

Abstract

The catalytic potential of palladium(II) complexes with chelating N-phosphanyl-N-heterocyclic carbenes featuring a saturated imidazolin-2-ylidene or tetrahydropyrimid-2-ylidene ring has been investigated in intermolecular alkyne hydroamination reactions. The complexes were found to be among the most active Pd-based catalysts for these processes and to enable the use of low reaction temperatures (40 °C) and of solventless conditions. The Pd complexes require activation by 2 equiv of a silver salt to remove chlorido ligands from the metal coordination sphere; they can however also be presynthesized in active form, which allows their use under silver-free conditions. The hydroamination reaction was found to efficiently proceed with terminal alkynes and different ring-substituted, primary arylamine substrates.
Introduction
The addition of molecules containing an N–H function across multiple C–C bonds of unsaturated organic molecules, commonly termed hydroamination, is a very useful synthetic tool for the preparation of complex nitrogen-containing compounds, including, for example, nitrogen heterocycles featured in several pharmaceuticals and agrochemicals of technological interest.1,2 This reaction is generally run under organometallic catalysis, as at least one of the reaction partners, the N–H containing substrate or the unsaturated substrate, needs to be activated for reaction in order to overcome the electronic repulsion between the C–C multiple bond and the nitrogen-containing functional group, which are both electron-rich moieties. Whereas hard metal centers preferentially coordinate and activate the N–H group, soft ones, typically late-transition metals, interact instead preferentially with the π system of the unsaturated substrate, depleting its electron density and favoring nucleophilic attack by the N–H moiety;1−3 the lower oxophilicity of late transition metal centers renders them also more tolerant toward functional groups present in the substrates. Although gold-based catalysts for this reaction class have become increasingly popular in the course of the last two decades,2,4 other late transition metals, in particular group 10 metals such as Pd and Pt,5−12 have been intensively investigated as well; palladium species that have been successfully employed as catalyst include simple palladium(II) salts and also palladium(II) complexes, such as the Pd(II) complexes with N-heterocyclic carbene (NHC) ligands successfully tested by several research groups.9−11,13
In recent years, we14,15 and others16,17 have reported on Pd complexes with a novel class of ligands, namely, stable carbene ligands N-functionalized with a phosphanyl moiety (Scheme 1). Such ligands, termed N-phosphanyl-N-heterocyclic carbenes (NHCPs) or N-phosphanyl acyclic diaminocarbenes, have been extensively investigated as catalysts for cross-coupling reactions.14,15 They were found to exhibit a high reactivity toward unreactive aryl chlorides but also to undergo decomposition in the course of the reaction. This instability has been tentatively attributed to the fact that these complexes feature small bite angle chelating ligands, which give rise to stable square planar complexes with palladium(II) but do not fit well in the tetrahedral coordination geometry of palladium(0); as the catalytic cycle for cross-coupling reactions relies on a Pd(0)/Pd(II) manifold and expectedly produces palladium(0) complexes, these complexes will be prone to ligand dissociation and decomposition. Consequently, in the frame of this work, we have extended our investigation on the catalytic performance of palladium(II) complexes with NHCP ligands to the intermolecular hydroamination of alkynes, which is generally considered to be a redox-neutral process under Pd catalysis (Figure 1).2
Scheme 1. General Structure of a Stable Carbene Ligand N-Functionalized with a Phosphanyl Moiety.
Figure 1.

Postulated mechanism for a palladium(II)-catalyzed hydroamination of an alkyne; S = solvent molecules.
Results and Discussion
In order to assess the catalytic potential of complexes with N-phosphanyl carbenes for hydroamination reactions, we have considered complexes 1–2, featuring a saturated imidazolin-2-ylidene or tetrahydropyrimid-2-ylidene ring, which were previously reported and thoroughly characterized by us14 (Scheme 2).
Scheme 2. Synthesis of Complexes 1–2.

Mes = mesityl; BN = benzonitrile.
Catalytic tests were run on the hydroamination of phenylacetylene with an aromatic primary amine such as mesitylamine (Scheme 3), and were aimed at establishing the best reaction conditions, particularly in terms of solvent and employed cocatalyst. Indeed, the palladium(II) complexes have to be activated for reaction upon removal of the chlorido ligands from their coordination sphere, which generates the catalytically competent, formally dicationic complex (Figure 1); no reaction is observed employing the starting palladium(II) complexes alone as catalyst. We purposely chose a notoriously rather difficult amine substrate for hydroamination reactions, such as the rather sterically encumbered mesitylamine, in order to have an engaging test reaction against which to optimize our catalytic system. The reaction produced the Markovnikov hydroamination product exclusively, as it is commonly the case with group 10 and 11 metal catalysts.2
Scheme 3. General Hydroamination Reaction Investigated in This Study.

