

Abstract
Nemaline myopathy (NM) is the most common form of congenital myopathy that results in hypotonia and muscle weakness. This disease is clinically and genetically heterogeneous, but three recently discovered genes in NM encode for members of the Kelch family of proteins. Kelch proteins act as substrate-specific adaptors for Cullin 3 (CUL3) E3 ubiquitin ligase to regulate protein turnover through the ubiquitin-proteasome machinery. Defects in thin filament formation and/or stability are key molecular processes that underlie the disease pathology in NM; however, the role of Kelch proteins in these processes in normal and diseases conditions remains elusive. Here, we describe a role of NM causing Kelch protein, KLHL41, in premyofibil-myofibil transition during skeletal muscle development through a regulation of the thin filament chaperone, nebulin-related anchoring protein (NRAP). KLHL41 binds to the thin filament chaperone NRAP and promotes ubiquitination and subsequent degradation of NRAP, a process that is critical for the formation of mature myofibrils. KLHL41 deficiency results in abnormal accumulation of NRAP in muscle cells. NRAP overexpression in transgenic zebrafish resulted in a severe myopathic phenotype and absence of mature myofibrils demonstrating a role in disease pathology. Reducing Nrap levels in KLHL41 deficient zebrafish rescues the structural and function defects associated with disease pathology. We conclude that defects in KLHL41-mediated ubiquitination of sarcomeric proteins contribute to structural and functional deficits in skeletal muscle. These findings further our understanding of how the sarcomere assembly is regulated by disease-causing factors in vivo, which will be imperative for developing mechanism-based specific therapeutic interventions.
Introduction
Muscle development is a multistep process that involves the specification of myogenic progenitors and myoblasts and subsequent fusion of myoblasts that generates syncytial, multinucleated myotubes (1–3). Muscle cells typically contain dozens of myofibrils, each consisting of many sarcomeres, the smallest functional contractile units of muscle. Consequently, mutations in genes encoding proteins that are critical for sarcomere assembly, and efficient coupling to the excitation-contraction machinery are associated with several forms of myopathies in humans (4–9). Despite a reasonable understanding of the mature sarcomere and myofibrils architecture, the assembly mechanism of protein complexes that leads to myofibril and sarcomere formation during muscle development remains less understood.
The process of sarcomeric assembly is mostly studied during myoblast differentiation or in cardiomyocytes in vitro and has led to several models of sarcomere assembly with a common premise that shows premyofibrils formation begins with the polymerization of actin monomers into thin filaments by Arp2/3 protein complex whose barbed ends (fast growing) are aligned and crossed linked by α-actinins in Z-lines, the borders of sarcomeres (3,10,11). The pointed ends (slow growing) extend toward the M-line, where they interdigitate with thick filaments and form premyofibrils. Mature myofibrils are formed through lateral fusion of premyofibrils with addition of proteins that stabilize the core and structure of developing myofibrils (12). While the role of a few central structural proteins during sarcomere assembly is established, factors and mechanism that regulate the dynamics and transition between different stages of myofibrillogenesis remain elusive. Recent studies have identified mutations in a number of genes encoding non-structural proteins contributing to defects in sarcomeric architecture and muscle weakness in skeletal muscle disorders (13–18). Determining the mechanism of sarcomeric assembly in vivo during vertebrate myogenesis by these factors is crucial for identifying the critical processes that control muscle development, and ultimately for determining how disease-causing variants in these genes contribute to muscle weakness in human myopathies.
Nemaline myopathy (NM) is a rare congenital disorder primarily affecting skeletal muscle function (19–21). Clinically, NM is a heterogeneous group of myopathies of variable severity. The ‘severe’ congenital form of NM presents with reduced or absent spontaneous movements in utero leading to severe contractures or fractures at birth and respiratory insufficiency leading to early mortality. NM is a genetically heterogeneous condition, and mutations in 11 different genes have been identified by us and others that are associated with dominant and/or recessive forms of this disease (4,5,14,15,22–28). While eight genes encode structural components of thin filaments in sarcomeres, the function of newly discovered genes encoding Kelch proteins KBTBD13, KLHL40 and KLHL41 in muscle development and function remains to be elucidated. Recent in vitro studies have shown that these Kelch proteins act as substrate specific adaptors for CUL3-E3 ubiquitin ligases that regulate the stability and turnover of specific target proteins by ubiquitination (29–32). CUL3 ubiquitin ligase requires BTB-Kelch proteins as substrate-specific adaptors to direct the ubiquitination of target proteins. A previous in vitro study has demonstrated that KLHL41-CUL3 interaction is crucial for CUL3-mediated protein ubiquitination of substrate proteins (32).
The protein turnover process is highly dynamic and provides a mechanism for the removal of unwanted proteins or changes in protein isoforms without compromising the structural integrity needed for the myofibril function (33). The proteome fidelity is mediated by the ubiquitin-proteasome system (UPS), which constitutes the central protein turnover machinery in cells. The specificity to UPS in development, diseases and, more importantly, a tissue-specific context is provided by E3 ubiquitin ligases. E3 ubiquitin ligases initially catalyze ubiquitin transfer from an E2-ligase to their target substrates and subsequent polyubiquitination from various linkage-specific E2s (34). One subgroup of E3 ligases are the ubiquitously expressed cullins that do not bind to their substrates directly but rely on an array of adaptor proteins such as Kelch proteins (35,36). Ubiquitination-mediated protein turnover has been the focus of intense investigation in the catabolic processes contributing to muscular atrophies and degeneration (37–40). On the contrary, there is very limited knowledge available about the ubiquitin-dependent machinery during skeletal muscle development and how such machinery regulates different aspects of myogenesis. As mutations in several Kelch protein-encoding genes result in skeletal muscle diseases, an understanding of the role of Kelch proteins in ubiquitination-mediated protein turnover during skeletal muscle development may provide novel therapeutic intervention in NM and related diseases.
Our current work has addressed these crucial points to understand the mechanism of KLHL41-mediated ubiquitination of a thin filament chaperone, nebulin related anchoring protein (NRAP), on regulating the skeletal muscle structure, function and disease pathogenesis of NM. We show that KLHL41 interacts with and destabilizes NRAP, a protein that is critical for premyofibril-myofibril transition. KLHL41 deficiency results in the accumulation of NRAP protein, which leads to myopathy in zebrafish. In KLHL41 deficiency, overexpression of NRAP prevents binding of KLHL40 to nebulin that is required to stabilize thin filaments in mature myofibers. Finally, downregulation of NRAP in KLHL41 deficiency improves disease pathophysiology. These studies highlight the role of KLHL41-regulated ubiquitination in skeletal muscle development and provide new insights on disease pathology in NM.
Results
Identification of in vivo substrates for CUL3-adaptor protein, KLHL41 in skeletal muscle
To identify the molecular interactors of KLHL41 in skeletal muscle and gain insights into pathophysiological mechanism of NM, we performed a yeast two-hybrid screening (Y2H) using the Kelch domain of KLHL41 protein as a bait against a human fetal and adult skeletal muscle library and identified 350 clones coding nebulin (NEB), NRAP, interferon-related developmental regulator 1 and RNA-binding motif protein 4 (Fig. 1A and Supplementary Material, Table S1). NM is primarily a thin filament disease (41). Recent work has shown that KLHL41 interacts with nebulin to stabilize thin filaments (32). However, implications of KLHL41-NRAP interactions in vivo are not known. Therefore, we focused our studies on the role of KLHL41-NRAP in skeletal muscle development and maintenance using nebulin as a positive control. Sequencing of different interacting clones revealed that KLHL41 interacted with nebulin super repeats in NRAP protein. KLHL41 also interacted with nebulin through interactions with different domains: nebulin repeats, nebulin super repeats, C-terminal serine-rich region and SH3 domain (Fig. 1A).
Figure 1.
