

Abstract
Heart failure is associated with hypertrophying of cardiomyocytes and changes in transcriptional activity. Studies from rapidly dividing cells in culture have suggested that transcription may be compartmentalized into factories within the nucleus, but this phenomenon has not been tested in vivo and the role of nuclear architecture in cardiac gene regulation is unknown. While alterations to transcription have been linked to disease, little is known about the regulation of the spatial organization of transcription and its properties in the pathological setting. In the present study, we investigate the structural features of endogenous transcription factories in the heart and determine the principles connecting chromatin structure to transcriptional regulation in vivo. Super-resolution imaging of endogenous RNA polymerase II clusters in neonatal and adult cardiomyocytes revealed distinct properties of transcription factories in response to pathological stress: neonatal nuclei demonstrated changes in number of clusters, with parallel increases in nuclear area, while the adult nuclei underwent changes in size and intensity of RNA polymerase II loci. Fluorescence in situ hybridization-based labeling of genes revealed locus-specific relationships between expression change and anatomical localization—with respect to nuclear periphery and heterochromatin regions, both sites associated with gene silencing—in the nuclei of cardiomyocytes in hearts (but not liver hepatocytes) of mice subjected to pathologic stimuli that induce heart failure. These findings demonstrate a role for chromatin organization and rearrangement of nuclear architecture for cell type-specific transcription in vivo during disease. RNA polymerase II ChIP and chromatin conformation capture studies in the same model system demonstrate formation and reorganization of distinct nuclear compartments regulating gene expression. These findings reveal locus-specific compartmentalization of stress-activated, housekeeping and silenced genes in the anatomical context of the endogenous nucleus, revealing basic principles of global chromatin structure and nuclear architecture in the regulation of gene expression in healthy and diseased conditions.
Keywords: transcriptional regulation, epigenomics, chromatin structure, heart disease
1. Introduction
Eukaryotic transcription requires coordination between DNA accessibility, transcription factor binding and recruitment of RNA polymerase machinery. The mechanisms of local gene regulation, as well as the global patterns of histone modifications associated with transcription, are increasingly well understood [1–3]—but how these processes are regulated within the three-dimensional environment of the nucleus remains to be determined.
The nucleus is arranged into structural and functional compartments that facilitate the cell type-specific utilization of genetic information [4, 5]. The genome itself is partitioned into chromosome territories [6, 7] and on the sub-chromosomal scale into topologically associating domains which themselves are associated with active and inactive compartments [7, 8]. These structural features can contribute to co-regulation at the transcriptional level, forming chromatin neighborhoods in which similar gene regulation regimes prevail. One such environment is a transcription factory, defined by enrichment with active RNA polymerases and permissiveness for transcription [9]. The nucleolus, which can be readily visualized with DAPI staining in many cell types, is a common example of a nuclear compartment that houses transcription factories for RNA polymerase I [10], which synthesizes ribosomal RNA. However, RNA polymerase II transcription factories, which produce mRNA, are less well understood with more undefined properties.
RNA polymerase II factories are hypothesized to allow for efficient organization of mRNA/ncRNA transcription, based on the observation that a portion of GFP-tagged RNA polymerase II molecules appears to be fixed within the nucleus [11] and labeling of nascent RNA in HeLa cells shows an increase in intensity rather than abundance of puncta over time [12, 13]. Molecular analyses have identified that transcription factories may bring together single or multiple co-regulated gene promoters with enhancer elements and RNA polymerase II [14]. Genes may alter their association with transcription factories upon activation [15] and while stoichiometric measurements of RNA polymerase II molecules support the notion of stable compartments [15], quantitative live-cell super-resolution imaging has suggested that polymerase molecules tend to act alone and demonstrate dynamic behavior, and do not cluster into super-molecular complexes [16].
Lacking from this body of work is a direct measurement of RNA polymerase II distribution and behavior in vivo. Furthermore, the dynamics of transcription have not been studied in post-mitotic cells. Lastly, most studies examine reporter genes and/or fusion protein constructs to understand endogenous chromatin dynamics during transcriptional activation: the dynamics of endogenous RNA polymerase II localization and activity, in the context of endogenous genetic loci following disease relevant stimuli, has not been explored.
To address this paucity of knowledge, we examined transcription factory behavior in the setting of heart failure, a condition in which the heart fails to meet the circulatory demands of the body. The cardiomyocyte is a terminally differentiated, post-mitotic cell that serves as a valuable model to understand endogenous structural features that facilitate transcription in the interphase nucleus. During the development of heart failure, cardiomyocytes—the contractile muscle cells of the heart—hypertrophy (i.e. they increase in size, but not number) and exhibit a pathological shift in their gene expression profile. Levels of nascent transcripts increase in hypertrophic cardiomyocytes [17], as do levels of elongating RNA polymerase II in animal models and heart failure patients [18, 19], all supportive of global transcriptional activation. The present investigation used the cardiomyocyte as a model to test the general hypothesis that in terminally differentiated cells in vivo, RNA polymerase II factories are dynamic entities into which genes are actively recruited in a locus specific manner. Two models were used: neonatal myocytes, which retain some proliferative potential and yet respond to disease stimuli, and adult cardiomyocytes, which do not proliferate and are centrally involved in disease pathogenesis. Our findings demonstrate global changes in endogenous RNA polymerase II localization and activity, resulting in coordinated, nucleus-wide partitioning of the genome into compartments of distinct transcriptional output.
2. Materials and Methods
2. 1. Cell Culture
Neonatal rat ventricular myocytes (NRVM) were isolated using enzymatic digestion, collected and plated with DMEM supplemented with 17% M199, 1× Penicillin-Streptomycin-Glutamine (Gibco), 10% Horse Serum (Gibco) and 5% Newborn Calf Serum (Gibco). For NRVM imaging, cells were plated on glass coverslips pre-coated with laminin (10μg/ml in PBS, Sigma). After 24hr plating, media was switched to serum-free (1% Penicillin/Streptomycin [Gibco] and 1:1000 ITS [BD] in DMEM). The next day cells were treated with 10μM phenylephrine for 48hr to induce hypertrophy.
2.2. Transcriptional Run-on Assay
Transcriptional run-on assay with 5’fluorouridine (5’FU, Sigma) was performed to label nascent RNA [20, 21]. NRVMs were treated in culture with 4mM 5’FU for the indicated times after which the cells were rinsed with 1×HEPEM (65mM PIPES, 30mM HEPES, 2mM MgCl2-6H2O, 10mM EGTA, pH 6.9). Cells were fixed and permeabilized (3.7% formaldehyde/1×HEPEM/0.5% Triton X-100), underwent a series of washes (1×HEPEM, PBS, 0.05% Tween20/PBS) and incubated with primary antibody (1:50 in PBS for BrdU, Sigma [1:100 for all other antibodies]). The cells were then washed with PBS and 0.05% Tween20/PBS, incubated with secondary antibody/DAPI/PBS (1:100) and mounted with Prolong Gold.
To examine RNA polymerase II/III activity, NRVMs were supplemented with 2μM CX-5461 (Selleckchem), an RNA polymerase I inhibitor [22], for 15min prior to the addition of 5’FU. The addition of 4mM 5’FU also included CX-5461. To differentiate nucleolar versus nucleoplasmic transcription, cells were colabeled with nucleolin to mark the nucleolus. 5’FU intensity analyses were carried out using FIJI/ImageJ software. Mann-Whitney tests were used for statistical analyses for comparing control and hypertrophic cardiomyocytes. To test the specificity of RNA labeling, samples were treated with 0.2mg/ml RNase A (in 0.2% Tween20/PBS, Thermo Scientific) for 45min and rinsed with PBS prior to the incubation with primary antibody.
2.3. Cell and Nuclear Size Analyses
For cell/nuclear size measurements, NRVMs were fixed with formalin and then washed with PBS and permeabilized with 0.1% Triton X-100/PBS. Cells were stained with Phalloidin/DAPI/PBS, washed with PBS and mounted using Prolong Gold. Area and circularity measurements were performed using FIJI/ImageJ and statistical analyses were performed using Mann-Whitney test. For correlations, statistical significance was determined using Pearson correlation coefficient.
2.4. Animal Models
All animal procedures were performed in compliance with NIH Guide for the Care and Use of Laboratory Animals and approved by the UCLA Animal Research Committee. Adult male C57/Bl6 mice (8weeks of age, Jackson Lab) were subjected to transverse aortic constriction (TAC) surgery until the development of heart failure, as indicated by cardiac dimensions and functional parameters monitored by echocardiography. Analyses were performed on animals in the heart failure state (~6weeks post surgery). Whole hearts and corresponding liver tissue from the same mice were fixed with formalin and paraffin-embedded to be used for imaging. For a subset of samples, isolated cardiomyocytes and homogenized brain and liver tissues were used for RNA, protein and ChIP analyses.