We performed first some blank experiments in acetonitrile in order to ascertain whether the silver salt cocatalyst could promote the reaction by itself, as there have been reports in the literature that hydroamination can be also catalyzed by Ag salts.18 Under the reaction conditions employed herein, though, the Ag salt alone was a poorly effective catalyst for hydroamination (4% yield), whereas it appeared a bit more active for alkyne hydration by traces of water introduced together with the silver salt; indeed, yields in acetophenone (the product of phenylacetylene hydration) were variable (1–10%) and correlated with the hygroscopicity of the silver salt.
Next, we evaluated the effect of different solvents on the reaction outcome using catalyst 1 (Table 1). As it is apparent from the table, the catalytic system exhibits a fair performance in acetonitrile, whereas a more coordinating solvent such as dimethyl sulfoxide (DMSO) suppresses catalytic activity. Activity in a less-coordinating, less-polar solvent such as toluene is also low, possibly because of poor solubility of the dicationic catalyst in this solvent. Use of a standard ionic liquid such as 1-butyl-2,3-dimethylimidazolium bis(trifluoromethanesulfonyl)imide as solvent enhances the catalytic performance, and even better results can be obtained with no solvent at all. It is interesting to remark that under neat condition the extent of formation of acetophenone (the hydration product) is lower than in organic solvents at comparable conversion, but there is a somewhat higher consumption of alkyne in competitive polymerization reactions, which decreases the reaction selectivity. Concerning the formation of the hydration product, control experiments performed without addition of the amine substrate in acetonitrile/water 20:1 have demonstrated that the activity of the palladium catalysts for the direct phenylacetylene hydration reaction is negligible, and consequently that acetophenone is mainly formed upon hydrolysis of the imine produced upon hydroamination.
Table 1. Solvent Effect on the Performance of Catalyst 1a.
| entry | solvent | alkyne conversion (%) | hydroamination yield (%) | hydration yield (%) |
|---|---|---|---|---|
| 1 | acetonitrile | 70 | 45 | 7 |
| 2 | toluene | 49 | 24 | 3 |
| 3 | ILb | 88 | 62 | 8 |
| 4 | DMSO | 0 | 0 | 0 |
| 5 | neat | >99 | 68 | 3 |
Reaction conditions: 1 mmol mesitylamine, 1 mmol phenylacetylene, 0.01 mmol (1 mol %) catalyst 1, 2 mol % AgPF6, 1 mL solvent, 80 °C, 25 h.
IL = 1-butyl-2,3-dimethylimidazolium bis(trifluoromethanesulfonyl)imide.
Having established that our catalysts allow to carry out an intermolecular alkyne hydroamination reaction under very sustainable conditions (neat conditions, stoichiometric quantities of the two reagents, 1 mol % catalyst), we then turned to the evaluation of the nature and amount of the silver salt cocatalyst on the catalytic performance (Table 2). Reactions were carried out using catalyst 1 in combination with different silver salts, both in acetonitrile (Table 2, entries 1–6) and under neat conditions (Table 2 entries 7–9); under the latter conditions, catalyst 2 was investigated as well (Table 2, entries 10–13).
Table 2. Counteranion Effect on the Catalyst Performance of Catalysts 1 and 2a.
| entry | catalyst | cocatalyst | solvent | conv.b (%) | HAc (%) | HYd (%) |
|---|---|---|---|---|---|---|
| 1 | 1 | AgPF6 | acetonitrile | 70 | 45 | 7 |
| 2 | 1 | AgPF6e | acetonitrile | 71 | 46 | 9 |
| 3 | 1 | AgNTf2 | acetonitrile | 65 | 41 | 5 |
| 4 | 1 | AgOTf | acetonitrile | 86 | 45 | 6 |
| 5 | 1 | AgOTff | acetonitrile | 51 | 31 | 2 |
| 6 | 1 | AgSbF6 | acetonitrile | 84 | 56 | 8 |
| 7 | 1 | AgSbF6 | neat | >99 | 58 | 11 |
| 8 | 1 | AgPF6 | neat | >99 | 68 | 3 |
| 9 | 1 | AgOTf | neat | 97 | 68 | 4 |
| 10 | 2 | AgOTf | neat | 94 | 83 | 3 |
| 11 | 2 | AgOTfg | neat | 95 | 87 | 3 |
| 12 | 2 | AgSbF6 | neat | 95 | 89 | <1 |
| 13 | 2 | AgSbF6g | neat | 97 | 89 | <1 |
Reaction conditions: 1 mmol mesitylamine, 1 mmol phenylacetylene, 0.01 mmol (1 mol %) catalyst, 2 mol % AgX, 1 mL solvent, 80 °C, 25 h.
