard curves were constructed using blank human serum (DC MASS SPECT GOLD MSG4000) from Golden West diagnostics (Temecula, CA) as described previously (22). Analyst software version 1.6.3 was used for data analysis.
Mouse serum RBP4 concentrations were quantified using mouse RBP4 ELISA kit (Abcam; Cambridge, UK) according to the manufacturer's instructions. Briefly, serum samples were diluted with the sample diluent buffer to make a 1:200,000 dilution. Diluted serum and calibration standards were incubated with antibody mixture in the ELISA microplate for 1 h with shaking at 4,000 rpm at room temperature. After the plate was washed three times with washing buffer, 3,3′,5,5′-tetramethylbenzidine substrate was added and incubated for 10 min followed by the addition of the stop solution. Absorbance was measured at 450 nm, and RBP4 concentrations were calculated using the calibration curve provided by the kit.
atRA pharmacokinetic studies
Female Cyp26a1−/− and Cyp26a1+/+ mice were weaned at 3 weeks of age, and Cyp26a1 knockout was induced at 4 weeks of age with tamoxifen injections as described above. At 30 weeks post-tamoxifen induction, one dose of atRA was administered i.p. at 10 mg/kg. Blood was collected from the mice at 0, 0.5, 1, 2, and 4 h, and plasma was separated. At the end of the study, mice were euthanized, and bone marrow cells and liver tissue were collected. atRA concentrations were analyzed by LC-MS/MS as follows. Mouse plasma (50 μl) and 5 μl of 1 μm atRA-d5 were added to a 1.5-ml Eppendorf tube followed by the addition of 75 μl of ice-cold acetonitrile. After vortexing, the samples were centrifuged at 18,000 × g at 4 °C for 30 min. The supernatant was transferred to an MS vial for LC-MS/MS analysis as described above. Samples were analyzed with the method described above for atRA measurement in mouse serum. Plasma concentration of atRA was plotted as a function of time. The pharmacokinetic analysis was performed using Phoenix (St. Louis, MO) and standard noncompartmental analysis. Maximum concentration (Cmax), area under the plasma concentration-time curve from time 0 to infinity (AUC0 → ∞), clearance (CL), and half-life (t½) values were calculated using standard noncompartmental analysis.
Pharmacokinetic modeling
To simulate the effect of elimination of Cyp26a1 activity on endogenous and exogenous atRA concentrations, a semi-physiologically-based model of atRA disposition was developed in using MATLAB and Simulink platform (R2018a; MathWorks, Natick, MA). The model structure and relevant model parameters are shown in Fig 10 and Table S1. Overall, the model includes four major compartments to describe the central circulation system, fast-distributing organs (e.g. visceral organs), slow-distributing tissues (e.g. skin, adipose, bone, and muscle), and liver, with all tissue/organ compartments assumed to be perfusion rate-limited. Endogenous formation of atRA was modeled in the slow-distributing tissue compartment and the liver compartment as a constant rate. Clearance of atRA was modeled in the slow-distributing tissue compartment as mediated by Cyp26b1 and in the liver compartment as mediated by Cyp26a1. The activity of both Cyp26a1 and Cyp26b1 is modeled to be saturable when atRA concentration exceeds their Km values. The intraperitoneal single dose was introduced to the liver compartment by a first-order absorption process governed by ka. When atRA was only formed endogenously, simulation was run until steady state with and without hepatic clearance mediated by Cyp26a1 to explore the effect of elimination of Cyp26a1 activity on endogenous atRA concentrations. When atRA was introduced by the intraperitoneal exogenous administration, simulation was run until 100 h with and without hepatic clearance mediated by Cyp26a1 to explore the effect of elimination of Cyp26a1 activity on exogenous atRA exposure.
Statistical analyses
The impact of genotype, age, and sex on body weight gain in the experimental groups was tested by a multivariate linear mixed effect analysis using R (R Foundation for Statistical Computing, Vienna, Austria). The multivariate mixed effect model treats Cyp26a1 gene status, mouse sex, and time as main effects, and incorporates the dependence of body weight measures from a given mouse with time. The model also accounts for missing data and intrinsic variability within an animal that originates from random sampling. The model tests for the impact of sex, genotype, and age on mouse body weight and enables statistical analysis of inference of Cyp26a1 gene status and mouse sex on the rate of mouse body weight gain (slopes) and the body weight at baseline (intercepts).