2.5. Adult Mouse Cardiomyocyte Isolation
Adult mouse cardiomyocytes were isolated using a Langendorff system [23]. Mice were injected with heparin and then anesthetized with pentobarbital, after which hearts were excised and cannulated. Hearts were perfused with Tyrode’s calcium-free solution (13mM NaCl, 0.54mM KCl, 0.06mM NaH2PO4, 0.1mM MgCl2, 1mM HEPES, 10mM glucose, pH 7.37), digested with 0.1mg/ml Protease (Sigma)/0.7mg/ml Collagenase, Type 2 (Worthington) prepared in Tyrode’s solution, then washed with KB solution (25mM KCl, 10mM KH2PO4, 2mM MgSO4, 5mM HEPES, 20mM glucose, 20mM taurine, 5mM creatine, 100mM glutamic acid-potassium salt, 10mM aspartic acid, 0.5mM EGTA, pH 7.18). Atria and right ventricle were removed, and left ventricular myocytes were dissociated into solution. Cells were spun and washed with PBS. Cell pellets were distributed for subsequent RNA, protein and ChIP analyses.
2.6. Immunolabeling (Tissue)
Formalin-fixed paraffin embedded tissues were deparaffinized and rehydrated with serial washes (xylene, 100% ethanol, 95%, 70%, dH2O, PBS), underwent heat-mediated antigen retrieval in 10mM sodium citrate/0.05% Tween20 buffer using vegetable steamer, then allowed to cool to room temperature. For immunofluorescence, samples were washed with PBS, blocked with 5% BSA/PBS and then incubated with primary antibody (prepared with 2.5% BSA; 1:100 dilution). Tissue was then washed with 0.05% Tween20/PBS, incubated with secondary antibody (1:100 in PBS), washed with PBS and mounted using Prolong Gold.
2.7. Generation of DNA FISH Probes
For labeling of genes, we generated DNA FISH probes as previously published, using hdfish.nl [24]. Gene coordinates (mm10 mouse genome) were used to obtain a list of PCR primers to be used to generate DNA products of ~200bp that tile the region of interest. For shorter genes, coordinates were expanded upstream and downstream, such that the probe covered at least 20kb. The following genomic sites were targeted: Nppa, gene Chr4:148,000,745–148,002,067, primers Chr4:147,997,910–148,021,634 (64 primer sets); Atp2a2, gene Chr5:122,453,512–122,502,225, primers Chr5:122,453,757–122,499,765 (51 primer sets); Gapdh, gene Chr6:125,161,851–125,165,773, primers Chr6:125,148,520–125,176,210 (60 primer sets); Nefl, gene Chr14:68,083,883–68,087,737, primers Chr14:68,069,935–68,102,872 (62 primer sets). After semi-quantitative PCR, products of primers that successfully produced specific amplicons of ~200bp were pooled, fluorescently labeled with AlexaFluor 647 using Ulysis Nucleic Acid Labeling Kit (Molecular Probes) and underwent column purification to remove excess label (Biorad). Labeled probes were reconstituted at 2ng/μl in hybridization buffer (1.7xSSC [saline sodium citrate buffer], 70% formamide, 50mM phosphate buffer [Na2HPO4/NaH2PO4], 10% dextran sulfate, 5x Denhardt’s solution, pH 7.5) and included 20ng/μl mouse cot-1 DNA (Invitrogen) and 8ng/μl salmon sperm DNA (Ambion).
2.8. 3D-DNA Fluorescence In Situ Hybridization
Formalin-fixed, paraffin-embedded tissue sections were deparaffinized and rehydrated. Samples were heated in Pretreatment Reagent (Cytocell) for 20min using a vegetable steamer, allowed to cool and washed with 2xSSC. Tissue was denatured with 60% formamide/2xSSC (70C on heat block). Concurrently, FISH probes were denatured (75C, 7min) and incubated at 37C for at least 1hr to pre - anneal. After pre-annealing, probes were added to tissue, sealed with rubber cement and allowed to hybridize at 37C (at least 24hr). Sections were washed with 50% formamide/2xSSC (42C, 2min) and 2xSSC (4×2min) and then counterstained with DAPI and WGA (Molecular Probes), washed with PBS and mounted with Prolong Gold.
2.9. RNA and Protein Preparation
Isolated cardiomyocytes were resuspended in either Trizol (Ambion) for RNA or lysis buffer (50mM Tris pH 7.4/10mM EDTA/1% SDS/0.1mM PMSF/0.2mM sodium orthovanadate/0.1mM sodium fluoride/10mM sodium butyrate/protease inhibitor cocktail tablets [Roche]). RNA was isolated using chloroform extraction, and preparation of cDNA was carried out using iScript cDNA Synthesis kit (Biorad), followed by quantitative PCR. For protein, lysates were sonicated and samples were diluted in Laemmli buffer for westerns.
2.10. Chromatin Conformation Capture Analysis
HiC data (significant interactions with q<0.01 at 40kb resolution) from isolated cardiomyocytes of sham and TAC mice was used to examine gene-gene contacts of differentially expressed genes [23]. There are 1566 differentially expressed genes in response to TAC surgery with q<0.05 (622 downregulated and 944 upregulated). For each of these genes, the number of interactions with other differentially expressed genes was recorded and subsetted based on whether they displayed increases or decreases in expression. The fractions of the gene-gene interactions for lost interactions (unique to the sham state) and gained interactions (specific to TAC) that showed up- or down-regulation were analyzed.
2.11. Chromatin Immunoprecipitation
For chromatin immunoprecipitation (ChIP) experiments, isolated adult mouse cardiomyocytes were fixed with 1% formaldehyde and quenched with 125mM glycine. Cells were then lysed using a dounce homogenizer and sonicated using both probe and bath sonications to obtain DNA fragments averaging ~500bp. Sonicates from 6 animals were pooled, and ChIP was performed using the ChIP -IT High Sensitivity kit (Active Motif), with 30μg chromatin for each IP reaction and input. Quantitation was determined by quantitative PCR by first normalizing to input and then determining fold enrichment from IgG.
2.12. Microscopy
Confocal imaging was carried out using a Nikon A1R system using 60x objective (100x was used for DNA FISH images). For DNA FISH imaging, nuclei from tissue were assessed by scanning through the z axis and 2D images were acquired selecting the plane giving the strongest FISH signal. For RNA polymerase II imaging, a 2-color in-house built stimulated emission depletion (STED) microscope was used. Samples were visualized with Atto647N (Active Motif) and Oregon Green 488 (Life Technologies) fluorophore-conjugated secondary antibodies and imaged using a 100x objective. We used 635nm excitation and 750nm depletion lasers for Atto647 visualization and 485nm excitation and 592nm depletion lasers for Oregon Green 488 labels.
2.13. Image Analysis
For RNA polymerase II distance and intensity quantifications, 2D images were processed by the following: application of a Gaussian filter, subtracting background noise and removing objects less than 50nm (based on STED microscope resolution). Imaris software (Bitplane) was then used to designate polymerase spots (clusters) and measure closest distances of center of the spots with respect to each other (Spots to Spots Closest Distance tool) as well as with the nuclear periphery (by generating a surface and using distance transformation function to calculate spot distance to surface). To determine the nuclear distribution of RNA polymerases for each nucleus (to normalize for differences in nuclear area as a complementary approach), the minimum and maximum cluster distances to the nuclear periphery were used to determine the distance range. This range was divided by 5 to generate bins, and clusters were then assigned their corresponding bin based on their distance to the periphery for analyses. Mean intensity values were also recorded. For the top and bottom most intense clusters, the spots were ranked by intensity for each nucleus and the top and bottom 20% values were used for subsequent analyses. The data plotted represents the average of means generated from individual nuclei.
For DNA FISH analyses, we carried out quantitation of nuclear gene positioning as described [25] to segment nuclei into 5 equal concentric areas using erosion image processing and determine localization of gene loci with respect to the nuclear periphery. For distance quantifications, Imaris software was used to create surfaces to demarcate the nucleus or heterochromatin (determined based on DAPI intensity). The closest distance from the center of the FISH signal to the nuclear periphery (inside of nuclear surface) or distance to heterochromatin (outside of heterochromatin surface) was calculated using the distance transformation function.
For statistical analyses of mean intensities or distances, a Mann-Whitney test was used to determine significance while a Chi-squared test was used to assess comparisons of distributions between control and treated groups.