Alkyne conversion.
Yield in hydroamination product.
Yield in hydration product.
4 mol % AgX.
1 mol % AgX.
3 mol % AgX.
The nature of the employed silver salt was found to have little influence on the yield in addition products (hydroamination + hydroamination-derived hydration), whereas it seems to have an influence on the overall conversion of the alkyne reagent, which is however difficult to rationalize. Thus, the yield in addition products appears to be quite insensitive to the different coordinating ability and Brønstedt basicity of the employed anions. This is important for an understanding of the reaction mechanism, as it suggests that the counteranion is not involved in the rate-determining step of the catalytic process, as it is has been found to be the case, for example, in related gold(I)-catalyzed reactions such as alkyne hydroalkoxylation,19 and hydration.20 It could be argued with reason that it is not so meaningful to draw kinetic inferences from reaction yields measured after a 24 h reaction time, as they could also more simply reflect the lack of influence of the counteranion on the stability of the catalyst. However, as it will be apparent below, additional experiments at a shorter reaction time support the former interpretation. Interestingly, conversions and yields decrease significantly when only 1 equiv of silver salt is employed as cocatalyst (Table 2, entry 5), whereas they remain largely unaffected when an excess of silver salt cocatalyst is present (Table 2, entries 2, 11, and 13). This result highlights once more the importance of catalyst activation by the silver salt as well as the negligible role of silver(I) centers in the catalytic event.
A limitation of the use of these catalysts is represented by the need for the silver salt cocatalyst to activate the Pd complex, which complicates the setup of the reaction and often causes unwanted formal alkyne hydration as side reaction because of the hygroscopic nature of the silver salt. Finally, it has been sometimes claimed that silver can interfere with the catalytic process (the so-called “silver effect”) not only by being catalytically active in its own right but also by assisting the catalytically competent centers.21 In order to evaluate this effect, we set out to preform complexes with less coordinating counteranions. Anion exchange with the corresponding silver salt in acetonitrile serves well for this purpose, and it allows to isolate the complexes as analytically pure compounds (Scheme 4).
Scheme 4. Preparation of Silver-Free Pd Complexes for Hydroamination Catalysis.

The complexes 1–SbF6, 2–OTf, and 2–SbF6 turned out to be fairly stable and could be stored for weeks in the solid state under air without noticeable decomposition. The reactivity of these complexes has been extensively evaluated and the results are reported in Table 3.
Table 3. Catalytic Performance of Preformed Pd Complexes without Halide Ligandsa.
| entry | catalyst | AgX | solvent | conv.b (%) | HAc (%) | HYd (%) |
|---|---|---|---|---|---|---|
| 1 | 1 | AgSbF6 | acetonitrile | 84 | 56 | 8 |
| 2 | 1–SbF6 | acetonitrile | 100 | 50 | 2 | |
| 3 | 1 | AgSbF6 | neat | 100 | 58 | 11 |
| 4 | 1–SbF6 | neat | 90 | 60 | 3 | |
| 5 | 2 | AgOTf | acetonitrile | 60 | 40 | 6 |
| 6 | 2–OTf | acetonitrile | 45 | 28 | 5 | |
| 7 | 2 | AgOTf | neat | 94 | 83 | 3 |
| 8 | 2–OTf | neat | 97 | 64 | 2 | |
| 9 | 2 | AgSbF6 | acetonitrile | 77 | 41 | 18 |
| 10 | 2–SbF6 | acetonitrile | 53 | 29 | 4 | |
| 11 | 2 | AgSbF6 | neat | 95 | 89 | 0 |
| 12 | 2–SbF6 | neat | 100 | 62 | 0 |
Reaction conditions: 1 mmol mesitylamine, 1 mmol phenylacetylene, 0.01 mmol (1 mol %) catalyst, 2 mol % AgX, 80 °C, 25 h.