The multivariate linear mixed effect model was as follows: body weight (kg) = a1 + b1 × time + b2 × gene 1 + b3 × gene 2 + b4 × sex + c1 × time × gene1 + c2 × time × gene 2 + c3 × sex × gene1 + c4 × sex × gene 2 + c5 × sex × time + c6 × sex × time × gene1 + c7 × sex × time × gene 2, where time is treated as a continuous variable with each value indicating the number of days after tamoxifen treatment; gene 1 and gene 2 are binary variables for the genotype of the individual animals (Cyp26a1+/+, Cyp26a1+/−, or Cyp26a1−/−); sex is a binary variable for male (1) or female (0). Each coefficient (b1–c7) was tested to evaluate whether its value is statistically significantly different from 0. The inference test of the linear combination coefficient was conducted when significance of the sum of two coefficients was queried. A nominal value of 0.05 was considered significant with a Bonferroni correction for multiple comparisons.
Significant differences of mRNA expression and retinoid concentrations in serum and tissues among the experimental groups were analyzed using two-way ANOVA with sex and knockout type as two independent variables. Significant differences of retinoid concentrations in spleen and testis were analyzed using one-way ANOVA as sex was not included as a biological variable for these organs. p values less than 0.05 were considered significant. Flow cytometry and CFU values are described using dispersion methods. Data are presented as mean ± S.D. Groups were compared using two-tailed unpaired Student's t test, and a p value of 0.05 was considered statistically significant.
Author contributions
G. Z., J. M. S., T. T., W. H., and J. L. data curation; G. Z., L. P., W. H., and N. I. formal analysis; G. Z., C. H., G. G., and N. I. supervision; G. Z., C. H., J. M. S., L. P., T. T., L. C. C., J. L., and G. G. investigation; G. Z., J. M. S., L. P., T. T., W. H., L. C. C., and J. L. methodology; G. Z., C. H., J. M. S., L. P., W. H., G. G., and N. I. writing-original draft; G. Z., C. H., J. M. S., L. P., G. G., and N. I. writing-review and editing; C. H., G. G., and N. I. conceptualization; C. H., J. M. S., T. T., L. C. C., J. L., and N. I. resources; C. H., G. G., and N. I. funding acquisition; J. M. S. visualization; W. H. software; G. G. and N. I. project administration.
Supplementary Material
Acknowledgments
We thank Dr. Jay Kirkwood for help in methods development for retinoid bioanalysis and Nathan Alade, Natalie Tjota, and Huaqing Xi for providing help in processing mouse tissues for retinoid measurements.
This work was supported by National Institutes of Health Grants R01GM111772 (to N. I. and C. H.) and K08HL127269 (to G. G.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Figs. S1–S5 and Table S1.
- atRA
- all-trans-retinoic acid
- CFU
- colony-forming unit
- RE
- retinyl ester
- ROL
- retinol
- ROL-d8
- all-trans-retinol–d8
- RP-d4
- all-trans-retinyl palmitate-d4
- ANOVA
- analysis of variance
- BM
- bone marrow
- MRM
- multiple reaction monitoring
- CRABP
- cellular retinoic acid–binding protein
- AUC
- area under the curve
- RP-d4
- all-trans-retinyl palmitate-d4
- IS
- internal standard
- ACN
- acetonitrile
- CL
- clearance
- dpp
- day post-partum
- Lrat
- lecithin:retinol acetyltransferase.