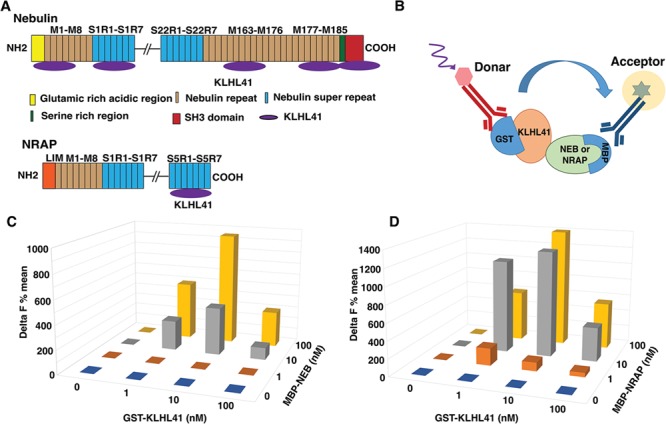
KLHL41 protein interactors in skeletal muscle. (A) Schematics of regions on nebulin and NRAP proteins that interact with KLHL41, identified by Y2H screening. (B) Principle of the HTRF method for detecting interaction between KLHL41-GST (Glutathione S-Transferase) and Nebulin or NRAP-MBP fusion proteins. Antibodies against GST or MBP tags were labeled donor or acceptor, respectively. The HTRF signals are generated on close proximity of each of the labeled antibodies through direct interaction of two proteins. (C) Quantification of the HTRF interaction between different concentrations of KLHL41 and nebulin and (D) NRAP interactor proteins; where KLHL41 is GST tagged and nebulin or NRAP are MBP tagged.
To validate these interactions, we employed homogeneous time-resolved fluorescence (HTRF). The HTRF technology is based on time-resolved fluorescence energy transfer that occurs between long-lived fluorophore europium or terbium as a donor and d2 as acceptors (Fig. 1B). The HTRF interaction between two proteins is quantified by calculating delta F (%) (described in methods) (42). Different interacting fragments of NEB and NRAP were expressed as MBP (Maltose Binding Protein) fusion proteins in BL21 E. coli cells and purified using affinity purification. Three different concentrations of interacting proteins (1, 10 and 100 nM) were tested in the HTRF assay that confirmed the interactions of KLHL41 with NEB and NRAP (Fig.1C and D). Under these conditions, best delta F% signal detected for KLHL41-NEB was 950 (10 nM KLHL41 and 100 nM NEB). The highest delta F% signal detected for KLHL41 (10 nM) and NRAP (100 mm) was 1390 (10 nM KLHL41 and 100 nM NRAP). Higher delta F% values were observed for KLHL41-NRAP interactions compared to KLHL41-NEB interactions suggesting a higher affinity of KLHL41-NRAP interactions in comparison to KLHL41-NEB interactions. In summary, our studies demonstrate that KLHL41 interacts with nebulin and NRAP through direct protein–protein interactions.
KLHL41 regulates the stability of thin-filament chaperone NRAP by ubiquitination
NRAP is expressed in differentiating myotubes at the onset of myoblast fusion and co-localizes with actin to promote myofibril assembly (43). Therefore, to validate the interaction of KLHL41 and NRAP in differentiating myotubes, KLHL41-FLAG and NRAP-V5 were over-expressed in C2C12 myoblasts and 12 h post transfection; myotubes were cultured in the differentiation media for 36 hours. Cell extracts were immunoprecipitated by an antibody against V5 (Fig. 2A) that resulted in co-immunoprecipitation of KLHL41-FLAG with NRAP-V5 but not with control mouse IgG, confirming that KLHL41 interacts with NRAP in myotubes. Kelch proteins interact with protein substrates for ubiquitination through E3 CUL3 ligases (44). Therefore, we hypothesized these protein–protein interactions may be required to regulate ubiquitination of the interacting partners such as NRAP. To investigate the ubiquitnation of NRAP by KLHL41, we overexpressed human KLHL41-FLAG with NRAP-V5 plasmids in C2C12 myotubes in the presence of ubiquitin-HA plasmid. The amounts of KLHL41 plasmid was varied (0–1.0 μg) whereas the amount of NRAP protein was kept constant (2.0 μg; Fig. 2B). 12 h post transfections, cells were grown in the differentiation media and cell lysates were prepared 48 h post transfections. NRAP was immunoprecipitated with a V5 antibody and analyzed by western blot analysis. Western blot analysis demonstrated that ubiquitination of NRAP was increased with an increase in KLHL41 concentration. Concurrently, we observed a significant reduction in total NRAP protein level with an increase in KLHL41 concentration. These results suggest that KLHL41-mediated ubiquitination resulted in the degradation of NRAP in differentiating myotubes, potentially through proteolysis by the proteasome degradation pathway.
Figure 2.
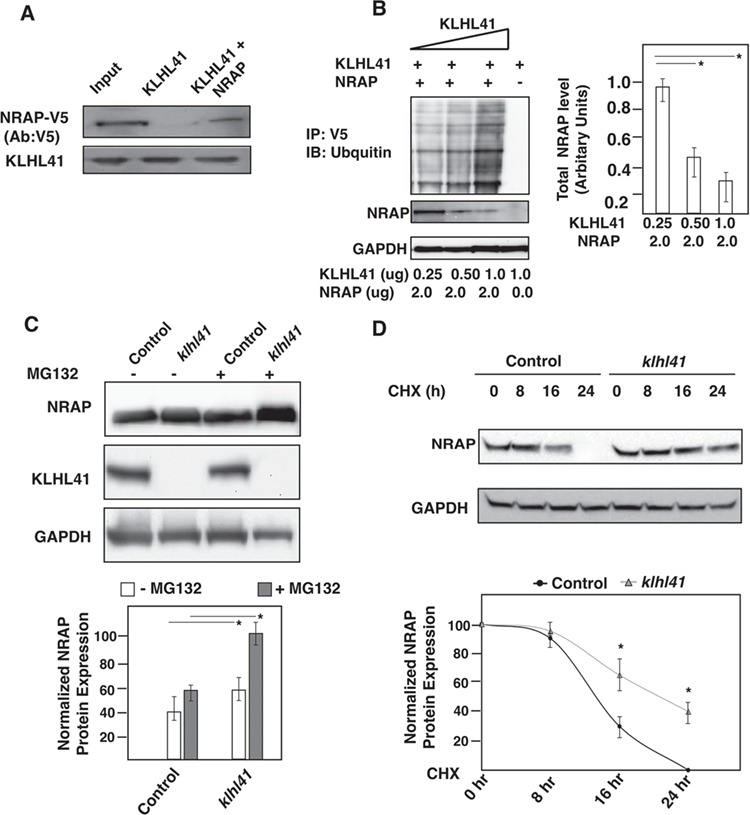
KLHL41-NRAP interaction results in ubiquitination and proteasome-mediated degradation of NRAP. (A) Co-immunoprecipitation in C2C12 cells transfected with KLHL41-FLAG and NRAP-V5 demonstrating interaction of the two proteins in the myogenic context. (B) Overexpression of KLHL41-FLAG and NRAP-V5 in the presence of ubiquitin-HA in C2C12 results in increased ubiquitination and decreased stability of NRAP. (C) Western blot analysis, quantification of NRAP mRNA and protein in control or Klhl41 knockout myotubes treated with MG132. (D) Western blot analysis and quantification of NRAP in control or Klhl41 knockout myoblasts treated with cycloheximide for different time intervals. The NRAP band intensity was normalized to GAPDH and then normalized to t = 0 hr controls (bottom panel). Data are shown as mean ± s.d. and blots are representative of three different experiments (n = 3). P-values were calculated using a two-tailed Mann–Whitney U-test, *P < 0.01.
KLHL41 deficiency results in increased NRAP protein
As KLHL41 is required for the ubiquitination and subsequent degradation of NRAP, we investigated the effect of KLHL41 deficiency on NRAP levels. To study the effect of KLHL41 deficiency on NRAP in a mammalian disease model, we created Klhl41 mouse C2C12 knockout cell lines using CRISPR technology. The effect of KLHL41 on NRAP protein stability was analyzed in control and Klhl41 C2C12 knockout differentiating myotubes (day 2 in the differentiation media). Klhl41 knockout myotubes exhibited increased levels of NRAP protein in comparison to control cells as observed by western blot (Fig. 2C). No changes in Nrap gene transcription were observed by q-PCR, validating that the effect of KLHL41-mediated degradation of NRAP is regulated at the post-translational level. Consistent with the role of ubiquitin-mediated proteasomal degradation, NRAP downregulation was mitigated by proteasome inhibitor, MG132, in control myotubes (Fig. 2C). The role of endogenous KLHL41 on endogenous NRAP protein turnover was further investigated by cycloheximide chase assay in control and Klhl41 knockout differentiated myotubes (day 2 in the differentiation media; Fig. 2D). Control myotubes exhibited increased turnover of NRAP, and no protein was detected in control myotubes at 24 h of cycloheximide treatment. Conversely, Klhl41 knockout cells exhibited a reduced turnover of NRAP protein in comparison to control cells, and a significant amount of NRAP protein accumulation was still observed at 24 h of the cycloheximide chase. Collectively, these data indicate that KLHL41 controls NRAP protein levels in skeletal muscle by ubiquitination-mediated proteasomal degradation.