Alkyne conversion.
Yield in hydroamination product.
Yield in hydration product.
Generally speaking, the catalytic performance of preformed catalysts in terms of yield of hydroamination product is comparable to (in the case of complex 1) or lower than (in the case of complex 2) that of systems formed in situ, which again rules out a possible catalytic role of the introduced Ag(I) beside halide removal from Pd, at least in the case of complex 1. The extent of formation of the formal alkyne hydration product is decreased, as expected, as no hygroscopic silver salt is introduced in the system. Furthermore, the nature of the counteranion seems to play no role, which confirms our findings reported in Table 2 using silver salts. Altogether, use of preactivated, silver-free catalysts appears to bring no significant advantage in the reaction outcome; hence, in the following, catalysts activated in situ by addition of silver salt have been employed.
We then considered the effect of changes in the reaction time and temperature on the outcome of the reaction (Table 4). We first established that 25 h of reaction time was not necessary in order to reach good hydroamination yields, and that respectable amounts of imine product were produced already after only 4 h of reaction time (Table 4, entries 1–5). Furthermore, use of silver triflate or hexafluoroantimonate as catalyst activator has a slight effect on the conversion of alkyne and on the incidence of formal alkyne hydration (probably due to the higher hygroscopicity of the hexafluoroantimonate salt) but not on the yield in the hydroamination product, which remains the same irrespective of the silver salt employed (Table 4, entries 2 and 3); this corroborates our previous assumption about the negligible influence of the nature of the non-coordinating anion on the performance of the catalyst.
Table 4. Effect of the Reaction Temperature and Time on the Catalytic Performance of the Pd Complexesa.
| entry | catalyst | cocatalyst | T (°C) | time (h) | conv.b (%) | HAc (%) | HYd (%) |
|---|---|---|---|---|---|---|---|
| 1 | 1 | AgOTf | 80 | 25 | 97 | 68 | 4 |
| 2 | 1 | AgOTf | 80 | 4 | 79 | 57 | 4 |
| 3 | 1 | AgSbF6 | 80 | 4 | 89 | 58 | 11 |
| 4 | 2 | AgOTf | 80 | 25 | 99 | 83 | 7 |
| 5 | 2 | AgOTf | 80 | 4 | 80 | 66 | 7 |
| 6 | 2 | AgSbF6 | 40 | 4 | 48 | 24 | 4 |
| 7 | 2 | AgSbF6 | 40 | 25 | 97 | 65 | 9 |
| 8 | 1 | AgSbF6 | 40 | 4 | 43 | 41 | 4 |
| 9 | 1 | AgSbF6 | 40 | 25 | 99 | 71 | 8 |
Reaction conditions: 1 mmol mesitylamine, 1 mmol phenylacetylene, 0.01 mmol (1 mol %) catalyst, 2 mol % AgX.
Alkyne conversion.
Yield in hydroamination product.
Yield in hydration product.
Gratifyingly, significant catalytic activity was recorded with both complexes also at 40 °C (Table 4, entries 6–9), which to the best of our knowledge represents the lowest temperature ever reported for an intermolecular alkyne hydroamination reaction promoted by a Pd-based catalyst. Furthermore, comparing the performance of our catalysts with that of other NHC–Pd catalysts previously reported in the literature for the same reaction,9−11 it is evident that the latter require significantly higher temperatures, in the range 100–120 °C, as well as an excess of alkyne (1.2–2 equiv) in order to operate efficiently, which highlights the advantage of using our compounds.
The results of the tests reported in Table 4 allow also to make a comparison between the performances of catalysts 1 and 2. A pictorial view of the same data is reported in the graph in Figure 2. It can be appreciated that at a lower temperature (40 °C) catalyst 1 appears slightly more active than catalyst 2 at the beginning of the reaction and that both catalysts remain active for several hours. On the other hand, at 80 °C catalytic activity decreases markedly for both catalysts after the first hours of reaction, and the decrease is much more pronounced for catalyst 1 compared to catalyst 2. We conclude that catalyst 1 is a more active catalyst for the intermolecular alkyne hydroamination reaction, but at the same time it is also less stable under the reaction conditions, especially at a higher temperature. The reason for this lower stability, which has been recorded also using the same complexes as catalysts for cross-coupling reactions, is in our opinion explained by the more strained nature of the chelate ring in complex 1 compared to complex 2.14
Figure 2.