2.14. Antibodies
ANF (Abcam ab91250); BrdU (Sigma B8434); Desmin (Sigma D1033); Gapdh (Millipore MAB374); H3K9me3 (Abcam ab8898); H3K27me3 (Abcam ab108245); IgG (Santa Cruz sc2027); Nefl (Abcam ab72997); Nucleolin (Abcam ab22758); RNA polymerase II Ser2P (Abcam ab5095); Serca2 (Abcam ab2861); secondary AlexaFluor antibodies (Life Technologies)
3. Results
3.1. Endogenous features of transcriptional activity in cardiomyocytes
Transcriptional activation is a feature of heart disease that contributes to increased abundance of pathological mRNA and protein—these factors in turn contribute to cell growth, a necessary step in disease progression [26, 27]. To understand the regulatory properties of the cardiomyocyte nucleus underpinning pathological gene expression, a transcriptional run-on assay using 5’fluorouridine (5’FU) incorporation [20] was used to obtain a direct readout of RNA polymerase activity. This assay allowed spatial and temporal measurement of nascent RNA transcripts, for example following treatment of neonatal rat ventricular myocytes (NRVM) with hypertrophic agonists (Figures 1A, S1A, S1B). Total nascent RNA content, measured by 5’FU intensity, increased by 42% in phenylephrine (PHE)-treated cardiomyocytes. RNA polymerase I mediated transcription also increased by 19% (measured by 5’FU intensity in nucleolar regions) (Figure 1B). Nucleolar area increased by 53% in these cells, as did the ratio of nucleolar to nuclear area, 0.05 in control to 0.07 after phenylephrine treatment (p<0.001), without change in number of nucleoli per nucleus (Figures 1B, S1C). This assay measured the transcriptional activity of all polymerases; because of the high transcriptional activity in the nucleolus due to RNA polymerase I, the signal observed is contributed primarily by nascent rRNA (Figure S1B). To discriminate the role of other polymerases, RNA polymerase I activity was blocked with CX-5461 [22], revealing increases in RNA polymerases II and III activity (up by 46%; Figures 1C, S1D, S1E). While previous reports demonstrated increases in transcription with hypertrophic stress in cardiac cells [17, 28, 29], these studies are the first to provide direct visualization of these changes in the anatomical context of the nucleus.
Figure 1. Endogenous transcriptional activity in cardiomyocytes following pathological stimulation.

A) Neonatal rat ventricular cardiomyocytes (NRVM) were treated with 5’fluorouridine (4mM) for the indicated times. There is a large enrichment of signal in the nucleolus (marked by nucleolin) that increases with time. The nucleolus is occupied by RNA polymerase I molecules, which carry out ribosomal RNA transcription and quench the 5’fluorouridine due to high transcriptional rates; signal is detected in the nucleolus within 5min of 5’fluorouridine treatment. B) NRVMs were treated with phenylephrine (10µM, 48hr) to induce hypertrophy and treated with 5’fluorouridine to assess transcriptional activity. Representative images of nascent transcripts in nuclei are shown (left). 5’Fluorouridine intensities were measured across the entire nucleus (total transcription) or within nucleolus (marked by nucleolin, RNA Pol I transcription) (center), with distribution of nuclear and nucleolar areas (right). Average nucleoli number is not affected with treatment (p=0.803). Control: n=60 nuclei; Phenylephrine: n=58 nuclei. C) RNA polymerase II and III actvities were measured by inhibiting RNA polymerase I activity with CX-5461 (2µM). We observe 45% and 46% increases in total and nucleoplasmic 5’fluorouridine intensities, respectively. Control: n=50 nuclei; Phenylephrine: n=47 nuclei. Data is representative of >3 biological replicates. D) The relationship between cell and nuclear areas were analyzed in hypertrophying cardiomyocytes; measurements were subsetted based on nuclei number. The increase in cell and nuclear areas, as well as reduction in nuclear:cell area ratio, was consistent in the mononucleated cell population. Binucleated cell populations had larger cell areas in hypertrophic cardiomyocytes, but total nuclear area did not differ between control and hypertrophic states. E) The nuclear versus cell sizes were plotted for the different cell populations. Mononucleated cells show significant correlation between nuclear versus cell sizes (control: r=0.655, p<0.001; phenylephrine: r=0.563, p<0.001). On the other hand, binucleated cells lost this association when treated with phenylephrine (control: r=0.506, p=0.023; phenylephrine: r=0.233, p=0.272). Scale bar = 5µm. * p<0.05; ** p<0.001 [Mann-Whitney].
Because both nuclear and total cell areas increase after hypertrophic stimulus (Figures 1B, 1C), we next examined their relationship (Figure S1F). The adrenergic agonist phenylephrine, which is analogous to endogenous ligands upregulated in vivo during heart failure and is sufficient to induce cardiomyocyte hypertrophy and pathologic gene expression [30, 31], was employed and induced a more robust response in comparison to other hypertrophic stimuli such as isoproterenol (Figures S1I, S1J). Hypertrophic cardiomyocytes (phenylephrine-treated) showed both significant increases in cell and nuclear sizes by 58% and 8% respectively without consistent gross changes to nuclear shape, indicated by circularity (Figure S1G). To determine whether this increase was proportional, we calculated the ratio of nuclear to cell areas, which was found to decrease (nuclear area dropping from 12% to 8% of the cell area), demonstrating that cell volume increases to a greater degree than nuclear volume (Figure S1G). Cardiomyocytes do not divide and their hypertrophic growth is associated with disease. Recent studies have suggested that nuclear content (most cardiomyocytes are binucleated) correlates with functionality of the cells during regenerative processes in disease [32, 33]. Interestingly, the change in nuclear to cell area ratio was consistent when cells are segregated based on nuclei number (Figure 1D). While there was an upward trend, there was no statistically significant difference in the fraction of binucleated cells in control and phenylephrine-treated cell populations (Figure S1H). In control cells, the nuclear area versus cell area relationships between mononucleated and binucleated cells were significant and consistent, with slopes of 8.22 (r=0.655, p<0.001) and 7.321 (r=0.506, p=0.023); phenylephrine altered these relationships to 11.20 (r=0.563, p<0.001) and 4.691 (r=0.233, p=0.272), respectively (Figure 1E). While the relationship between cell and nuclear area was maintained—although different—in control and phenylephrine-treated mononucleated cells (significant correlations but different slopes), this relationship was lost in binucleated cells with phenylephrine treatment. While hypertrophy was associated with increases in both cell size and transcriptional activity, these variables showed little correlation with each other in either control or hypertrophic states (Figure S1K).
3.2. Developmental state differences in stress response of transcription factories
Although neonatal cells (NRVMs) exhibit transcriptional responses reminiscent of those seen in the adult heart during disease, neonatal and adult cells are quite different in terms of morphology, metabolism, myofilament function and—linking, and perhaps driving, these other differences—gene expression. No previous analyses have examined real-time transcription in cardiac cells, and to our knowledge, no characterization of the effects of developmental stage and terminal differentiation on the endogenous transcriptional environment of any cell lineage has been reported. To investigate how terminal differentiation influences transcription in the anatomical context of the nucleus, we directly measured RNA polymerase II localization. Previous studies using live cell imaging of RNA polymerase II have demonstrated a subpopulation of polymerases are transcriptionally active, engaged in the formation of so-called transcription factories [11]. The punctate nature of the labeling patterns of 5’FU provided support for the organization of such transcription factories in cardiomyocytes. We next sought to characterize the properties of these transcription sites in healthy neonatal and adult cardiomyocytes, as well as those in the setting of disease. Because previous studies have reported the size of transcription factories to be <200nm (below the resolution of conventional microscopy) [9, 34], super-resolution stimulated emission depletion (STED) microscopy, which improves the resolution down to 50nm, was used to characterize features of cardiac transcription factories, as marked by actively elongating RNA polymerase II (phosphorylated at serine 2 of the C terminal domain) (Figures 2A, S2A, S2B). Hypertrophic NRVMs had a higher number of detected puncta (averaging 988 in control and 1400 in phenylephrine-treated nuclei) but were comparable to control cells when normalized to nuclear area (Figure 2B). The cluster size and average intensity of polymerase puncta also did not differ between control and phenylephrine-treated cells (Figures 2B, 2C). The intensity ranges, minimum and maximum values across nuclei, between groups were also comparable (Figure S2C). To examine the distribution of clusters within the nucleus for determination of spatial organization, the clusters were ranked based on closest distance to the nuclear periphery. The maximum and minimum distances were then used to determine the range of cluster distribution across each nucleus, which was used to generate 5 bins (Figure 2D); this segmentation generates bins of equal distances to the periphery. The fraction of total clusters per nucleus across each bin did not differ between groups (p=0.999), nor did the average mean intensity of the clusters (p=0.861; Figure 2E). These findings suggest that RNA polymerase II distribution is tightly controlled within the developmentally immature cardiomyocyte.
Figure 2. High transcriptional activity correlates with increases in cluster number in neonatal myocytes and increases in mean intensity and size in adult cardiomyocytes.