NRAP accumulation leads to myopathy in zebrafish
As KLHL41 deficiency results in accumulation of NRAP in C2C12 Klhl41 differentiating myotubes, we investigated the in vivo impact of NRAP accumulation on skeletal muscle structure and function. We created transgenic NRAP overexpressing zebrafish under the control of skeletal muscle-specific myosin promoter (mylz; Fig. 3A) (45). Human NRAP was integrated into the zebrafish genome utilizing the Tol2 transposon-based system (46). Genomic integration and transgene expression were monitored using a GFP (Green Fluorescent Protein) reporter protein linked to the coding sequence of NRAP (mylz:NRAP-gfp). Germline transmission of the NRAP transgene was confirmed by PCR-based genotyping. The percentage of transgene transmission for F0 founders varied from 2.39% to 28.58%. F0 zebrafish lines with the highest transgene transmission rate were raised and crossed with wild-type zebrafish (AB strain) to identify the rate of transgene transmission and estimate the copy number of the transgene. The transmission rate of one of the mylz:NRAP-gfp lines followed normal mendelian ratios for a single insertion, with 50% of the progeny inheriting the transgene. Therefore, this line was selected for the follow-up studies. Whole-mount immunofluorescence with GFP antibody revealed that exogenous NRAP co-localized with actin on thin filaments as reported previously (Supplementary Material, Fig. S1). To investigate if NRAP overexpression affects the structure and function of skeletal muscle, transgenic zebrafish larvae were analyzed at 3 dpf by brightfield microscopy. Zebrafish larvae expressing NRAP transgene demonstrated extensive hypotonia and dorsal curvature indicative of a myopathic phenotype (Fig. 3B and C). To test if the morphological abnormalities in the mylz:NRAP-gfp transgenic fish are a consequence of structural defect in skeletal muscle, birefringence analysis was performed. Visualization of mylz:NRAP-gfp transgenic fish under polarized lenses showed a significant reduction in birefringence in comparison to the control indicative of structural disorganization of skeletal muscle (Fig 3D and E). To determine the specific defects in skeletal muscle structure due to NRAP overexpression, immunofluorescence and electron microscopy were performed in the mylz:NRAP-gfp transgenic and control zebrafish (3 dpf). Whole-mount immunofluorescence of skeletal muscle by phalloidin staining revealed thinner myofibers and gaps between adjacent myofibers in mylz:NRAP-gfp zebrafish (Fig. 3F and G). Additionally, visualization of skeletal muscle ultrastructure by electron microscopy also showed disorganized sarcomeres in the mylz:NRAP-gfp transgenic fish in comparison to the control (Fig. 3H and I, arrow). Evaluation of the skeletal muscle function by swimming behavior analysis also revealed a blunted motor activity in mylz:NRAP-gfp transgenic fish compared to controls (Fig. 3J). Transient overexpression of NRAP mRNA in wild-type zebrafish embryos resulted in phenotypic and motor function defects as observed in the mylz:NRAP-gfp transgenic fish (Supplementary Material, Fig. S2). This suggests that phenotypes observed in mylz:NRAP-gfp transgenic fish are due to NRAP overexpression and not a consequence of the potential disruption of another gene or GFP fusion protein. Together, these data provide evidence that overexpression of NRAP prevents the formation of mature myofibers and results in reduced motor function in affected skeletal muscle.
Figure 3.
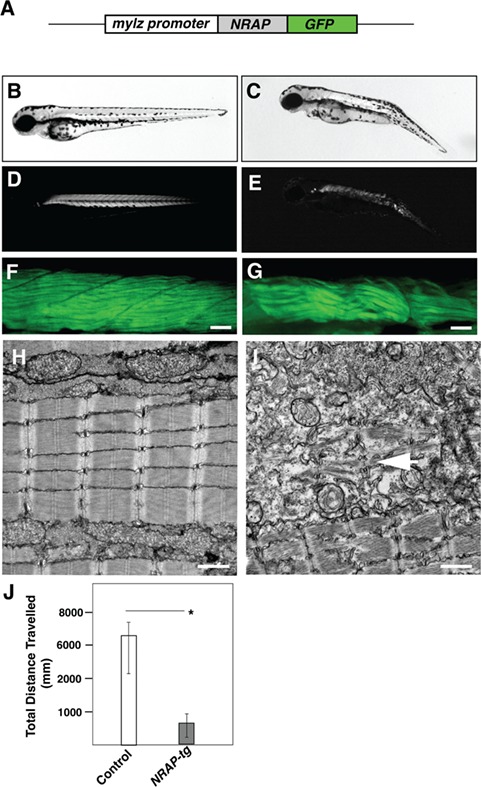
Transgenic NRAP zebrafish exhibit structural and functional deficits. (A) Schematics of NRAP transgenic construct. Human NRAP cDNA was cloned in-frame with GFP reporter and downstream of the zebrafish myosin light chain promoter using tol2-transgenesis. (B and C) Tg (mylz:NRAP-gfp) fish exhibit dorsal curvature and leaner bodies indicative of a myopathic phenotype (3dpf) (D and E) Visualization of Tg (mylz:NRAP-gfp) under polarized light revealed reduced birefringence compared to control (3 dpf). (F and G) Whole-mount phalloidin staining of Tg (mylz:NRAP-gfp) larvae (3 dpf) exhibiting fewer myofibers and smaller myotomes in comparison to controls. Scale bar: 50 μm. (H and I) Transmission electron microscopy demonstrating mature myofibers in control fish whereas Tg (mylz:NRAP-gfp) fish exhibit presence of immature sarcomeres (white arrow) and lacked matured myofibrils (arrow). Scale bar: 500 nm. (J) Quantification of the distance traveled by control (n = 55–75) and Tg (mylz:NRAP-gfp) (n = 28–44), acquired by an automated infra-red imager. Data are shown as mean ± s.d. from three different experiments. P-values were calculated using a two-tailed Mann–Whitney U-test, *P < 0.01.
NRAP prevents KLHL40-nebulin interaction in KLHL41 deficiency
During skeletal muscle development, NRAP depletion corresponds to lateral fusion of premyofibrils and assembly of proteins complexes by scaffolding on KLHL41 resulting in mature thin filaments in sarcomeres. In addition to KLHL41, closely related NM-causing Kelch protein, KLHL40, interacts with nebulin and NRAP in skeletal muscle and stabilizes mature thin filaments (31). Therefore, in KLHL41 deficiency, we investigated the impact of NRAP on complex formation between KLHL40 and nebulin to evaluate the functional redundancy between closely related KLHL40 and KLHL41. Co-expression of equimolar amounts of KLHL40-FLAG, NEB (C-terminal or N-terminal) and NRAP was performed in klhl41 deficient C2C12 myoblasts (Fig. 4A and B). Immunoprecipitation of KLHL40-FLAG with FLAG antibody and western blotting revealed that KLHL40 co-immunoprecipitated with either NRAP or NEB, validating the interaction between these proteins. Interestingly, in the presence of both NRAP and NEB, KLHL40 preferentially co-immunoprecipitated with NRAP, and no interaction of KLHL40 was observed with NEB. These studies suggest that NRAP exhibit a high affinity for proteins in comparison to nebulin, such as KLHL40 and KLHL41, that are essential for the assembly of proteins required in the formation of mature myofibrils.
Figure 4.
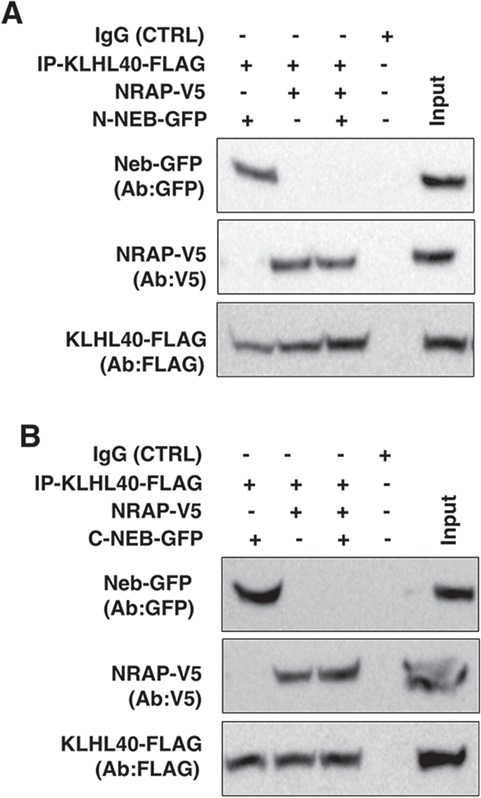
NRAP inhibits KLHL40-NEB interaction. KLHL40 and NRAP or nebulin were overexpressed in Klhl41 C2C12 myoblasts. (A) Co-expression of KLHL40 and N-terminal nebulin (N-NEB) or NRAP in C2C12 myoblasts revealed protein–protein interactions between KLHL40/N-NEB or KLHL40-NRAP. In the presence of nebulin, KLHL40 demonstrated preferential binding to NRAP. (B) Similarly, overexpression in C2C12 and Western blot analysis revealed that KLHL40 interacts with C-terminal nebulin (C-NEB), and this interaction is blocked in the presence of NRAP.