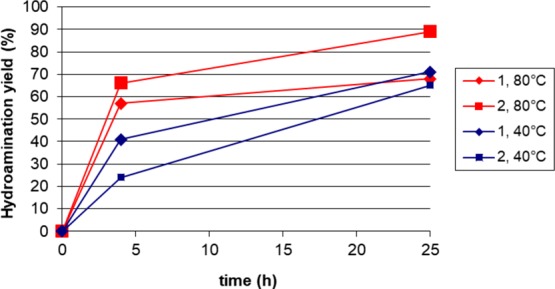
Graphic representation of the catalytic performance of complexes 1 and 2.
Finally, the generality of the intermolecular alkyne hydroamination reaction employing catalysts 1 and 2 has been evaluated with several different alkynes and aromatic amines. The yields in hydroamination product are reported in Table 5; they have been recorded after a 4 h reaction time at 80 °C and are therefore not optimized. It can be appreciated that the reactivity of the two Pd complexes is not limited to phenylacetylene and mesitylamine; other terminal alkynes and differently ring-substituted primary arylamines can be employed. Yields are somewhat lower with the p-substituted anilines compared with mesitylamine, and are found not to significantly depend on the nature of the para-substituent. This suggests that the electronic properties of the amine do not influence the reaction yield to a great extent, whereas steric effects are more important; possibly the steric bulk of the amine promotes faster protonolysis of the vinylpalladium intermediate and/or dissociation of the coordinated enamine product, thereupon accelerating the reaction. On the other hand, the reaction fails altogether when internal alkynes (such as phenylpropyne) or secondary arylamines (such as N-methylaniline) are employed as substrates. Consequently, the substrate scope of these catalysts appears somewhat narrower compared with the Pd complex with an iminophosphane ligand recently reported by us,12 which exhibits notable reactivity also with internal alkynes.
Table 5. Substrate Screening for the Catalytic Performance of the Pd Complexes in Hydroaminationa.

Reaction conditions: 1 mmol alkyne, 1 mmol amine, 0.01 mmol (1 mol %) catalyst, 2 mol % AgOTf, 80 °C, 4 h.
Conclusions
NHCP palladium(II) complexes 1 and 2 have been found to be among the most active Pd-based catalysts reported to date for the intermolecular hydroamination of alkynes. Use of these catalysts enables the use of low reaction temperatures (40 °C) and of solventless conditions, and allows to carry out the reaction with stoichiometric quantities of the reagents, thus rendering the whole process very sustainable. The complexes can be activated in situ by addition of 2 equiv of a silver salt, or prepared in advance in active, silver-free form. Finally, the complexes were able to activate terminal alkynes and different ring-substituted primary arylamine substrates for the reaction. Work in progress aims at extending the application of these complexes as catalysts to other reactions, and to further optimize the catalytic performance of the complexes by acting on the NHCP ligand structure.
Experimental Section
All manipulations of air- and moisture-sensitive compounds were carried out using standard Schlenk techniques or in a glovebox under an atmosphere of dinitrogen. The reagents were purchased from Aldrich as high-purity products and generally used as received. All solvents were purified and dried by standard methods. Nuclear magnetic resonance (NMR) spectra were recorded on Bruker AVANCE spectrometers working at 300 MHz (300.1 MHz for 1H, 75.5 MHz for 13C, and 121.5 MHz for 31P); chemical shifts (δ) are reported in units of ppm relative to the residual solvent signals and to external 85% H3PO4 (for 31P). Elemental analyses were carried out with a Fisons EA 1108 CHNS–O apparatus or with a Carlo Erba analyzer.
Synthesis of Catalysts 1–SbF6, 2–OTf, 2–SbF6
Complex 1 or 2 (50 mg) was placed in a Schlenk tube under an inert atmosphere and dissolved in 2 mL of anhydrous acetonitrile. A solution of 2 equiv silver(I) triflate or hexafluoroantimonate in 2 mL of anhydrous acetonitrile was added in one portion, and the resulting mixture was stirred at room temperature for 30 min. The reaction mixture was filtered over Celite and the resulting clear solution was evaporated to dryness. The product was washed with diethylether and dried under vacuum. Isolated yields were in the range 45–55%.