A) NRVMs were labeled with RNA polymerase II for STED imaging of cardiac transcription factories which were quantified using Imaris (non-nuclear signal was subtracted as background). Scale bar = 2µm. B) The number of clusters increases while the cluster density (spot number normalized to nuclear area) or size indicate no difference. C) Mean cluster intensity also does not differ. Together, these data suggest that increases in transcriptional activity are due to recruitment and activation of polymerases in the NRVM nucleus after agonist treatment. D) To examine the spatial distribution of puncta, the nucleus was divided into 5 bins based on the maximum and minimum distances to the periphery for each nucleus and then assessed cluster number and cluster intensity at each bin (1 is closest to and 5 is furthest from periphery). Scale bar = 2µm. E) The distribution of percent of total clusters per nucleus found within each bin is plotted (top) along with the mean intensities of the clusters (bottom). Treatment does not affect intensity distribution (p=0.861, two-way ANOVA). F) RNA polymerase II labeling was performed in adult mouse heart tissue sections in animals subjected to sham or TAC surgery, and cardiomyocyte nuclei from the fixed tissue sections were used for analyses (nuclei were selected based on co-labeling with desmin to ensure non-cardiomyocyte nuclei were not incorporated into analyses). Representative nuclei of RNA polymerase II-labeled nuclear are shown with respective magnifications and examples of subsequent cluster analyses. Scale bar = 3µm (1µm for magnification). G) RNA polymerase II total cluster number is not affected, but the mean cluster area does increase after pressure overload stress. H) Mean intensity of total active RNA polymerase II puncta is higher in nuclei from failing hearts. I) The distribution of spots across the nucleus does not differ between sham or TAC mice while the increase in intensity occurs uniformly across the nucleus as indicated by average of mean cluster intensity at each bin (p<0.001, two-way ANOVA). Control: n=10 nuclei; Phenylephrine: n=11 nuclei. Sham: n=21 nuclei; TAC: n=20 nuclei. (Imaging experiments, with different sets of control/phenylephrine -treated cells and sham/TAC hearts, were repeated >3 times. Representative dataset of analyses is presented here.) * p<0.05; ** p<0.001 [Mann-Whitney; for cluster distribution, Chi-squared]. Error bars represent SEM.
We next tested this phenomenon in adult cardiomyocytes in situ within heart tissue sections (Figure S2D). Adult mice were subjected to transverse aortic constriction surgery (TAC), which induces pressure overload to the heart and causes heart failure, marked by a reduction in left ventricular ejection fraction and increased cross-sectional cardiomyocyte area [31]. Heart tissue sections were used to examine cardiomyocyte nuclei from the left ventricle labeled for endogenous RNA polymerase II using STED imaging. Interestingly, unlike the neonatal cardiomyocytes treated with phenylephrine, the number of RNA polymerase II puncta in adult mouse cardiomyocytes from TAC animal heart sections did not show significant change when compared to heart tissue sections from mice subjected to sham control (731 in sham vs 938 in TAC nuclei), but the cluster size increased with heart failure (Figures 2F, 2G, S3G). Furthermore, the mean intensity of the puncta also increased (Figures 2F, 2H, S3I). Again, when examining the cluster properties across the nucleus, the fraction of the clusters across each bin remained consistent (p=0.989) with a uniform increase in intensity across nuclei after TAC stress (p<0.001; Figures 2I, S3J, S3K, S3L). These findings reveal the behavior of endogenous transcription factories in adult myocytes (which appear to target all RNA polymerase clusters, rather than a discrete subset), highlighting previously unrecognized differences in transcription factory function.
To further investigate this observation, we examined the spatial organization of the RNA polymerase II puncta with respect to the nuclear envelope and with respect to each other. The distribution of all of the clusters as measured to the nuclear envelope increased in NRVMs, from 1625nm to 2036nm (Figures 3A, S3A), as did the absolute distances for the most intense clusters (top 20% of clusters, as ranked per nucleus; Figures 3B, S3B, S3C). The most intense puncta were more centralized in the nucleus, and this pattern was unchanged with phenylephrine treatment (Figures S3B, S3C). Spacing between them, measured as the average distance to a spot’s closest neighbor, did not differ in NRVMs (139.6nm vs 140.3nm; Figure 3C). Transcription factories were also measured by 5’FU incorporation (Figures 3D, S3D): while the number of 5’FU clusters increased with phenylephrine treatment, the density of clusters (number of puncta normalized to area) was unchanged between control and treated nuclei (Figure 3E). The distance of a 5’FU spot to the nuclear periphery increased without change to spacing between 5’FU clusters (Figures 3F, S3E, S3F), consistent with observations made with active RNA polymerase II. To confirm this observation, the distance of each RNA polymerase II cluster to the closest 5’FU was measured and found to be unchanged (134.9nm in control and 133.2nm in hypertrophic cells; Figure 3G). Additionally, we also examined colocalization between RNA polymerase II and 5’FU signals and found no changes in average correlation coefficient, determined by intensities for each channel (0.206 in control vs 0.199 in hypertrophy, p=0.697).
Figure 3. Distinct spatial and intensity properties of RNA polymerase II factories in neonatal and adult cardiomyocytes.
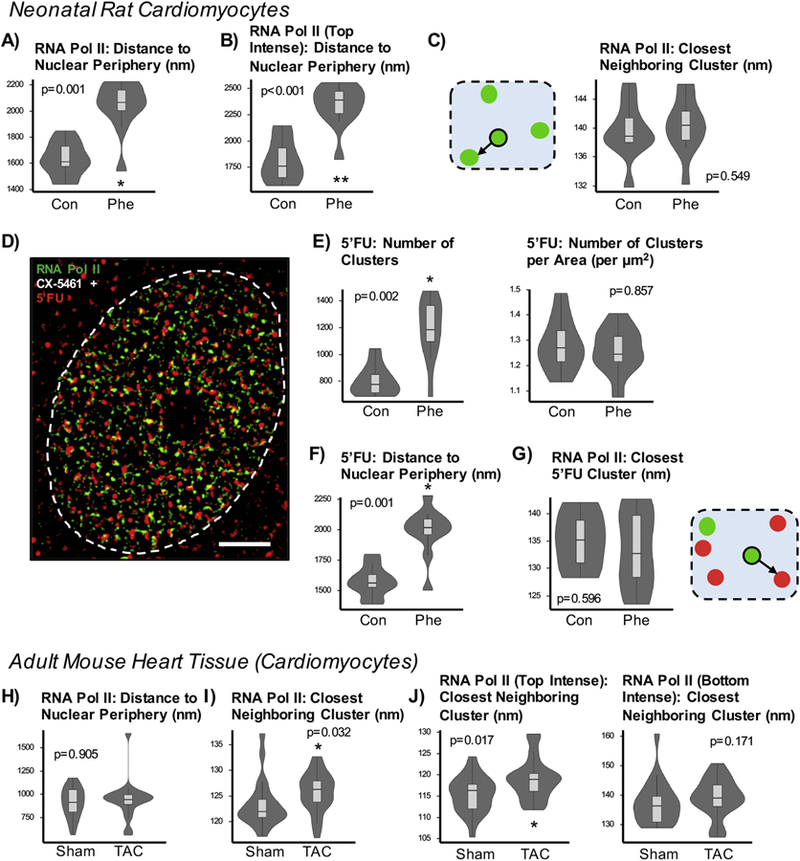
A) In neonatal cardiomyocytes, the distances from the center of each cluster to the nuclear periphery increases. B) For each nucleus, the clusters were ranked by mean intensity and the top 20% were used for analysis. The absolute distance to the periphery of the top-ranked clusters increases (highly intense clusters found more centrally). C) To assess anatomical distribution of cardiac transcription factories, we analyzed the distances to the closest neighboring spot. The cartoon illustrates RNA polymerase II clusters in green. For each spot, the distance to the nearest neighbor is recorded. The mean spacing between RNA polymerase II clusters remains similar between control and hypertrophic NRVMs. D) Analyses integrating 5’fluorouridine measurements, a direct readout of RNA polymerase activity, were performed. Neonatal myocytes were treated with CX-5461 and then supplemented with 5’fluorouridine for 30min. Scale bar = 2µm. E) Like RNA polymerase II, the absolute number of 5’fluorouridine clusters increases though not when normalized to nuclear area. F) The distance of 5’fluorouridine clusters to the nuclear periphery increases, consistent with measurement patterns of RNA polymerase II. G) The distance of closest neighboring 5’fluorouridine (red) with respect to RNA polymerase II cluster (green) does not significantly change, although the trend is towards a decrease. H) In adult cardiomyocytes from sham and pressure overload sections, the distance of clusters to the nuclear envelope does not change after TAC surgery. I) Unlike in neonatal cardiomyocytes, the average distances between closest neighboring RNA polymerase II clusters show change in spacing in the adult cardiomyocyte in the disease setting (123.5nm to 125.8nm). J) Examination of top intense clusters also shows increased spacing between close RNA polymerase II neighbors (115.2nm to 118.8nm) while the least intense clusters do not show changes in spacing between neighboring puncta. Control: n=10 nuclei; Phenylephrine: n=11 nuclei. Sham: n=21 nuclei; TAC: n=20 nuclei. * p<0.05; ** p<0.001 [Mann - Whitney].
Adult cardiomyocytes analyzed from diseased hearts showed no change in distance of clusters to the nuclear periphery compared with sham hearts (Figures 3H, S3H). On the other hand, the spacing between clusters increased during disease, from 123.5nm to 125.8nm (Figures 2F, 3I). This increased distance was pronounced in the cohort of most intense puncta, where the increase was 3.2%, from 115.2nm to 118.8nm (Figure 3J). These analyses reveal distinct features between developing and mature cardiomyocytes in the response of endogenous transcription clusters to stress. In the neonatal cardiomyocyte, there is an increase in the number of RNA polymerase II factories without a change in individual cluster intensity, maintaining the global organization and spacing between clusters in response to hypertrophic stress. In contrast, adult cardiac nuclei exhibit recruitment and accumulation of active RNA polymerase II (indicated by increases in size and intensity of the clusters without a change in number).