References
- 1. Rhinn M., and Dollé P. (2012) Retinoic acid signalling during development. Development 139, 843–858 10.1242/dev.065938 [DOI] [PubMed] [Google Scholar]
- 2. Gudas L. J. (2012) Emerging roles for retinoids in regeneration and differentiation in normal and disease states. Biochim. Biophys. Acta 1821, 213–221 10.1016/j.bbalip.2011.08.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Clagett-Dame M., and Knutson D. (2011) Vitamin A in reproduction and development. Nutrients 3, 385–428 10.3390/nu3040385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kistler A. (1986) Hypervitaminosis A: side-effects of retinoids. Biochem. Soc. Trans. 14, 936–939 10.1042/bst0140936 [DOI] [PubMed] [Google Scholar]
- 5. McCaffery P. J., Adams J., Maden M., and Rosa-Molinar E. (2003) Too much of a good thing: retinoic acid as an endogenous regulator of neural differentiation and exogenous teratogen. Eur. J. Neurosci. 18, 457–472 10.1046/j.1460-9568.2003.02765.x [DOI] [PubMed] [Google Scholar]
- 6. Hall J. A., Grainger J. R., Spencer S. P., and Belkaid Y. (2011) The role of retinoic acid in tolerance and immunity. Immunity 35, 13–22 10.1016/j.immuni.2011.07.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ross A. C., and Zolfaghari R. (2011) Cytochrome P450s in the regulation of cellular retinoic acid metabolism. Annu. Rev. Nutr. 31, 65–87 10.1146/annurev-nutr-072610-145127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hogarth C. A., and Griswold M. D. (2013) Retinoic acid regulation of male meiosis. Curr. Opin. Endocrinol. Diabetes Obes. 20, 217–223 10.1097/MED.0b013e32836067cf [DOI] [PubMed] [Google Scholar]
- 9. Geelen J. A. (1979) Hypervitaminosis A induced teratogenesis. CRC Crit. Rev. Toxicol. 6, 351–375 10.3109/10408447909043651 [DOI] [PubMed] [Google Scholar]
- 10. Duester G. (2008) Retinoic acid synthesis and signaling during early organogenesis. Cell 134, 921–931 10.1016/j.cell.2008.09.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Altucci L., Leibowitz M. D., Ogilvie K. M., de Lera A. R., and Gronemeyer H. (2007) RAR and RXR modulation in cancer and metabolic disease. Nat. Rev. Drug Discov. 6, 793–810 10.1038/nrd2397 [DOI] [PubMed] [Google Scholar]
- 12. Napoli J. L. (2012) Physiological insights into all-trans-retinoic acid biosynthesis. Biochim. Biophys. Acta 1821, 152–167 10.1016/j.bbalip.2011.05.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Shirakami Y., Lee S. A., Clugston R. D., and Blaner W. S. (2012) Hepatic metabolism of retinoids and disease associations. Biochim. Biophys. Acta 1821, 124–136 10.1016/j.bbalip.2011.06.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. O'Byrne S. M., Wongsiriroj N., Libien J., Vogel S., Goldberg I. J., Baehr W., Palczewski K., and Blaner W. S. (2005) Retinoid absorption and storage is impaired in mice lacking lecithin:retinol acyltransferase (LRAT). J. Biol. Chem. 280, 35647–35657 10.1074/jbc.M507924200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kedishvili N. Y. (2013) Enzymology of retinoic acid biosynthesis and degradation. J. Lipid Res. 54, 1744–1760 10.1194/jlr.R037028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Thatcher J. E., and Isoherranen N. (2009) The role of CYP26 enzymes in retinoic acid clearance. Expert Opin. Drug Metab. Toxicol. 5, 875–886 10.1517/17425250903032681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Arnold S. L., Kent T., Hogarth C. A., Griswold M. D., Amory J. K., and Isoherranen N. (2015) Pharmacological inhibition of ALDH1A in mice decreases all-trans retinoic acid concentrations in a tissue specific manner. Biochem. Pharmacol. 95, 177–192 10.1016/j.bcp.2015.03.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Amory J. K., Muller C. H., Shimshoni J. A., Isoherranen N., Paik J., Moreb J. S., Amory D. W. Sr., Evanoff R., Goldstein A. S., and Griswold M. D. (2011) Suppression of spermatogenesis by bisdichloroacetyldiamines is mediated by inhibition of testicular retinoic acid biosynthesis. J. Androl. 32, 111–119 10.2164/jandrol.110.010751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Stevison F., Hogarth C., Tripathy S., Kent T., and Isoherranen N. (2017) Inhibition of the all trans-retinoic acid hydroxylases CYP26A1 and CYP26B1 results in dynamic, tissue-specific changes in endogenous atRA signaling. Drug Metab. Dispos. 