1–SbF6
1H NMR (CD3CN): δ 1.60 (d, JP–H = 19 Hz, 18H, tBu), 2.26 (s, 6H, Me), 2.32 (s, 3H, Me), 4.12 (t, J = 10 Hz, 2H, CH2), 4.33 (t, J = 10 Hz, 2H, CH2), 7.10 (s, 2H, CHAr). 13C NMR (CD3CN): δ 17.0 (s, CH3), 20.2 (s, CH3), 27.2 (d, JC–P = 4 Hz, CH3), 40.6 (d, J = 11 Hz, CP), 50.7 (d, JC–P = 5 Hz, CH2), 52.8 (d, J = 4 Hz, CH2), 129.7 (s, CH), 135.9 (s, C), 140.8 (s, C), 137.5; carbene carbon not detected. 31P NMR (CD3CN): δ 99.1. Anal. Calcd (%) for C20H33F12N2PPdSb2·CH3CN·H2O: C, 24.94; H, 4.57; N, 3.97. Found: C, 24.71; H, 4.37; N, 4.57.
2–OTf
1H NMR (CD3CN): δ 1.63 (d, J = 19 Hz, 18H, tBu), 2.27 (s, 6H, Me), 2.28 (m, 2H, CH2), 2.33 (s, 3H, Me), 3.67 (m, 2H, CH2), 3.72 (m, 2H, CH2), 7.08 (s, 2H, CH). 13C NMR (CD3CN): δ 17.0 (s, CH3), 20.2 (s, CH3), 20.3 (d, J = 2 Hz, CH2), 27.8 (d, J = 4 Hz, CH3), 40.5 (d, J = 12 Hz, CP), 44.9 (d, J = 6 Hz, CH2), 49.9 (s, CH2), 129.8 (s, CH), 135.0 (s, C), 138.8 (s, C), 140.3 (s, C), 158.2 (d, J = 13 Hz, NCN). 31P NMR (CD3CN): δ 58.3. Anal. Calcd (%) for C23H35F6N2O6PPdS2·5H2O: C, 34.04; H, 5.46; N, 4.76; S, 7.27. Found: C, 33.90; H, 5.18; N, 4.73; S, 7.90.
2–SbF6
1H NMR (CD3CN): δ 1.63 (d, J = 19 Hz, 18H, tBu), 2.26 (s, 6H, Me), 2.29 (m, 2H, CH2), 2.32 (s, 3H, Me), 3.66 (m, 2H, CH2), 3.71 (m, 2H, CH2), 7.07 (s, 2H, CH). 13C NMR (CD3CN): δ 17.0 (s, CH3), 20.2 (s, CH3), 20.4 (d, J = 3 Hz, CH2), 28.1 (d, J = 4 Hz, CH3), 40.5 (d, J = 14 Hz, CP), 44.3 (d, J = 4 Hz, CH2), 49.0 (s, CH2), 129.7 (s, CH), 135.0 (s, C), 139.4 (s, C), 139.7 (s, C), 166.2 (d, J = 16 Hz, NCN). 31P NMR (CD3CN): δ 58.5. Anal. Calcd (%) for C21H35F12N2PPdSb2·CH3CN: C, 28.61; H, 3.97; N, 4.35. Found: C, 28.30; H, 3.33; N, 4.56.
General Procedure for Catalytic Hydroaminations
In a Schlenk tube equipped with a magnetic stirring bar were placed under an inert atmosphere 10 μmol Pd complex and optionally 20–80 μmol silver salt cocatalyst. The tube was degassed and put under an inert atmosphere. Aniline (1.00 mmol), 1.00 mmol alkyne, and optionally 1 mL of dry solvent were then injected into the Schlenk tube. The flask was immediately placed in an oil bath preheated at the reaction temperature and the reaction mixture was vigorously stirred for the given reaction time. Conversions and yields were determined as the average of two runs by 1H NMR. 1,4-Bis-trimethylsilylbenzene (22 mg 0.10 mmol) was dissolved as an internal standard in the reaction mixture and a sample thereof was diluted in CDCl3 for the measurement.
2,4,6-Trimethyl-N-(1-phenylethylidene)aniline
1H NMR (CDCl3): δ 2.03 (s, 6H, CH3), 2.10 (s, 3H, CH3), 2.32 (s, 3H, CH3), 6.90 (s, 2H, ArH), 7.50 (br s, 3H, ArH), 8.05 (d, J = 8 Hz, 2H, ArH).
4-Methoxy-N-(1-phenylethylidene)aniline
1H NMR (CDCl3): δ 2.26 (s, 3H, CH3), 3.82 (s, 3H, CH3), 6.75 (d, J = 9 Hz, 1H, ArH), 6.92 (t, J = 9 Hz, 2H, ArH), 7.10 (m, 1H, ArH), 7.45 (m, 3H, ArH), 7.97 (m, 2H, ArH).