3.3. Nuclear positioning of genes is a mechanism for differential gene expression
It has been suggested that physical positioning of genes within the nucleus influences expression, with central localization associated with expression and peripheral localization associated with silencing [35, 36]. Whether this phenomenon is true for endogenous gene loci, and in the setting of adult terminally differentiated cells, has never been examined. To assess this hypothesis, DNA fluorescence in situ hybridization (FISH) was used to map endogenous gene loci within the cardiac nucleus in tissue sections from healthy and diseased mouse hearts (Figure 4A) [24]. Nuclei were segmented into concentric annuli of equal areas (or bins) by erosion processing, thereby giving the measured gene locus an equal probability of falling in any one of the analyzed areas [25]. This approach served to normalize for variability in nuclear size and shape. Atp2a2, a downregulated gene in the failing heart [26], underwent a significant change in nuclear positioning, shifting towards the nuclear periphery (bin 1, 0.5% to 4.4% of measured loci) and away from the center (bin 5, 46.3% to 36.7%) (Figures 4B, S4). A second gene, Nppa, whose expression increases with disease, exhibited the antithetical pattern of change in distribution, shifting away from the periphery (7.5% to 1.1% of loci; Figures 4C, S4) [26]. These changes in gene localization were specific and not reflective of wholesale chromatin reorganization: the control genes Gapdh (housekeeping gene that remains actively expressed in both states) and neuronal Nefl (a neuronal gene, which remains silent in healthy and diseased hearts [37]) did not show changes in distribution (Figures 4D, 4E, S4). Interestingly, genes that can be expressed in the heart (Atp2a2, Nppa and Gapdh) tended to exhibit an overall localization pattern favoring the center of the nucleus, whereas Nefl, which is never expressed in the heart, lacks preferential distribution and is uniformly distributed (Figure 4). When examining the absolute distances (rather than relative distances, as was done in Figure 4) of gene loci to the nuclear envelope, the same trend of localization was observed, although the effect was muted (differences in distance were minimal and not significant for Atp2a2 and Nppa; the differences were significant for Gapdh and Nefl; Figure 5A). We attribute this effect to variation in three-dimensional nuclear morphology. As a further control, we examined gene localization in the liver. This choice is notable because the liver is centrally involved in heart failure [38], yet these target genes undergo altered expression in the heart (and not the liver), allowing us to test the hypothesis that systemic stresses that impact multiple organs and cells have cell type-specific effects due to prevailing chromatin environments in the given cell. The data indicate that expression and positioning of these genes (Atp2a2, Nppa, Gapdh and Nefl) were unaffected in liver (Figures S4, S5) supporting a model in which cell type-specific chromatin enables distinct spatial organization of transcription.
Figure 4. Cardiac gene localization is associated with differential expression in the failing heart.

A) DNA FISH measurements to the nuclear periphery were normalized for shape and size between nuclei by using DAPI to bin the nucleus into 5 equal area annuli using erosion processing, allowing for an equal probability of finding a locus in any given bin [25]. Loci were assigned a bin location (1 near the periphery to 5 at the center) for analysis. Scale bar = 3µm. B-E) DNA FISH was performed in adult mice, either control or those that had developed heart failure. Heart tissue sections were labeled for the following genes Atp2a2 (B), Nppa (C), Gapdh (D) and Nefl (E). The distribution of nuclear positioning is shown, with bins 1 (Periphery) and 5 (Center) highlighted on the right. Quantitation is compiled from different labeling experiments from 2–3 different hearts per treatment. Atp2a2: Sham n=167 nuclei/205 loci, TAC n=220/297; Nppa: Sham n=179/213, TAC n=145/188; Gapdh: Sham n=178/215, TAC n=177/208; Nefl: Sham n=152/192, TAC n=177/224. Scale bar = 5µm. * p<0.05 [Chi-squared].
Figure 5. Localization of genes with respect to nuclear heterochromatin domains associate with patterns of differential expression.
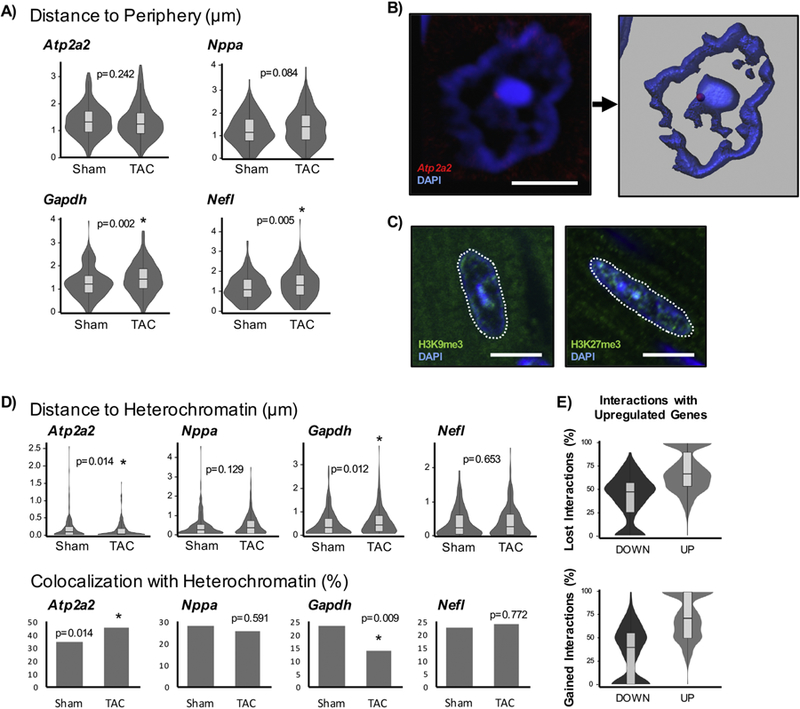
A) In addition to performing binned assessments of radial positioning of genes, the absolute distances to the nuclear periphery were examined (note that these are absolute distances and do not account for differences in nuclear shape or area). B) To measure the distance to constitutive heterochromatin, a DAPI intensity threshold was used in Imaris to create a surface. The closest distance of the FISH spot to DAPI surface was measured. If loci were found inside the surface (within the heterochromatin), they were assigned a distance of 0µm. Scale bar = 5µm. C) The intense DAPI signal corresponds to constitutive heterochromatin, marked by H3K9me3, and is separate from facultative heterochromatin, indicated by H3K27me3. H3K9me3 colocalizes well with DAPI while H3K27me3 is found throughout the nucleus. Scale bar = 10µm. D) The distribution of absolute distances to heterochromatin are shown on top. The fractions of loci that colocalize (have distances of 0µm and associate directly with heterochromatin), are shown on bottom. Atp2a2: Sham n=205 loci, TAC n=298; Nppa: Sham n=216, TAC n=190; Gapdh: Sham n=226, TAC n=212; Nefl: Sham n=192, TAC n=227. E) HiC data was used to examine gene-gene interactions from isolated sham and TAC cardiomyocytes [23]. The lost and gained interactions for significant differentially expressed genes (DOWN or UP) with other differentially expressed genes are described. Genes that are upregulated associate with other upregulated genes while those that are downregulated show the inverse relationship, providing support for genes with similar expression behavior tend to congregate together. * p<0.05 [Mann-Whitney; for co-localization, Chi-squared].
3.4. Heterochromatin surfaces are structural features of cell type-specific nuclear architecture and gene expression
Adult mouse cardiomyocyte nuclei exhibit a unique nuclear architecture visible with DAPI staining: a large heterochromatic feature is commonly observed at the center of the nucleus, in addition to a ring of heterochromatin at the periphery. Thus, there is the possibility that gene silencing may also be occurring at this centrally located heterochromatin island (Figure 5B). To test this possibility, we performed a complementary analysis in which the distance of a gene locus was measured relative to the nearest region of constitutive heterochromatin. We observed that the aforementioned DAPI patterns closely overlapped with H3K9me3, a mark for constitutive heterochromatin, and not H3K27me3 (a facultative heterochromatin mark that is found across the nucleus), and thus measured the distances between a FISH locus for an individual gene and the nearest intense DAPI surface (Figures 5B, 5C). The Atp2a2 loci exhibited a significant decrease in average distance to heterochromatin surfaces (182nm in sham and 128nm in diseased cells) whereas the same distances for Nppa increased from 392nm to 453nm (Figure 5D), in agreement with the changes in gene expression and nuclear localization described above. Furthermore, the number of Atp2a2 loci that colocalize with DAPI (that is, those with a distance equal to 0nm) also increased (35% to 45% of loci) in disease. Gapdh displays the opposite behavior, moving away from constitutive heterochromatin surfaces, again in agreement with changes in expression and the measurements in total nuclear orientation.