45, 846–854 10.1124/dmd.117.075341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Nelson C. H., Buttrick B. R., and Isoherranen N. (2013) Therapeutic potential of the inhibition of the retinoic acid hydroxylases CYP26A1 and CYP26B1 by xenobiotics. Curr. Top. Med. Chem. 13, 1402–1428 10.2174/1568026611313120004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Stoppie P., Borgers M., Borghgraef P., Dillen L., Goossens J., Sanz G., Szel H., Van Hove C., Van Nyen G., Nobels G., Vanden Bossche H., Venet M., Willemsens G., and Van Wauwe J. (2000) R115866 inhibits all-trans-retinoic acid metabolism and exerts retinoidal effects in rodents. J. Pharmacol. Exp. Ther. 293, 304–312 [PubMed] [Google Scholar]
- 22. Zhong G., Kirkwood J., Won K.-J., Tjota N., Jeong H.-Y., and Isoherranen N. (2019) Characterization of vitamin A metabolome in human livers with and without NAFLD. J. Pharmacol. Exp. Ther. 370, 92–103 10.1124/jpet.119.258517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Niederreither K., Abu-Abed S., Schuhbaur B., Petkovich M., Chambon P., and Dollé P. (2002) Genetic evidence that oxidative derivatives of retinoic acid are not involved in retinoid signaling during mouse development. Nat. Genet. 31, 84–88 10.1038/ng876 [DOI] [PubMed] [Google Scholar]
- 24. Beedle M.-T., Stevison F., Zhong G., Topping T., Hogarth C., Isoherranen N., and Griswold M. D. (2019) Sources of all-trans retinal oxidation independent of the aldehyde dehydrogenase 1A isozymes exist in the postnatal testis. Biol. Reprod. 100, 547–560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Abu-Abed S., Dollé P., Metzger D., Beckett B., Chambon P., and Petkovich M. (2001) The retinoic acid-metabolizing enzyme, CYP26A1, is essential for normal hindbrain patterning, vertebral identity, and development of posterior structures. Genes Dev. 15, 226–240 10.1101/gad.855001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Sakai Y., Meno C., Fujii H., Nishino J., Shiratori H., Saijoh Y., Rossant J., and Hamada H. (2001) The retinoic acid-inactivating enzyme CYP26 is essential for establishing an uneven distribution of retinoic acid along the anterio-posterior axis within the mouse embryo. Genes Dev. 15, 213–225 10.1101/gad.851501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lutz J. D., Dixit V., Yeung C. K., Dickmann L. J., Zelter A., Thatcher J. E., Nelson W. L., and Isoherranen N. (2009) Expression and functional characterization of cytochrome P450 26A1, a retinoic acid hydroxylase. Biochem. Pharmacol. 77, 258–268 10.1016/j.bcp.2008.10.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Topletz A. R., Thatcher J. E., Zelter A., Lutz J. D., Tay S., Nelson W. L., and Isoherranen N. (2012) Comparison of the function and expression of CYP26A1 and CYP26B1, the two retinoic acid hydroxylases. Biochem. Pharmacol. 83, 149–163 10.1016/j.bcp.2011.10.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Zhong G., Ortiz D., Zelter A., Nath A., and Isoherranen N. (2018) CYP26C1 is a hydroxylase of multiple active retinoids and interacts with cellular retinoic acid binding proteins. Mol. Pharmacol. 93, 489–503 10.1124/mol.117.111039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Pennimpede T., Cameron D. A., MacLean G. A., Li H., Abu-Abed S., and Petkovich M. (2010) The role of CYP26 enzymes in defining appropriate retinoic acid exposure during embryogenesis. Birth Defects Res. A. Clin. Mol. Teratol. 88, 883–894 10.1002/bdra.20709 [DOI] [PubMed] [Google Scholar]
- 31. Taimi M., Helvig C., Wisniewski J., Ramshaw H., White J., Amad M., Korczak B., and Petkovich M. (2004) A novel human cytochrome P450, CYP26C1, involved in metabolism of 9-cis and all-trans isomers of retinoic acid. J. Biol. Chem. 279, 77–85 10.1074/jbc.M308337200 [DOI] [PubMed] [Google Scholar]