4-Chloro-N-(1-phenylethylidene)aniline
1H NMR (CDCl3): δ 2.24 (s, 3H, CH3), 6.75 (d, J = 8 Hz, 2H, ArH), 7.31 (d, J = 8 Hz, 2H, ArH), 7.47 (d, J = 8 Hz, 3H, ArH), 7.97 (d, J = 8 Hz, 2H, ArH).
N-(1-Phenylethylidene)aniline
1H NMR (CDCl3): δ 2.26 (s, 3H, CH3), 6.83 (m, 2H, ArH), 7.12 (m, 1H, ArH), 7.37 (m, 2H, ArH), 7.48 (m, 3H, Ar), 8.01 (m, 2H, ArH).
2,4,6-Trimethyl-N-(octan-2-ylidene)aniline
1H NMR (CDCl3): δ 0.92 (m, 3H, CH3), 1.16–1.62 (m, 10H, CH2), 1.98 (s, 6H, CH3), 2.19 (s, 3H, CH3), 2.27 (s, 3H, CH3), 2.48 (m 2H, CH2), 6.84 (s, 2H, ArH).
Acknowledgments
Financial support from the University of Padova is gratefully acknowledged (P-DiSC BIRD 2017 UNIPD).
The authors declare no competing financial interest.
References
- Müller T. E.; Hultzsch K. C.; Yus M.; Foubelo F.; Tada M. Hydroamination: direct addition of amines to alkenes and alkynes. Chem. Rev. 2008, 108, 3796–3892. 10.1021/cr0306788. [DOI] [PubMed] [Google Scholar]
- Huang L.; Arndt M.; Gooßen K.; Heydt H.; Gooßen L. J. Late Transition Metal-Catalyzed Hydroamination and Hydroamidation. Chem. Rev. 2015, 115, 2596–2697. 10.1021/cr300389u. [DOI] [PubMed] [Google Scholar]
- Hartwig J. F.Organotransition Metal Chemistry; University Science Books: Sausalito, CA, 2010. [Google Scholar]
- Alyabayev S. B.; Beletskaya I. P. Gold as a catalyst. Part I. Nucleophilic addition to the triple bonds. Russ. Chem. Rev. 2017, 86, 689–749. 10.1070/rcr4727. [DOI] [Google Scholar]
- Kadota I.; Shibuya A.; Lutete L. M.; Yamamoto Y. Palladium/Benzoic Acid Catalyzed Hydroamination of Alkynes. J. Org. Chem. 1999, 64, 4570–4571. 10.1021/jo990498r. [DOI] [PubMed] [Google Scholar]
- Shimada T.; Yamamoto Y. Palladium-Catalyzed Intermolecular Hydroamination of Alkynes: A Dramatic Rate-Enhancement Effect of o-Aminophenol. J. Am. Chem. Soc. 2002, 124, 12670–12671. 10.1021/ja027683y. [DOI] [PubMed] [Google Scholar]
- Shimada T.; Bajracharya G. B.; Yamamoto Y. Aquapalladium Complex: A Stable and Convenient Catalyst for the Intermolecular Hydroamination of Alkynes. Eur. J. Org. Chem. 2005, 59–62. 10.1002/ejoc.200400568. [DOI] [Google Scholar]
- Shaffer A. R.; Schmidt J. A. R. Palladium(II) 3-Iminophosphine Complexes as Intermolecular Hydroamination Catalysts for the Formation of Imines and Enamines. Organometallics 2008, 27, 1259–1266. 10.1021/om701106q. [DOI] [Google Scholar]
- Yuan D.; Tang H.; Xiao L.; Huynh H. V. CSC-pincer versus pseudo-pincer complexes of palladium(II): a comparative study on complexation and catalytic activities of NHC complexes. Dalton Trans. 2011, 40, 8788–8795. 10.1039/c1dt10269a. [DOI] [PubMed] [Google Scholar]
- Chen Q.; Lv L.; Yu M.; Shi Y.; Li Y.; Pang G.; Cao C. Simple, efficient and reusable Pd–NHC catalysts for hydroamination. RSC Adv. 2013, 3, 18359–18366. 10.1039/c3ra42990c. [DOI] [Google Scholar]
- Bernhammer J. C.; Huynh H. V. Benzimidazolin-2-ylidene Complexes of Palladium(II) Featuring a Thioether Moiety: Synthesis, Characterization, Molecular Dynamics, and Catalytic Activities. Organometallics 2014, 33, 1266–1275. 10.1021/om500083r. [DOI] [Google Scholar]