3.5. Role of chromatin features to establish nuclear architecture and transcription factories
To investigate the role of chromatin features to establish gene expression microenvironments, we examined chromatin immunoprecipitation-DNA sequencing (ChIP-seq) data for RNA polymerase II enrichment from healthy and diseased hearts [39], confirming behavior of specific genes by ChIP-PCR in isolated adult cardiomyocytes (Figure S6). Activation of Nppa expression in disease was associated with enrichment of active RNA polymerase II at its promoter, whereas the converse was observed with Atp2a2, which is downregulated and showed reduced occupancy. Gapdh (constitutively expressed) had substantial enrichment of RNA polymerase II along its gene body, whereas Nefl (not expressed) had no enrichment. Our results from isolated cardiomyocytes were comparable to results from whole hearts (which are composed of a heterogeneous cell population); the qualitative differences may be explained by the contributions from non-cardiac cells in the ChIP-seq experiment. To test the involvement of specific chromatin domains determined by sequencing, data from chromosome conformation capture studies (Hi-C) performed in the same model [23] were used to investigate interactions amongst genomic regions constituting a unique transcription factory. For each up- or down-regulated gene, the corresponding interactions with other differentially expressed genes were examined. Of the other genes interacting with differentially expressed genes, the majority of them also change expression in the same direction: that is, upregulated genes tend to have a greater fraction of interactions with other upregulated genes and vice versa (Figure 5E). This was a dynamic process: upregulated genes gain interactions with other differentially expressed genes, 72.7% of which are also upregulated, whereas 65.8% of gained interactions of downregulated genes are also downregulated. These results indicate that genes that are co-regulated tend to congregate with each other in nuclear environments tailored for transcription and demonstrate a mechanism for coordinated transcription during stress that involves physical reorientation of genomic regions to designated sites for transcription.
4. Discussion
Our data support a model in which stress-induced changes in global transcription are mediated by recruitment or loss of RNA polymerase II molecules at transcription sites in the adult mouse cardiomyocyte nucleus, with corresponding movement of differentially expressed genes to and from these factories (Figure 6). During diseases like cancer and heart failure, there is a global shift in DNA packaging towards a more plastic phenotype as evinced by changes in histone isoforms and modifications [31, 40, 41]. Super-resolution imaging of histone H3 proteins previously provided evidence of global structural reorganization of chromatin domains in hypertrophic cardiomyocytes [30]. Recently, chromatin capture studies in adult cardiomyocytes have demonstrated changes to genomic interactions in the setting of heart failure [23] as well as cardiac development [42]. How these global arrangements affect transcriptional output is unclear and the spatial contributions to gene expression have not been examined.
Figure 6. Gene expression is regulated by spatial positioning at the nuclear scale.
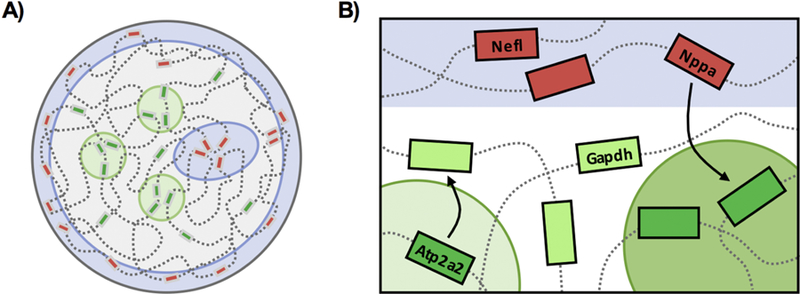
A) The nucleus is compartmentalized into active and inactive compartments. Inactive regions (red boxes) are found in heterochromatic regions (blue), which are heavily enriched at the nuclear lamina and centrally at chromocenters while highly transcribed regions (green boxes) are found at transcription factories (green). The cardiac nucleus contains stable RNA polymerase II transcription compartments enriched with active genes while genes outside of these sites in the nucleoplasm are expressed at low levels or poised for activation. Upon pathological stress, strength of transcription factory activity increases, without significant changes to number, through the recruitment of elongating RNA polymerase II molecules. B) The differential changes in gene expression are mediated by changes in gene positioning with respect to active (transcription factories, green) and inactive (heterochromatin, blue) compartments. Increases in gene expression correlate with the concentration of upregulated genes and RNA polymerase II molecules at discrete transcription sites.
In various cell models—but to our knowledge, not tested in mammalian tissues—it has been shown that gene silencing can occur at the nuclear envelope, such that recruitment represses expression and release from the lamina towards the center of the nucleus promotes expression. Imaging approaches have utilized reporter genes to complement sequencing-based DamID analyses, which have mapped genomic interactions with the nuclear lamina [35], and shown gene silencing occurs at the nuclear envelope, with anchoring mechanisms dependent on histone methylation and histone deacetylase proteins [43–45].
Our results also point to a clear role for distinct nuclear orientation of genes, with respect to nuclear periphery and chromatin microenvironments, that positions genes in the appropriate physical space for distinct transcriptional responses. Additionally, our findings support distinct mechanisms of transcriptional regulation at different stages of development and in response to pathological stress during disease—these features may be reflective of altered chromatin plasticity during cellular maturation. These novel features of transcription are reflective of cell type-specific nuclear architecture, insofar as the systemic stresses of disease did not affect gene positioning (or expression) in the liver. This study provides support for the compartmentalization of gene expression in vivo and quantifies the relationship between gene expression and chromatin architecture.
Consistent with earlier studies of cellular hypertrophy [17], we observed increased RNA production in hypertrophic cardiomyocytes, extending previous observations by (i) quantifying the relative contributions of different RNA polymerase isoforms and (ii) determining the anatomical compartments of the nucleus responsible for transcription in distinct developmental stages. We analyzed the relationship between global transcription and cell hypertrophy and characterized the structural organization of cardiac transcription in the mature cardiomyocyte nucleus. Increases in nuclear size are correlated with cell size in a state specific manner (ratio of nuclear to cell size areas differs between control and hypertrophic states) and is dependent on nuclear content: the correlation is lost in binucleated cells in hypertrophy, although it is unknown if these cells underwent mitosis without cytokinesis as a result of phenylephrine treatment. While there was an increase in the number of binucleated cells, it was not statistically significant, suggesting nucleation is independent of regulation of cell size. Interestingly, a direct correlation between transcript abundance, measured by total 5’FU intensity, and cell area was not observed in either control or hypertrophic states. Nevertheless, these findings suggest that mononuclear and binuclear cells exhibit distinct transcriptional programs that may contribute to the different functional capabilities of these cells [32]. In a time-course study of mouse skeletal muscle hypertrophy, an inverse relationship was observed between transcription and cell size at the peak of transcriptional activation in myofibers, whereas no association between transcriptional activity and cell size was found in control conditions [46]. It is intriguing to speculate that our measurements reflect a new state in which the cells have stopped responding to the phenylephrine and reached a steady cell size—future experiments would be required to test whether the relationship between transcription and cell size was different at a more acute time point after hypertrophic stimulation.
Characterization of the spatial organization of active RNA polymerase II clusters revealed different mechanisms of transcriptional remodeling. In hypertrophic neonatal cardiomyocytes, new RNA polymerase II/5’FU clusters formed, ostensibly to accommodate increases in transcriptional demand as the nucleus gets larger. On the other hand, adult cardiac transcription factories physically expanded in size, indicated by increases in area and intensity of RNA polymerase II puncta. This observation suggests recruitment of polymerase molecules to established factories in adult cardiomyocytes, in agreement with previous evidence from sequencing studies that indicates RNA polymerase II elongation increases with disease [18, 19]. Single molecule imaging of RNA polymerase II reveal that the majority of polymerases exist as individual molecules, are dynamic and form transient interactions at the basal state without perturbation [16, 47]. However, RNA polymerase clusters have increased lifetime and enhanced clustering upon transcriptional activation induced by serum stimulation [47, 48], which is correlated with increased mRNA production [49]. These support that the pathological stress induced by TAC surgery affects the stabilization and properties of RNA polymerase II clusters in the cardiomyocytes. The significant changes observed with the pressure overload model of disease in vivo, in comparison to phenylephrine treatment in the neonatal cells, could be reflective of the severity of stress as well as varied genome plasticity in these different developmental states. During differentiation to parietal endoderm, changes in transcription rates (based on labeling of nascent RNAs) are associated with changing nuclear volumes, maintaining the activity and distribution of transcription factories across the nucleus [50]. This prior observation is supported by our data on RNA polymerase II patterning in the neonatal cell model of hypertrophy, whereas size and intensity of the polymerase clusters are enhanced in the adult cardiomyocyte. The neonatal cardiomyocyte is still maturing and has residual proliferative potential (both characteristics that are absent in the adult cardiomyocyte): these features may be the result of a nuclear architecture that maintains greater potential for dynamism in the formation of transcription factories and the reorganization of genes. In the heart, targeting of TFIIB, important for the recruitment of RNA polymerase II to disease-associated genes, can prevent pathological hypertrophy [51]. Our studies extend this observation to map the physical orientation of these transcriptional events within the nucleus.