Downregulation of NRAP improves structure and function of Klhl41 deficient zebrafish
Our previous studies have shown Klhl41 deficient fish recapitulates clinical and pathological hallmarks of NM as observed in human patients (15). Therefore, we hypothesized that reducing NRAP levels in KLHL41 deficiency may rescue the pathophysiology observed in Klhl41-related NM. To test if reduced NRAP level in KLHL41-NM zebrafish model may rescue the structural and functional deficits in NM, we used antisense morpholino approach to knockdown nrap in zebrafish.
To optimize the morpholino (MO) treatment, two different splice sites (exon2-intron2 and exon3-intron3) at different concentrations (1–5 ng) were used to knockdown zebrafish nrap in wild-type control fish. With increasing concentration of nrap morpholinos, reduced amounts of nrap mRNA were observed in control fish (Supplementary Material, Fig. S3). Overexpression of NRAP mRNA rescued the motor function deficits in the exon2-intron2 (ex2-in2) or exon3-intron3 (ex3-in3) targeted nrap morphant fish (5.0 ng) indicating the specificity of nrap morpholinos (Fig. 5A and B). Downregulation of nrap by either splice site targeting morpholinos resulted in similar changes in zebrafish; therefore, we used nrap morpholino targeting exon2-intron2 for the follow-up rescue studies in klhl41 fish. As lower concentrations of nrap morpholinos (1–2.5 ng) resulted in an increase in the mis-spliced nrap mRNA without any phenotypic or functional defects in wild-type zebrafish, we used lower concentrations of morpholino (1.0 and 2.5 ng) to downregulate nrap in wild-type control and klhl41 morphant fish. No improvement in muscle function was observed in klhl41 morphant fish on injection with lowest morpholino amounts (1.0 ng) as evaluated by the quantification of the motor function by swimming behavior analysis. Injections with increased amounts of morpholino resulted in a significant improvement in muscle function in klhl41 morphants as analyzed by the swimming behavior (Fig. 5C). We previously showed that ultrastructure of Klhl41 deficient fish exhibit highly disorganized skeletal muscle with accumulation of nemaline bodies. Ultrastructure evaluation of nrap morphant fish (2.5 ng morpholino) by electron microscopy showed that reduction of nrap in wild-type fish did not result in any obvious defects in skeletal muscle ultrastructure as compared to the wild-type control. klhl41 morphant fish exhibited extensive sarcomeric disorganization with numerous nemaline bodies (arrows). Injection of nrap MO in klhl41 morphant fish resulted in an overall restoration of sarcomeric organization as evident by the presence of mature myofibrils (Fig. 5D). While the organization of sarcomere improved and no nemaline bodies were observed in klhl41 and nrap double morphant fish, the overall size of sarcomeres was significantly smaller in the double morphant fish in comparison to controls (Wild-type or nrap morphants). These data show that downregulation of nrap in KLHL41 deficiency leads to an improvement in sarcomeric organization and function of skeletal muscle.
Figure 5.
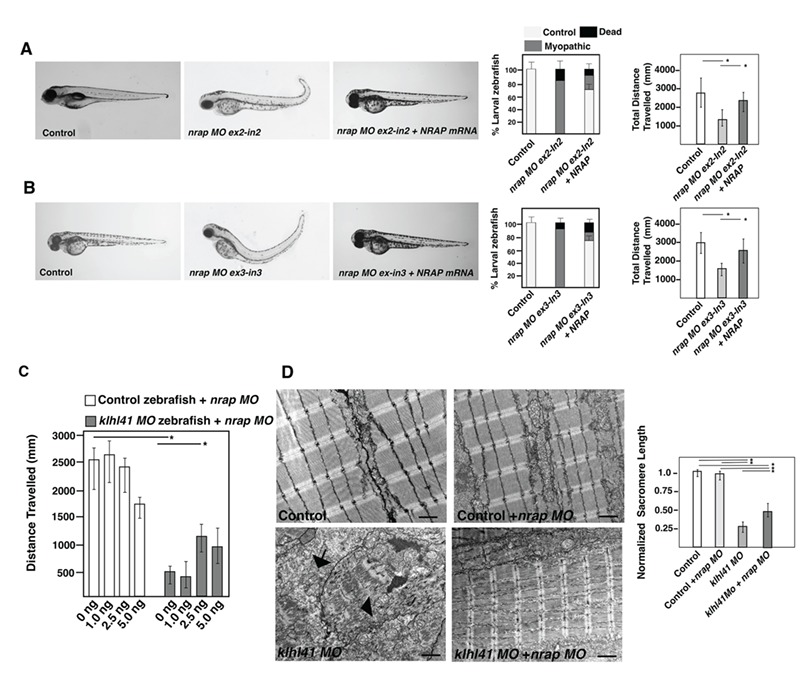
Downregulation of NRAP levels rescues the pathophysiology of KLHL41-NM. (A) Live larval fish at 3 dpf injected with control or nrap ex2-in2 morpholino (5 ng). nrap knockdown resulted in reduced motor function in the morphant fish that was rescued by overexpression of NRAP mRNA (100 ng). (B) Live larval fish at 3 dpf injected with control or nrap ex3-in3 morpholino (5 ng). nrap knockdown resulted in reduced motor function in the morphant fish that was rescued by overexpression of NRAP mRNA (100 ng). (C) Quantification of the swimming behavior in control or klhl41 morphant fish larval fish (3 dpf) injected with different amount nrap (ex2-in2) morpholino. All experiments were done in triplicate and 34–72 zebrafish embryos using independent zebrafish clutches. (D) Skeletal muscle ultrastructure of control and klhl41 morphant zebrafish targeted with nrap morpholino (2.5 ng) (3 dpf); n = 4–5. Quantification of sarcomere length was performed in three to four images from each fish. Scale bar: 500 nm. Data are shown as mean ± s.d. Kruskal Wallis one-way analysis on variance on ranks was performed and pairwise multiple comparison procedures were done according to Dunn’s method. *P < 0.01.
Discussion
The UPS pathway is highly conserved in vertebrates and a critical regulator of protein turnover that is required for normal functioning of skeletal muscle. However, little is known regarding the mechanism of protein turnover dynamics during muscle growth and impact of this process on skeletal muscle development and maintenance. In this work, we provide evidence that KLHL41 regulates sarcomere assembly through ubiquitination and proteasome-mediated degradation of the thin filament chaperone, NRAP. The degradation of NRAP that acts in the early steps of sarcomere assembly would comprise a timely and effective negative feedback mechanism to restrain abnormal accumulation of NRAP, thereby regulating the dynamics of myofibril formation. This regulation is critical for disease pathogenesis as abnormal accumulation of NRAP results in myopathic muscle. Therefore, our studies identify NRAP as a potential therapeutic target in NM and uncovers an intriguing crosstalk between the UPS and sarcomere assembly process.
We show that KLHL41 interacts with NRAP through C-terminal Kelch domain, and this interaction results in ubiquitination and subsequent degradation of NRAP. Studies have previously identified KLHL41 as an interactor of NRAP; however, functional implications of these interactions in skeletal muscle development and function were not known (47). Our study provides substantial evidence for the destabilizing effect of KLHL41 on NRAP by ubiquitination-mediated proteasomal degradation suggesting selective degradation of proteins may act as a mechanism to regulate the dynamics of sarcomeric proteins that play specific roles during skeletal muscle development. This mode of regulation of skeletal muscle development by ubiquitination is emerging as a critical regulatory mechanism to control protein dynamics and levels. Previous studies have shown that UPS-mediated degradation of key myogenic proteins such as Pax7 and MyoD is required for muscle differentiation (48,49). During muscle differentiation, myoblasts produce increased levels of reactive oxygen species leading to an elevated level of oxidized proteins that may negatively affect the muscle differentiation (50). Removal of such proteins by UPS ensures effective muscle differentiation. These studies not only demonstrate the extensive regulation of different molecular processes leading to muscle formation by the UPS but also uncover the therapeutic potential of targeting these processes in neuromuscular diseases.