- 32. Tay S., Dickmann L., Dixit V., and Isoherranen N. (2010) A comparison of the roles of peroxisome proliferator-activated receptor and retinoic acid receptor on CYP26 regulation. Mol. Pharmacol. 77, 218–227 10.1124/mol.109.059071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Thatcher J. E., Zelter A., and Isoherranen N. (2010) The relative importance of CYP26A1 in hepatic clearance of all-trans retinoic acid. Biochem. Pharmacol. 80, 903–912 10.1016/j.bcp.2010.05.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Jing J., Nelson C., Paik J., Shirasaka Y., Amory J. K., and Isoherranen N. (2017) Physiologically based pharmacokinetic model of all-trans-retinoic acid with application to cancer populations and drug interactions. J. Pharmacol. Exp. Ther. 361, 246–258 10.1124/jpet.117.240523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Grizotte-Lake M., Zhong G., Duncan K., Kirkwood J., Iyer N., Smolenski I., Isoherranen N., and Vaishnava S. (2018) Commensals suppress intestinal epithelial cell retinoic acid synthesis to regulate interleukin-22 activity and prevent microbial dysbiosis. Immunity 49, 1103–1115.e6 10.1016/j.immuni.2018.11.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Thatcher J. E., Buttrick B., Shaffer S. A., Shimshoni J. A., Goodlett D. R., Nelson W. L., and Isoherranen N. (2011) Substrate specificity and ligand interactions of CYP26A1, the human liver retinoic acid hydroxylase. Mol. Pharmacol. 80, 228–239 10.1124/mol.111.072413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Nelson C. H., Peng C.-C., Lutz J. D., Yeung C. K., Zelter A., and Isoherranen N. (2016) Direct protein-protein interactions and substrate channeling between cellular retinoic acid binding proteins and CYP26B1. FEBS Lett. 590, 2527–2535 10.1002/1873-3468.12303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Laue K., Pogoda H. M., Daniel P. B., van Haeringen A., Alanay Y., von Ameln S., Rachwalski M., Morgan T., Gray M. J., Breuning M. H., Sawyer G. M., Sutherland-Smith A. J., Nikkels P. G., Kubisch C., Bloch W., et al. (2011) Craniosynostosis and multiple skeletal anomalies in humans and zebrafish result from a defect in the localized degradation of retinoic acid. Am. J. Hum. Genet. 89, 595–606 10.1016/j.ajhg.2011.09.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Morton J. E., Frentz S., Morgan T., Sutherland-Smith A. J., and Robertson S. P. (2016) Biallelic mutations in CYP26B1: a differential diagnosis for Pfeiffer and Antley-Bixler syndromes. Am. J. Med. Genet. A 170, 2706–2710 10.1002/ajmg.a.37804 [DOI] [PubMed] [Google Scholar]
- 40. Nilsson O., Isoherranen N., Guo M. H., Lui J. C, Jee Y. H., Guttmann-Bauman I., Acerini C., Lee W., Allikmets R., Yanovski J. A., Dauber A., and Baron J. (2016) Accelerated skeletal maturation in disorders of retinoic acid metabolism: a case report and focused review of the literature. Horm. Metab. Res. 48, 737–744 10.1055/s-0042-114038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Wen J., Lopes F., Soares G., Farrell S. A., Nelson C., Qiao Y., Martell S., Badukke C., Bessa C., Ylstra B., Lewis S., Isoherranen N., Maciel P., and Rajcan-Separovic E. (2013) Phenotypic and functional consequences of haploinsufficiency of genes from exocyst and retinoic acid pathway due to a recurrent microdeletion of 2p13.2. Orphanet J. Rare Dis. 8, 100 10.1186/1750-1172-8-100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Chenery A., Burrows K., Antignano F., Underhill T. M., Petkovich M., and Zaph C. (2013) The retinoic acid-metabolizing enzyme Cyp26b1 regulates CD4 T cell differentiation and function. PLoS ONE 8, e72308 10.1371/journal.pone.0072308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Hogarth C. A., Evans E., Onken J., Kent T., Mitchell D., Petkovich M., and Griswold M. D. (2015) CYP26 enzymes are necessary within the postnatal seminiferous epithelium for normal murine spermatogenesis. Biol. Reprod. 93, 19 10.1095/biolreprod.115.129718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Alonso S., Jones R. J., and Ghiaur G. (2017) Retinoic acid, CYP26, and drug resistance in the stem cell niche. Exp. Hematol. 54, 17–25 10.1016/j.exphem.2017.07.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Ghiaur G., Yegnasubramanian S., Perkins B., Gucwa J. L., Gerber J. M., and Jones R. J. (2013) Regulation of human hematopoietic stem cell self-renewal by the microenvironment's control of retinoic acid signaling. Proc. Natl. Acad. Sci. 110, 16121–16126 10.1073/pnas.1305937110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Treuting P. M., and Snyder J. M. (2015) Mouse necropsy. Curr. Protoc. Mouse Biol. 5, 223–233 10.1002/9780470942390.mo140296 [DOI] [PubMed] [Google Scholar]
- 47. Livak K. J., and Schmittgen T. D. (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2(−ΔΔC(T)) Method. Methods 25, 402–408 10.1006/meth.2001.1262 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.