These experiments represent the first demonstration of the endogenous localization of distinct cohorts of genes in the adult cardiomyocyte nucleus, quantifying how this localization contributes to gene expression during disease. Genes expressed in the heart were more centrally located, whereas those not expressed were randomly distributed. Surprisingly, the liver also displays similar basal localization patterns to those observed in the heart, although unlike the heart, these distributions were unaffected in an in vivo model of heart failure. These patterns could be explained by a number of reasons: (i) the gene locus is situated near large heterochromatin segments on the linear chromosome. In general, centromeres and telomeres converge at constitutive heterochromatin foci found centrally (chromocenters) or at the periphery. The distance of the genes on the linear chromosome to these domains could restrict the region of localization in three dimensions; (ii) the associating genomic regions in the basal setting could also determine where the gene is found, a guilt by association model. For example, if Nppa is localized in a permissive chromatin environment in the basal state, this could enable its more ready activation even in a state where it is off (a type of structural transcriptional poising); (iii) chromatin structure is largely determined by developmental stage, such that once chromosome territories and nuclear architecture have been established on a large scale, nuanced reorganization (such as that observed with pressure overload hypertrophy in vivo) may be sufficient to alter association of genes with sites of transcription and/or heterochromatin regions. Unlike stem cell to cardiomyocyte differentiation, the changes in positioning with respect to the nuclear lamina are subtler during disease progression and point to complementary influences by local epigenetic mechanisms.
Genes with similar transcriptional behavior were found to preferentially interact in the context of the endogenous nucleus. Strikingly, 70% of the gene-gene interactions were between genes of increased expression, supporting the model wherein chromatin dynamics bring together coregulated genes in three dimensions in the cardiomyocyte. Our findings also demonstrate that a gene’s local environment is important for “priming” the locus for transcriptional activation or repression, which can then be regulated by transcription factors. Analysis of transcription factories targeted by TNFα signaling showed evidence of hierarchical regulation, such that disruption of the dominant gene (by cleaving the gene to affect gene looping) had an outsized effect on the expression of other genes in the transcription factory as contrasted with disruption of more subordinate genes (which had minimal or no effect on transcription at the factory) [52]. Another approach disrupted gene activity through knockdown of regulatory ERα transcription factor, which indirectly affected the expression of interacting promoters at the shared transcription factory [14]. Identification of the specific genomic components of cardiac transcription factories will be key to understanding the features—including shared epigenetic regulation or binding of a common set of transcription factors—that recruit and stabilize genes and regulatory elements. The mechanisms that drive shifts in gene positioning are still not well understood because most studies either measure one or a few chromatin marks with great specificity in terms of genome binding (i.e. via ChIP -seq) but with little or no data on where these marks and their associated genes localize within the anatomical context of the nucleus (as is provided by studies like those in the current paper) or relative to each other (e.g. using chromatin capture technologies).
What are the factors involved in the structural dynamics observed in this study? Decondensation of chromatin is sufficient to move gene loci away from the nuclear periphery, independent of transcription [25]. H3K9 methylation is an important signal for the recruitment and anchoring of heterochromatic loci to the nuclear lamina [53]. HDAC3 has been demonstrated to be important in keeping loci at the nuclear lamina in fibroblasts [43]. More recently, HDAC3 was shown to be a regulator of cardiac differentiation; loss of HDAC3 was sufficient for the release of cardiac genes resulting in premature cardiomyocyte differentiation [54]. In the cardiomyocyte, overexpression of histone deacetylase, HDAC4, has been shown to decrease the expression of a subset of genes, which was also linked to their displacement from the nuclear pores [55]. These observations provide support for the importance of three dimensional physical orientation, as regulated by histone-modifying enzymes, to determine regulation of gene expression.
Pathological transcription during heart failure exhibits at least two distinct behaviors: upregulation through pause-release of RNA polymerase II (employed for housekeeping genes), and de novo recruitment of RNA polymerase II (employed for disease-associated loci) [39]. A new frontier is now to understand how these categories of genes are physically localized in the nucleus using imaging approaches. The present investigation sets the foundation to search for cell type-specific chromatin features that have evolved for bespoke transcriptional requirements of different cells and to understand how spatial organization of genes regulates their function in health and disease.
Supplementary Material
Supplemental Figure 1: Characterization of transcription factory structure and function in cardiomyocytes. A) 5’Fluorouridine (5’FU) is a uracil analogue that can permeate live cells and be incorporated into synthesizing RNA. Sites of 5’FU incorporation can then be detected using immunofluorescence. Representative image of neonatal rat ventricular myocytes treated with 4mM 5’FU for 30min is shown (left). RNase A treatment, post-5’FU incorporation, shows absence of 5’FU signal, demonstrating specificity of labeling to RNA (right). Scale bar: 5μm. B) RNA polymerase I activity quenches 5’FU signal into the nucleolus while treatment with CX-5461 allows for selective labeling of transcriptional activity in the nucleoplasm, which is associated with sites of active RNA polymerase II (phosphorylated at serine 2) occupancy, indicated by co-localization (yellow). Scale bar = 5µm. C) The distribution of nucleoli number does not differ with hypertrophic stress. D) NRVMs were treated with CX-5461, an RNA polymerase I inhibitor (2µM, 15min), prior to the addition of 5’FU. Thus, 5’FU labeling is reflective of RNA polymerase II and III activity. Note the absence of signal in the nucleolus, marked by nucleolin. Scale bar = 5µm. E) CX-5461 treatment efficiency in inhibiting RNA polymerase I was confirmed by measuring ribosomal RNA expression, ITS (premature) and 18S (mature) at 15min (before addition of 5’FU) and 45min (time point at which cells were fixed for immunostaining). 15min: n=4; 45min: n=6. Error bars represent SEM. F) NRVMs were treated with 10µM phenylephrine for 48hr, after which they were fixed and labeled with phalloidin and DAPI for cell and nuclear size measurements. Scale bar = 25µm. G) Quantifications of cell and nuclear sizes show significant increases in cell (60% increase) and total nuclear (8% increase) areas. The ratio of total nuclear area to cell area was significantly reduced in the hypertrophic cardiomyocytes, highlighting greater growth of the cell versus nucleus (nuclear occupancy is reduced to 8% of the cell from 12%). The increase in nuclear size does not affect nuclear shape, indicated by circularity (a perfect circle has circularity value of 1). Control: n=259 cells (279 nuclei); Phenylephrine: n=255 cells (279 nuclei). H) Nuclei number after phenylephrine treatment is unaltered. Control: n=323 cells, Phenylephrine: n=372 cells. I) NRVMs treated with isoproterenol (1µM for 48hr) show minimal hypertrophy response. Scale bar = 25µm. J) Phenylephrine treatment induces robust pathological gene expression. K) The relationship between mean 5’FU intensity and cell area were compared. There was no significant correlation between nascent transcript levels and cell size (control: r=0.177, p=0.133; phenylephrine: r=0.083, p=0.515). Control: n=73 cells; Phenylephrine: n=64 cells. * p<0.05; ** p<0.001 [Mann-Whitney; for nucleoli number distribution, Chi-squared].
Supplemental Figure 2: Super-resolution mapping of cardiac transcription factories. A) NRVMs treated with phenylephrine were labeled for active RNA polymerase II, phosphorylated at serine 2, and 5’fluorouridine incorporation (2µM CX-546, 4mM 5’FU) and imaged with STED microscopy. Left image shows the confocal image of a control nucleus with its corresponding super-resolution image on the right. Boxed regions highlight the level of detail that can be resolved with the STED microscope. Scale bar = 2µm (1µm for magnification). B) Nuclear area increases with hypertrophy in the cells used for these analyses. C) RNA polymerase II clusters are mapped and color-coded based on mean cluster intensity. Analyses examining the intensity ranges (measuring the minimum and maximum intensities per nucleus) demonstrate no difference with hypertrophic agonist treatment. Control: n=10 nuclei; Phenylephrine: n=11 nuclei. Scale bar = 5µm. D) Adult cardiomyocyte transcription factories were analyzed in situ from heart tissue sections. Cardiac nuclei were distinguished from non-cardiomyocyte nuclei using desmin labeling. Scale bar = 50µm (5µm for magnification). ** p<0.001 [Mann-Whitney].