NM is a rare skeletal muscle disorder that is caused by mutations in 11 genes, most of which encode for thin filament proteins. We have previously identified mutations in a gene encoding CUL3 E3 ligase adaptor, KLHL41, that result in a severe form of NM (15). Mutations in genes encoding closely related Kelch family members, KLHL40 and KBTBD13, also result in recessive and dominant forms of NM, respectively (14,16). Skeletal muscle biopsies of these patients demonstrate extensive sarcomeric disorganization and abnormal accumulation of protein aggregates or nemaline bodies suggesting a potential defect in the protein turnover process. KLHL41-mediated ubiquitination plays dual roles in regulation of sarcomeric proteins. A previous study has shown that autoubiquitination of KLHL41 is critical for stabilization of nebulin, the major structural constituent of thin filaments in mature myofibers (32). Our work demonstrates that KLHL41 is critical for ubiquitination and subsequent degradation of thin filament chaperone, NRAP, a step that ensures that dynamics of proteins in maturing premyofibrils is maintained.
Our work points to a tightly controlled mechanism by which KLHL41 binds to NRAP and promotes its ubiquitination and subsequent degradation by UPS. Consequently, an increase in NRAP protein is observed in KLHL41 deficiency. The in vivo relevance of increased NRAP levels on muscle pathology is evident from mylz:NRAP-gfp transgenic zebrafish that exhibit structural and functional deficient in skeletal muscles. While the abnormal accumulation of NRAP has been previously reported in skeletal muscle biopsies in nemaline and myofibrillar myopathy, the functional significance of this abnormal accumulation in disease pathology was not known (51). Our studies suggest that pathologically increased levels of NRAP could be contributing to muscle dysfunction in NM and related forms of myopathies. We further demonstrate that NRAP exhibits high affinity for KLHL41 and related family member, KLHL40, and prevents KLHL40-NEB interaction that is required to form mature myofibrils. Similarly, this abnormally accumulated NRAP may interact with other muscle protein complexes that are required for mature myofibril formation (Fig. 4) and perturb their function (Fig. 6). Further studies may be able to identify those factors in NM. Previous work has shown that nemaline bodies are formed by different mechanisms and may affect skeletal muscle pathology differently (52). Therefore, identification of different sarcomeric proteins and mechanism contributing to protein aggregates is crucial for a precise understanding of disease biology in NM (Fig. 6). Downregulation of NRAP resulted in a significant improvement in skeletal muscle pathophysiology in klhl41-deficient zebrafish, thus providing direct evidence for reduction of NRAP as a potential therapeutic strategy in KLHL41-related NM. While complete loss of NRAP may lead to skeletal muscle defects, partial downregulation of NRAP may provide an improvement in KLHL41 and related myopathies. Of note, two recent studies have described loss of function mutations in NRAP as the cause of underlying disease pathology in dilated cardiomyopathy (53,54). As asymptomatic sibling also shared the same genotype as the proband, further studies are needed to conclude the pathogenicity of NRAP in human disorders. Downregulation of NRAP mRNA by morpholinos was not able to completely rescue structural and functional deficits observed in KLHL41 deficiency. Future design of therapeutics on targeting abnormal protein rather than mRNA to reduce protein amounts to physiological levels may further provide alternative therapeutic strategy without affecting the basal NRAP transcript levels that are required for normal muscle function. Finally, studies on other in vivo targets of KLHL41-CUL3 may provide new insights into suitable pathways and targets that may lead to better therapeutic design in combination therapies based on NRAP downregulation.
Figure 6.
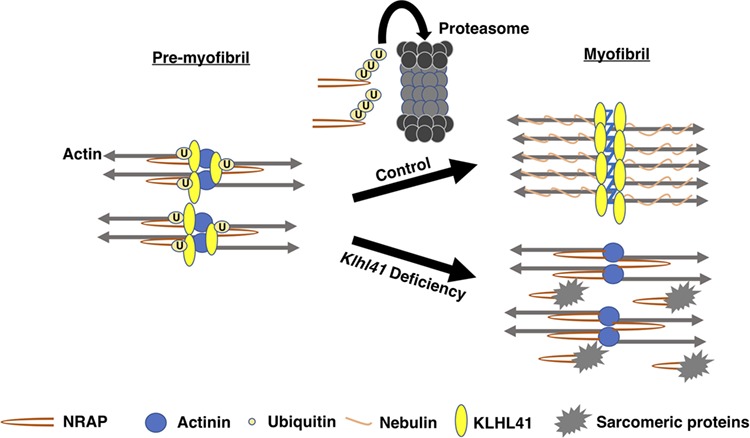
Model of KLHL41 function during sarcomere assembly. KLHL41 binds to NRAP in pre-myofibrils, and this interaction leads to ubiquitination and subsequent degradation of NRAP by proteasomes. In control skeletal muscle, removal of NRAP from developing pre-myofibrils results in their fusion and formation of mature myofibrils. In the absence of Klhl41, abnormal accumulation of NRAP exhibit high affinity to sarcomeric proteins that are otherwise required to form mature myofibers leading to weak muscles.
In summary, KLHL41 degrades NRAP by ubiquitination-mediated proteolytic degradation with subsequent recruitment and stabilization of nebulin. These studies propose a paradigm that degradation of skeletal muscle chaperones is required to restrict their function to a narrow temporal window. The identification of a role of KLHL41 in coordinating the premyofibril-myofibril transition offers a unique opportunity to explore the physiological and pathological basis of NM.
Materials and Methods
Zebrafish lines
Fish were bred and maintained using standard methods as described (55). All procedures were approved by the Institute Animal Care and Use Committee. Wild-type embryos were obtained from Oregon AB line and staged by hours (h) or days (d) post fertilization at 28.5°C. Zebrafish embryonic (0–2 dpf) and larval stages (3–5 dpf) have been defined as described previously (56).
Y2H
The Y2H screen was performed by Hybrigenics S.A.S., Paris, France. Human KLHL41 cDNA (C-terminal domain) was cloned into pB27 as a C-terminal fusion to LexA (N-LexA-KLHL41-C) and used as a bait for screening a human adult and fetal skeletal muscle cDNA library. The LexA bait construct was used to screen 61 million clones (6-fold coverage of the library). The prey fragments of the positive clones were confirmed for interaction, amplified by PCR, sequenced and identified using the Genbank database (NCBI).
Time-resolved FRET assay (HTRF)
Full-length human KLHL41 protein was expressed as GST-fusion tag protein in E.coli, BL21. Human NEB (amino acids 5647–5849) and NRAP (amino acids 1471–1591) were expressed as MBP-HA fusion proteins in E.coli, BL21 (Hybrigenics, S.A.S., Paris, France). Cells transfected with fusion plasmids were grown to an optical density of 0.8 (wavelength: 600 nM) and induced with IPTG Isopropyl beta-D-1-thiogalactopyranoside. Cell lysates were prepared by sonication and purified using affinity chromatography. Sonication buffer used for GST tag-fusion protein contained 1X PBS (Fluorescence-Activated Cell Sorting) (pH 7.4), 1% Triton X-100 and 10% Glycerol. For KLHL41-GST, Glutathione Sepharose 4B (GE Life Sciences, USA) was used for affinity purification, and bound fractions were eluted in the presence of 40 mm glutathione. The sonication buffer used for MBP tag-fusion protein contained 20 mm (Phosphate Buffer Saline) PBS (pH 7.4), 200 mm NaCl, 0.5% Triton X-100, 1 mm EDTA, 2 mm DTT and 10% Glycerol. Amylose affinity resin (New England Biolabs, USA) was used for affinity purification, and bound fractions were eluted in the presence of 10 mm maltose. For HTRF assay, dilutions of both interactor proteins and antibodies were freshly prepared and incubated together for 2 h at 4°C. Following this 10 μL anti-GST-EuK and/or anti MBP-d2, antibodies were added (1:400 dilution). The interaction between two proteins was detected by fluorescence transfer (excitation at 337 nm, emission at 665 nm). The emission at 620 nm occurs regardless of the interaction and used for normalizing the assay. The interaction between two proteins was quantified as DeltaF (%) as
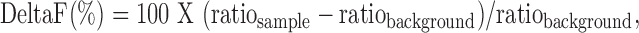 |
where ‘ratio’ is the 665/620 fluorescence ratio, ‘sample’ is the signal in the presence of protein interactors and antibodies, whereas, ‘background’ denotes the HTRF antibodies only. Fusion tags without interactor proteins were used as negative controls.