- Marchenko A.; Koidan G.; Hurieva A.; Kostyuk A.; Franco D.; Baron M.; Biffis A. PdII Complexes with N-(Diadamantylphosphanyl)diaminocarbene and Related Ligands: Synthesis and Catalytic Applications in Intermolecular Alkyne Hydroaminations. Eur. J. Inorg. Chem. 2018, 652–658. 10.1002/ejic.201701342. [DOI] [Google Scholar]
- For a recent review on the application of N-heterocyclic carbene ligands in catalysis, seePeris E. Smart N-Heterocyclic Carbene Ligands in Catalysis. Chem. Rev. 2017, 118, 9988–10031. 10.1021/acs.chemrev.6b00695. [DOI] [PubMed] [Google Scholar]
- Marchenko A.; Koidan G.; Hurieva A. N.; Vlasenko Y.; Kostyuk A.; Biffis A. Chelate Palladium(II) Complexes with Saturated N-Phosphanyl-N-Heterocyclic Carbene Ligands: Synthesis and Catalysis. Organometallics 2016, 35, 762–770. 10.1021/acs.organomet.5b01019. [DOI] [Google Scholar]
- Marchenko A.; Koidan G.; Hurieva A.; Vlasenko Y.; Kostyuk A.; Biffis A. Palladium(II) complexes with chelating N-phosphanyl acyclic diaminocarbenes: synthesis, characterization and catalytic performance in Suzuki couplings. Dalton Trans. 2016, 45, 1967–1975. 10.1039/c5dt02250a. [DOI] [PubMed] [Google Scholar]
- Nägele P.; Herrlich U.; Rominger F.; Hofmann P. Transition Metal Complexes of Bulky, Electron-Rich N-Phosphanyl-Substituted N-Heterocyclic Carbenes (NHCP Ligands). Small Bite Angle Four-Membered (κ-C,κ-P) Chelate Structures. Organometallics 2012, 32, 181–191. 10.1021/om300963t. [DOI] [Google Scholar]
- Ai P.; Gourlaouen C.; Danopoulos A. A.; Braunstein P. Novel Di- and Trinuclear Palladium Complexes Supported by N,N′-Diphosphanyl NHC Ligands and N,N′-Diphosphanylimidazolium Palladium, Gold, and Mixed-Metal Copper–Gold Complexes. Inorg. Chem. 2016, 55, 1219–1229. 10.1021/acs.inorgchem.5b02382. [DOI] [PubMed] [Google Scholar]
- Zhang X.; Yang B.; Li G. Z.; Shu X.; Mungra D. C.; Zhu J. Highly Regioselective AgNTf2-Catalyzed Intermolecular Hydroamination of Alkynes with Anilines. Synlett 2012, 23, 622–626. 10.1055/s-0031-1290341. [DOI] [Google Scholar]
- Trinchillo M.; Belanzoni P.; Belpassi L.; Biasiolo L.; Busico V.; D’Amora A.; D’Amore L.; Del Zotto A.; Tarantelli F.; Tuzi A.; Zuccaccia D. Extensive Experimental and Computational Study of Counterion Effect in the Reaction Mechanism of NHC-Gold(I)-Catalyzed Alkoxylation of Alkynes. Organometallics 2016, 35, 641–654. 10.1021/acs.organomet.5b00925. [DOI] [Google Scholar]
- Gatto M.; Belanzoni P.; Belpassi L.; Biasiolo L.; Del Zotto A.; Tarantelli F.; Zuccaccia D. Solvent-, Silver-, and Acid-Free NHC-Au-X Catalyzed Hydration of Alkynes. The Pivotal Role of the Counterion. ACS Catal. 2016, 6, 7363–7376. 10.1021/acscatal.6b01626. [DOI] [Google Scholar]
- Wang D.; Cai R.; Sharma S.; Jirak J.; Thummanapelli S. K.; Akhmedov N. G.; Zhang H.; Liu X.; Petersen J. L.; Shi X. “Silver Effect” in Gold(I) Catalysis: An Overlooked Important Factor. J. Am. Chem. Soc. 2012, 134, 9012–9019. 10.1021/ja303862z. [DOI] [PubMed] [Google Scholar]