RT-PCR (Real Time-Polymerase Chain Reaction) assay
To detect nrap mRNA expression in C2C12 myoblasts, RNA was prepared from control or Klhl41 knockout myotubes (day 2) in using Trizol reagent (Thermo Fisher Scientific, USA) according to manufacturer’s instructions. cDNAs were synthesized from 2 to 5 μg of total RNA using Superscript II Reverse Transcriptase (Thermo Fisher Scientific) and random hexamers. Quantitative PCR amplification of cDNAs was performed on Light-Cycler 480 and Light-Cycler 24 instruments (Roche Applied Sciences, Basel, Switzerland) using 58°C as melting temperature. Gapdh gene expression was used as control. Primers used were: nrap (F: GCTGCATAGATCAGCCACAA, R: CCCAATGGAGAACGGATTTA) and Gapdh (F: AGCTTTCCAGAGGGGCCATCCACA, R: CCAGTATGACTCCACTCACGGCAA).
Co-immunoprecipitation
Human KLHL41-pEZYFLAG, human NRAP-V5, C-terminal NEB-GFP and N-terminal NEB-GFP were used for co-immunoprecipitation assay in C2C12 cells. Equimolar concentration of KLHL41-pEZYFLAG, NRAP-V5 or NEB-GFP and control empty plasmids were transfected in C2C12 cells. Forty-eight hours post transfection, cell lysates were prepared in RIPA (Radioimmunoprecipitation Assay Buffer) buffer and precleared with mouse IgG and Protein A/G agarose beads for 1 h at 4°C. V5-agarose (Thermo Fisher Scientific) or FLAG-agarose (Sigma, St. Louis, MO, USA) beads were incubated with the supernatant (4°C, overnight) and washed three times with PBS and once with high stringency buffer (300 mm NaCl + PBS buffer). For all experiments, two negative controls consisted of a sample lacking the primary antibody (beads) and a sample incubated with IgG. Resulting protein complexes were eluted in 1X LDS buffer (Thermo Fisher Scientific) and analyzed by SDS-PAGE and Western blotting using KLHL41 antibody, (1:500, AV38732, Millipore Sigma, USA), FLAG antibody (1:500, F1804, Millipore Sigma), V5 antibody (1:500, R960CUS, Thermo Fisher Scientific) and GFP antibody (1:250, sc-9996, Santa Cruz Biotechnology, USA).
Whole-mount phalloidin staining
Zebrafish larvae (3 dpf) were fixed in 4% Paraformaldehyde (PFA) overnight at 4°C, then washed as follows: 2 × 10 min in PBS, 2 × 10 min in PBS-T (0.1% Tween-20), 1 × 60 min in PBS-TR (2% Triton X) and 2 × 5 min in PBS-T. Embryos were blocked in PBS-T containing 5% goat serum for 1 h at RT and incubated with phalloidin (1:40, Thermo Fisher Scientific, A12379). Subsequently, larvae were washed for 4 × 15 min in PBS-T, mounted in 70% glycerol and visualized using a Perkin Elmer UltraVIEW VoX spinning disk confocal microscope.
C2C12 myoblasts culture
Mouse C2C12 cells were cultured in a growth medium consisting of DMEM (Dulbecco Modified Eagle Medium) supplemented with 20% fetal bovine serum. To create Klhl41 knockout lines, Cas9-GFP plasmid was co-transfected with sgRNAs targeting exon1 of mouse Klhl41 gene. Twenty-four hours post transfection, GFP-positive cells were selected by FACS (Fluorescence-Activated Cell Sorting), and single cells were plated in each well of a 96 well plate in the C2C12 conditioned media. Clonal expansion was performed, and loss of function mutations were validated by Sanger sequencing and Western blot analysis.
Ubiquitination studies
C2C12 cells were transfected with different amounts of KLHL41-pEZYFLAG, NRAP-V5 and HA-ubiquitin plasmids (57). MG132 (10 uM) was added at 40 h post transfection, and cells were harvested at 48 h post transfection. Cell lysates were prepared in RIPA buffer. To reduce the non-specific binding during immunoprecipitation, supernatant was incubated with mouse IgG and Protein A/G agarose beads for 1 h at 4°C followed by overnight incubation with V5-agarose at 4°C. Beads were washed with 1X PBS buffer (3 times) and boiled in 1X LDS sample buffer to elute proteins. SDS-PAGE and western blotting were performed as previously described (58). To analyze proteasome mediated degradation of NRAP, control or Klhl41 knockout C2C12 differentiating myotubes were grown in the presence or absence of proteasome inhibitor (MG132, 10 μM) for 48 h, and proteins were analyzed by western blot analysis. For cycloheximide chase assay, control or Klhl41 knockout C2C12 cells (5 X 105) were plated in 35 mm plates. After 12 h, replace with fresh medium containing 100 μg/ml cycloheximide. Samples were collected at different time points, protein lysates were prepared and Western blotting was performed.
NRAP overexpression and knockdown in zebrafish
Human NRAP was integrated in to zebrafish genome utilizing the Tol2 transposon-based system (46). A GFP reporter protein was linked to the coding sequence of NRAP under the control of mylz, a skeletal muscle specific promoter (mylz:NRAP-gfp) using Gateway multisite cloning. 25 ng of the transgenic plasmid was co-injected with 10 ng of transposase mRNA in to 1-cell zebrafish embryos. The percentage of transgene transmission for F0 founders varied from 2.39% to 28.58%. Two F0 zebrafish lines with the highest transgene transmission rate (25.11 and 28.58%) were raised and crossed with wild-type zebrafish (AB strain) to identify the rate of transgene transmission and estimate the copy number of the transgene. The transmission rate of one of the mylz:NRAP-gfp lines followed normal mendelian ratios for a single insertion, with 50% of the progeny inheriting the transgene. Therefore, this line was selected for the follow-up studies. Germline transmission of the NRAP transgene in F1 generation was confirmed by PCR-based genotyping using primers for GFP: forward, 5′-AAGCTGACCCTGAAGTTCATCTGC-3′, and reverse, 5′-CTTGTAGTTGCCGTCGTCCTTGAA-3′.
Two antisense MO targeting the exon-intron junction of zebrafish nrap gene (NM_001324547) were designed to knockdown the zebrafish nrap transcript. The MO sequences were designed to target exon-intron junctions; exon2-intron2: 5′-TGTGCCAGTTCTGAAAGAGAAAAGA-3′ and exon3-intron3: 5′-GGTGTAAACAAAACCTCACCTCACT-3′.
MO against human β-globin, which is not homologous to any sequence in the zebrafish genome by BLAST search, was used as a negative control for all injections (5′-CCTCTTACCTCAGTTACAATTTATA-3′). klhl41 morpholino sequences have been described previously (15). Sequences of RT-PCR primers used for zebrafish nrap gene is: forward, 5′- CGCCTTGCTGTTCCTATACC-3′ and reverse, 5′GCCTCTGATTGCTTCTTTGC3′. MOs were dissolved in 1X Danieau buffer with 0.1% phenol red and 1–2 nl (1–10 ng) injected into the yolk of 1-cell stage embryos. Klhl41 genes were knockdown in zebrafish using morpholinos as described previously (15). Klhl41 knockdown in larval fish was quantified by RT-PCR analysis and described (15).
Zebrafish locomotion assay
Zebrafish swimming behavior was quantified by an infra-red tracking activity monitoring system (DanioVision, Noldus, Leesburg, VA, USA). Control, transgenic or morphant larval zebrafishes were placed individually into each well of a 24 well plate in dark for 10 min. The activity of these larvae was recorded during a follow-up light exposure of 20 min. Four independent blind trials were performed, and mean velocity, total distance and cumulative duration of movement were recorded. Reported values reflect an average of 30–35 larval fishes.
Quantification and statistical analysis
All samples were blinded till final analyses. Statistical analyses were non-parametic and performed using GraphPad Prism. Values are presented as the mean ± s.d. and were compared by using Student’s t-test. The Wilcoxon signed rank test was used for comparisons between related samples, the nonparametric Mann–Whitney U test for comparisons between two groups and the Kruskal–Wallis one-way analysis of variance for comparisons between more than two groups. When statistical significance between groups was obtained, the Dunn’s post-test with correction for multiple comparisons was performed.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
Conflict of Interest statement. None declared.
Funding
Miles Shore Fellowship for Scholars in Medicine and Brigham and Women’s Hospital Career Development Award; Foundation Building Strength Grant to V.A.G. Sequencing reactions were carried out with an ABI3730xl DNA analyzer at the DNA Resource Core of Dana-Farber/Harvard Cancer Center (funded in part by NCI (National Cancer Institute) Cancer Center support grant 2P30CA006516-48).
References
- 1. Abmayr S.M. and Pavlath G.K. (2012) Myoblast fusion: lessons from flies and mice. Development, 139, 641–656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Sampath S.C., Sampath S.C. and Millay D.P. (2018) Myoblast fusion confusion: the resolution begins. Skelet. Muscle, 8, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Sanger J.W., Wang J., Fan Y., White J., Mi-Mi L., Dube D.K., Sanger J.M. and Pruyne D. (2017) Assembly and maintenance of myofibrils in striated muscle. Handb. Exp. Pharmacol., 235, 39–75. [DOI] [PubMed] [Google Scholar]
- 4. Nowak K.J., Wattanasirichaigoon D., Goebel H.H., Wilce M., Pelin K., Donner K., Jacob R.L., Hubner C., Oexle K., Anderson J.R. et al. (1999) Mutations in the skeletal muscle alpha-actin gene in patients with actin myopathy and nemaline myopathy. Nat. Genet., 23, 208–212. [DOI] [PubMed] [Google Scholar]
- 5. Pelin K., Hilpela P., Donner K., Sewry C., Akkari P.A., Wilton S.D., Wattanasirichaigoon D., Bang M.L., Centner T., Hanefeld F. et al. (1999) Mutations in the nebulin gene associated with autosomal recessive nemaline myopathy. Pro. Natl. Acad. Sci. U. S. A., 96, 2305–2310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Tajsharghi H., Thornell L.E., Lindberg C., Lindvall B., Henriksson K.G. and Oldfors A. (2003) Myosin storage myopathy associated with a heterozygous missense mutation in MYH7. Ann. Neurol., 54, 494–500. [DOI] [PubMed] [Google Scholar]
- 7. Hackman P., Vihola A., Haravuori H., Marchand S., Sarparanta J., De Seze J., Labeit S., Witt C., Peltonen L., Richard I. et al. (2002) Tibial muscular dystrophy is a titinopathy caused by mutations in TTN, the gene encoding the giant skeletal-muscle protein titin. Am. J. Hum. Genet., 71, 492–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Zhang Y., Chen H.S., Khanna V.K., De Leon S., Phillips M.S., Schappert K., Britt B.A., Browell A.K. and MacLennan D.H. (1993) A mutation in the human ryanodine receptor gene associated with central core disease. Nat. Genet., 5, 46–50. [DOI] [PubMed] [Google Scholar]
- 9. Laporte J., Hu L.J., Kretz C., Mandel J.L., Kioschis P., Coy J.F., Klauck S.M., Poustka A. and Dahl N. (1996) A gene mutated in X-linked myotubular myopathy defines a new putative tyrosine phosphatase family conserved in yeast. Nat. Genet., 13, 175–182. [DOI] [PubMed] [Google Scholar]
- 10. Chopra A., Kutys M.L., Zhang K., Polacheck W.J., Sheng C.C., Luu R.J., Eyckmans J., Hinson J.T., Seidman J.G., Seidman C.E. et al. (2018) Force generation via beta-cardiac myosin, titin, and alpha-actinin drives cardiac sarcomere assembly from cell-matrix adhesions. Dev. Cell, 44, 87–96. e85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Weitkunat M., Brasse M., Bausch A.R. and Schnorrer F. (2017) Mechanical tension and spontaneous muscle twitching precede the formation of cross-striated muscle in vivo. Development, 144, 1261–1272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Crawford G.L. and Horowits R. (2011) Scaffolds and chaperones in myofibril assembly: putting the striations in striated muscle. Biophys. Rev., 3, 25–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Majczenko K., Davidson A.E., Camelo-Piragua S., Agrawal P.B., Manfready R.A., Li X., Joshi S., Xu J., Peng W., Beggs A.H. et al. (2012) Dominant mutation of CCDC78 in a unique congenital myopathy with prominent internal nuclei and atypical cores. Am. J. Hum. Genet., 91, 365–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ravenscroft G., Miyatake S., Lehtokari V.L., Todd E.J., Vornanen P., Yau K.S., Hayashi Y.K., Miyake N., Tsurusaki Y., Doi H. et al. (2013) Mutations in KLHL40 are a frequent cause of severe autosomal-recessive nemaline myopathy. Am. J. Hum. Genet., 93, 6–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Gupta V.A., Ravenscroft G., Shaheen R., Todd E.J., Swanson L.C., Shiina M., Ogata K., Hsu C., Clarke N.F., Darras B.T. et al. (2013) Identification of KLHL41 mutations implicates BTB-Kelch-mediated ubiquitination as an alternate pathway to myofibrillar disruption in nemaline myopathy. Am. J. Hum. Genet., 93, 1108–1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Sambuughin N., Yau K.S., Olive M., Duff R.M., Bayarsaikhan M., Lu S., Gonzalez-Mera L., Sivadorai P., Nowak K.J., Ravenscroft G. et al. (2010) Dominant mutations in KBTBD13, a member of the BTB/Kelch family, cause nemaline myopathy with cores. Am. J. Hum. Genet., 87, 842–847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Patel N., Smith L.L., Faqeih E., Mohamed J., Gupta V.A. and Alkuraya F.S. (2014) ZBTB42 mutation defines a novel lethal congenital contracture syndrome (LCCS6). Hum. Mol. Genet., 23, 6584–6593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Di Costanzo S., Balasubramanian A., Pond H.L., Rozkalne A., Pantaleoni C., Saredi S., Gupta V.A., Sunu C.M., Yu T.W., Kang P.B. et al. (2014) POMK mutations disrupt muscle development leading to a spectrum of neuromuscular presentations. Hum. Mol. Genet., 23, 5781–5792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Gonorazky H.D., Bonnemann C.G. and Dowling J.J. (2018) The genetics of congenital myopathies. Handb. Clin. Neurol., 148, 549–564. [DOI] [PubMed] [Google Scholar]
- 20. Malfatti E. and Romero N.B. (2016) Nemaline myopathies: state of the art. Rev. Neurol. (Paris), 172, 614–619. [DOI] [PubMed] [Google Scholar]
- 21. North K.N. (2011) Clinical approach to the diagnosis of congenital myopathies. Semin. Pediatr. Neurol., 18, 216–220. [DOI] [PubMed] [Google Scholar]
- 22. Donner K., Ollikainen M., Ridanpaa M., Christen H.J., Goebel H.H., de Visser M., Pelin K. and Wallgren-Pettersson C. (2002) Mutations in the beta-tropomyosin (TPM2) gene—a rare cause of nemaline myopathy. Neuromuscul. Disord., 12, 151–158. [DOI] [PubMed] [Google Scholar]
- 23. Laing N.G., Wilton S.D., Akkari P.A., Dorosz S., Boundy K., Kneebone C., Blumbergs P., White S., Watkins H., Love D.R. et al. (1995) A mutation in the alpha tropomyosin gene TPM3 associated with autosomal dominant nemaline myopathy. Nat. Genet., 9, 75–79. [DOI] [PubMed] [Google Scholar]
- 24. Johnston J.J., Kelley R.I., Crawford T.O., Morton D.H., Agarwala R., Koch T., Schaffer A.A., Francomano C.A. and Biesecker L.G. (2000) A novel nemaline myopathy in the Amish caused by a mutation in troponin T1. Am. J. Hum. Genet., 67, 814–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Yuen M., Sandaradura S.A., Dowling J.J., Kostyukova A.S., Moroz N., Quinlan K.G., Lehtokari V.L., Ravenscroft G., Todd E.J., Ceyhan-Birsoy O. et al. (2014) Leiomodin-3 dysfunction results in thin filament disorganization and nemaline myopathy. J. Clin. Invest., 124, 4693–4708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Winter J.M., Joureau B., Lee E.J., Kiss B., Yuen M., Gupta V.A., Pappas C.T., Gregorio C.C., Stienen G.J., Edvardson S. et al. (2016) Mutation-specific effects on thin filament length in thin filament myopathy. Ann. Neurol., 79, 959–969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Agrawal P.B., Greenleaf R.S., Tomczak K.K., Lehtokari V.L., Wallgren-Pettersson C., Wallefeld W., Laing N.G., Darras B.T., Maciver S.K., Dormitzer P.R. et al. (2007) Nemaline myopathy with minicores caused by mutation of the CFL2 gene encoding the skeletal muscle actin-binding protein, cofilin-2. Am. J. Hum. Genet., 80, 162–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Malfatti E., Bohm J., Lacene E., Beuvin M., Romero N.B. and Laporte J. (2015) A premature stop codon in MYO18B is associated with severe nemaline myopathy with cardiomyopathy. J. Neuromuscul. Dis., 2, 219–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Spence H.J., Johnston I., Ewart K., Buchanan S.J., Fitzgerald U. and Ozanne B.W. (2000) Krp1, a novel kelch related protein that is involved in pseudopod elongation in transformed cells. Oncogene, 19, 1266–1276. [DOI] [PubMed] [Google Scholar]
- 30. Sambuughin N., Swietnicki W., Techtmann S., Matrosova V., Wallace T., Goldfarb L. and Maynard E. (2012) KBTBD13 interacts with Cullin 3 to form a functional ubiquitin ligase. Biochem. Biophys. Res. Commun., 421, 743–749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Garg A., O'Rourke J., Long C., Doering J., Ravenscroft G., Bezprozvannaya S., Nelson B.R., Beetz N., Li L., Chen S. et al. (2014) KLHL40 deficiency destabilizes thin filament proteins and promotes nemaline myopathy. J. Clin. Invest., 124, 3529–3539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ramirez-Martinez A., Cenik B.K., Bezprozvannaya S., Chen B., Bassel-Duby R., Liu N. and Olson E.N. (2017) KLHL41 stabilizes skeletal muscle sarcomeres by nonproteolytic ubiquitination. Elife, 6, e26439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Goldberg A.L. (2003) Protein degradation and protection against misfolded or damaged proteins. Nature, 426, 895–899. [DOI] [PubMed] [Google Scholar]
- 34. Kleiger G. and Mayor T. (2014) Perilous journey: a tour of the ubiquitin-proteasome system. Trends. Cell. Biol., 24, 352–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Dubiel W., Dubiel D., Wolf D.A. and Naumann M. (2018) Cullin 3-based ubiquitin ligases as master regulators of mammalian cell differentiation. Trends. Biochem. Sci., 43, 95–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Xu L., Wei Y., Reboul J., Vaglio P., Shin T.H., Vidal M., Elledge S.J. and Harper J.W. (2003) BTB proteins are substrate-specific adaptors in an SCF-like modular ubiquitin ligase containing CUL-3. Nature, 425, 316–321. [DOI] [PubMed] [Google Scholar]
- 37. Glass D.J. (2005) Skeletal muscle hypertrophy and atrophy signaling pathways. Int. J. Biochem. Cell. Biol., 37, 1974–1984. [DOI] [PubMed] [Google Scholar]
- 38. Witt S.H., Granzier H., Witt C.C. and Labeit S. (2005) MURF-1 and MURF-2 target a specific subset of myofibrillar proteins redundantly: towards understanding MURF-dependent muscle ubiquitination. J. Mol. Biol., 350, 713–722. [DOI] [PubMed] [Google Scholar]
- 39. Franch H.A. and Price S.R. (2005) Molecular signaling pathways regulating muscle proteolysis during atrophy. Curr. Opin. Clin. Nutr. Metab. Care, 8, 271–275. [DOI] [PubMed] [Google Scholar]
- 40. Assereto S., Piccirillo R., Baratto S., Scudieri P., Fiorillo C., Massacesi M., Traverso M., Galietta L.J., Bruno C., Minetti C. et al. (2016) The ubiquitin ligase tripartite-motif-protein 32 is induced in Duchenne muscular dystrophy. Lab. Invest., 96, 862–871. [DOI] [PubMed] [Google Scholar]
- 41. de Winter J.M. and Ottenheijm C.A.C. (2017) Sarcomere dysfunction in nemaline myopathy. J. Neuromuscul. Dis., 4, 99–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Leblanc V., Delaunay V., Claude Lelong J., Gas F., Mathis G., Grassi J. and May E. (2002) Homogeneous time-resolved fluorescence assay for identifying p53 interactions with its protein partners, directly in a cellular extract. Anal. Biochem., 308, 247–254. [DOI] [PubMed] [Google Scholar]
- 43. Lu S., Borst D.E. and Horowits R. (2008) Expression and alternative splicing of N-RAP during mouse skeletal muscle development. Cell. Motil. Cytoskeleton, 65, 945–954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Gupta V.A. and Beggs A.H. (2014) Kelch proteins: emerging roles in skeletal muscle development and diseases. Skelet. Muscle, 4, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Pakula A., Lek A., Widrick J., Mitsuhashi H., Bugda Gwilt K.M., Gupta V.A., Rahimov F., Criscione J., Zhang Y., Gibbs D. et al. (2018) Transgenic zebrafish model of DUX4 misexpression reveals a developmental role in FSHD pathogenesis. Hum. Mol. Genet., 28, 320–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Kwan K.M., Fujimoto E., Grabher C., Mangum B.D., Hardy M.E., Campbell D.S., Parant J.M., Yost H.J., Kanki J.P. and Chien C.B. (2007) The Tol2kit: a multisite gateway-based construction kit for Tol2 transposon transgenesis constructs. Dev. Dyn., 236, 3088–3099. [DOI] [PubMed] [Google Scholar]
- 47. Lu S., Carroll S.L., Herrera A.H., Ozanne B. and Horowits R. (2003) New N-RAP-binding partners alpha-actinin, filamin and Krp1 detected by yeast two-hybrid screening: implications for myofibril assembly. J. Cell Sci., 116, 2169–2178. [DOI] [PubMed] [Google Scholar]
- 48. Bustos F., de la Vega E., Cabezas F., Thompson J., Cornelison D.D., Olwin B.B., Yates J.R. and Olguin H.C. (2015) NEDD4 regulates PAX7 levels promoting activation of the differentiation program in skeletal muscle precursors. Stem Cells, 33, 3138–3151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Feng Q., Jagannathan S. and Bradley R.K. (2017) The RNA surveillance factor UPF1 represses myogenesis via its E3 ubiquitin ligase activity. Mol. Cell, 67, 239–251. e236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Uchida T., Sakashita Y., Kitahata K., Yamashita Y., Tomida C., Kimori Y., Komatsu A., Hirasaka K., Ohno A., Nakao R. et al. (2018) Reactive oxygen species upregulate expression of muscle atrophy-associated ubiquitin ligase Cbl-b in rat L6 skeletal muscle cells. Am. J. Physiol. Cell Physiol., 314, C721–c731. [DOI] [PubMed] [Google Scholar]
- 51. Kley R.A., Maerkens A., Leber Y., Theis V., Schreiner A., van der Ven P.F., Uszkoreit J., Stephan C., Eulitz S., Euler N. et al. (2013) A combined laser microdissection and mass spectrometry approach reveals new disease relevant proteins accumulating in aggregates of filaminopathy patients. Mol. Cell Proteomics, 12, 215–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Sztal T.E., Zhao M., Williams C., Oorschot V., Parslow A.C., Giousoh A., Yuen M., Hall T.E., Costin A., Ramm G. et al. (2015) Zebrafish models for nemaline myopathy reveal a spectrum of nemaline bodies contributing to reduced muscle function. Acta Neuropathol., 130, 389–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Truszkowska G.T., Bilinska Z.T., Muchowicz A., Pollak A., Biernacka A., Kozar-Kaminska K., Stawinski P., Gasperowicz P., Kosinska J., Zielinski T. et al. (2017) Homozygous truncating mutation in NRAP gene identified by whole exome sequencing in a patient with dilated cardiomyopathy. Sci. Rep., 7, 3362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. D'Avila F., Meregalli M., Lupoli S., Barcella M., Orro A., De Santis F., Sitzia C., Farini A., D'Ursi P., Erratico S. et al. (2016) Exome sequencing identifies variants in two genes encoding the LIM-proteins NRAP and FHL1 in an Italian patient with BAG3 myofibrillar myopathy. J. Muscle Res. Cell Motil., 37, 101–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Westerfield M. (2000) The Zebrafish Book: A Guide for the Laboratory Use of Zebrafish (Danio Rerio). Institute of Neuroscience. University of Oregon.
- 56. Kimmel C.B., Ballard W.W., Kimmel S.R., Ullmann B. and Schilling T.F. (1995) Stages of embryonic development of the zebrafish. Dev. Dyn., 203, 253–310. [DOI] [PubMed] [Google Scholar]
- 57. Peng Y., Wang E., Peng G. and Lin S.-Y. (2016) Ubiquitination assay for mammalian cells. Bio Protoc., 6, e1880. [Google Scholar]
- 58. Bennett A.H., O'Donohue M.F., Gundry S.R., Chan A.T., Widrick J., Draper I., Chakraborty A., Zhou Y., Zon L.I., Gleizes P.E. et al. (2018) RNA helicase, DDX27 regulates skeletal muscle growth and regeneration by modulation of translational processes. PLoS Genet., 14, e1007226. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.