Supplemental Figure 3: Properties of RNA polymerase II factories in hypertrophic and failing cardiomyocytes. A) The normalized distance to the nuclear envelope shows no change in neonatal cardiomyocytes (normalized to radial distance to account for differences in nuclear size). B) The average of intensity of the top 20% intensity ranked clusters does not differ. When normalizing peripheral distance, there is a decrease in distance from the periphery though not significant. C) The positioning across the nucleus of the most intense clusters shows no change in distribution after hypertrophic stress (left). Enrichment was determined by comparing fraction of the most intense clusters to the fraction of total clusters for each bin (right). D) 5’Fluorouridine intensity is significantly reduced in hypertrophic myocytes. E) The average spacing between nearest neighboring 5’fluorouridine puncta does not change. F) The normalized distance of 5’fluorouridine clusters to the nuclear periphery decreases. G) Cluster number was normalized to nuclear area and indicated no difference with pressure overload in adult mouse cardiac nuclei. The nuclei that were analyzed had similar areas. Nucleoplasmic regions were determined based on presence of RNA polymerase II signal. Regions devoid of signal were regions associated with nucleoli or characteristic cardiomyocyte heterochromatin chromocenters. The ratio of nucleoplasm to nuclear area does not change. H) Cluster distances to the periphery were normalized to area and are not different between groups. I) The mean intensity of the highly ranked clusters significantly increases with TAC stress. The bottom 20%-ranked clusters have increases in mean intensity without changes to spatial organization in the nucleus (distance to nuclear envelope, p=0.689). J) To normalize nuclear distances, the difference between maximum and minimum distances to the nuclear periphery was used to generate 5 bins. Scale bar = 3µm. K) The top 20% most intense clusters were examined. The distribution of intensity shows that these clusters are found across the nucleus without preferential localization as indicated by the comparable mean intensity at each bin across the nucleus (p<0.001, two-way ANOVA). L) Examination of cluster enrichment (fraction of top intense clusters divided by fraction of total clusters per bin) suggests that there is a reduction in highly intense clusters from the nuclear periphery (right). Sham: n=21 nuclei; TAC: n=20 nuclei. * p<0.05; ** p<0.001 [Mann-Whitney; for cluster distribution, Chi-squared]. Error bars represent SEM.
Supplemental Figure 4: Validation of cardiac-specific changes in gene expression. A) RNA expression levels of four genes to be further analyzed by DNA FISH are validated in isolated adult cardiomyocytes. These genes were chosen to represent four categories of expression responses due to stress (activation, silencing, remain active and remain silent) and serve as models to understand genomic organization in cardiomyocytes. Nppa and Atp2a2 expression (normalized to Gapdh) occur in a cardiac-specific manner; the liver expression is shown for comparison (i). Nefl expression, a neuronal gene, serves as a negative control in both heart and liver tissues (normalized to Gapdh expression) (ii). Changes in expression of Nppa and Atp2a2 after TAC are shown (iii), along with raw expression of Gapdh (iv). Error bars represent SEM. B) The changes in expression on the level of RNA reflect the changes in protein abundance. Quantifications of western blots are shown on right. Molecular weights for ANF ~17kDa, SERCA2 ~110kDa, NEFL ~68kDa, GAPDH ~37kDa.
Supplemental Figure 5: Distribution of DNA FISH loci in liver nuclei is unaffected after pressure - overload stress. DNA FISH labeling in liver tissue from mice with heart failure was used to assess tissue-specificity of genomic architecture. Furthermore, based on DAPI patterns, liver nuclei have different nuclear shape and architecture. The distributions of Atp2a2 (A), Nppa (B), Gapdh (C) and Nefl (D) are not affected by TAC surgery. Quantitation is compiled from different labeling experiments from 2 different mice per treatment. Atp2a2: Sham n=78 nuclei/95 loci, TAC n=107/138; Nppa: Sham n=102/119, TAC n=102/125; Gapdh: Sham n=98/116, TAC n=112/119; Nefl: Sham n=99/111, TAC n=104/122. Scale bar = 5µm. P values were calculated using Chi-squared.
Supplemental Figure 6: RNA polymerase II occupancy confirms relationship between gene expression and association with transcription factories. The coordinates of DNA FISH primers were converted from mm10 to mm9 (using liftOver tool) to compare to published RNA polymerase II ChIP - seq (active RNA polymerase II, phosphorylated at serine 2) performed from whole heart lysates of sham and TAC mice (4 days after surgery) [39]. The mapped signal from ChIP-seq (sham in blue, TAC in red) at the regions targeted by DNA FISH is shown on left. Each red bar represents location of 200bp fragments that constitute DNA FISH probe tiling genomic loci; dotted boxes mark gene bodies. Fragment density indicates the number of mapped fragments per bin. ChIP -PCR was performed on isolated cardiomyocytes pooled from 6–7mice/group that were in heart failure (~6 weeks after TAC; right). IPs were normalized to input and fold enrichment over IgG is shown (n=3 technical replicates, data is representative of 4 independent IPs). Locations of the primers used for promoter and gene body regions are indicated by arrows. These results show increased enrichment of RNA polymerase II at the Nppa gene body, decreased enrichment at Atp2a2 and Gapdh genes and no enrichment at the Nefl gene. Error bars represent SEM. P values were calculated using two-tailed t-test.
Acknowledgements
This project was supported by the UCLA Cardiovascular Theme and NIH grants UL1TR000124 (Steven Dubinett), HL 105699 (TMV), HL 115238 (TMV), and HL 129639 (YW and TMV). EK was supported by an AHA Pre-doctoral Fellowship.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
The authors declare that they have no conflicts of interest related to this manuscript.
References
- [1].Gillette TG, Hill JA, Readers, writers, and erasers: chromatin as the whiteboard of heart disease, Circ Res 116(7) (2015) 1245–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Mathiyalagan P, Keating ST, Du XJ, El-Osta A, Chromatin modifications remodel cardiac gene expression, Cardiovasc Res 103(1) (2014) 7–16. [DOI] [PubMed] [Google Scholar]
- [3].Olson EN, Backs J, McKinsey TA, Control of cardiac hypertrophy and heart failure by histone acetylation/deacetylation, Novartis Found Symp 274 (2006) 3–12; discussion 13–9, 152–5, 272–6. [PubMed] [Google Scholar]
- [4].Schneider R, Grosschedl R, Dynamics and interplay of nuclear architecture, genome organization, and gene expression, Genes Dev 21(23) (2007) 3027–43. [DOI] [PubMed] [Google Scholar]
- [5].Cremer T, Cremer C, Chromosome territories, nuclear architecture and gene regulation in mammalian cells, Nat Rev Genet 2(4) (2001) 292–301. [DOI] [PubMed] [Google Scholar]
- [6].Cremer T, Cremer M, Chromosome territories, Cold Spring Harb Perspect Biol 2(3) (2010) a003889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Lieberman-Aiden E, van Berkum NL, Williams L, Imakaev M, Ragoczy T, Telling A, Amit I, Lajoie BR, Sabo PJ, Dorschner MO, Sandstrom R, Bernstein B, Bender MA, Groudine M, Gnirke A, Stamatoyannopoulos J, Mirny LA, Lander ES, Dekker J, Comprehensive mapping of long-range interactions reveals folding principles of the human genome, Science 326(5950) (2009) 289–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Dixon JR, Selvaraj S, Yue F, Kim A, Li Y, Shen Y, Hu M, Liu JS, Ren B, Topological domains in mammalian genomes identified by analysis of chromatin interactions, Nature 485(7398) (2012) 376–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Rieder D, Trajanoski Z, McNally JG, Transcription factories, Front Genet 3 (2012) 221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Hozak P, Cook PR, Schofer C, Mosgoller W, Wachtler F, Site of transcription of ribosomal RNA and intranucleolar structure in HeLa cells, J Cell Sci 107 ( Pt 2) (1994) 639–48. [DOI] [PubMed] [Google Scholar]
- [11].Kimura H, Sugaya K, Cook PR, The transcription cycle of RNA polymerase II in living cells, J Cell Biol 159(5) (2002) 777–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Jackson DA, Hassan AB, Errington RJ, Cook PR, Visualization of focal sites of transcription within human nuclei, EMBO J 12(3) (1993) 1059–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Iborra FJ, Pombo A, Jackson DA, Cook PR, Active RNA polymerases are localized within discrete transcription “factories’ in human nuclei, J Cell Sci 109 ( Pt 6) (1996) 1427–36. [DOI] [PubMed] [Google Scholar]
- [14].Li G, Ruan X, Auerbach RK, Sandhu KS, Zheng M, Wang P, Poh HM, Goh Y, Lim J, Zhang J, Sim HS, Peh SQ, Mulawadi FH, Ong CT, Orlov YL, Hong S, Zhang Z, Landt S, Raha D, Euskirchen G, Wei CL, Ge W, Wang H, Davis C, Fisher-Aylor KI, Mortazavi A, Gerstein M, Gingeras T, Wold B, Sun Y, Fullwood MJ, Cheung E, Liu E, Sung WK, Snyder M, Ruan Y, Extensive promoter-centered chromatin interactions provide a topological basis for transcription regulation, Cell 148(1–2) (2012) 84–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Osborne CS, Chakalova L, Brown KE, Carter D, Horton A, Debrand E, Goyenechea B, Mitchell JA, Lopes S, Reik W, Fraser P, Active genes dynamically colocalize to shared sites of ongoing transcription, Nat Genet 36(10) (2004) 1065–71. [DOI] [PubMed] [Google Scholar]
- [16].Zhao ZW, Roy R, Gebhardt JC, Suter DM, Chapman AR, Xie XS, Spatial organization of RNA polymerase II inside a mammalian cell nucleus revealed by reflected light-sheet superresolution microscopy, Proc Natl Acad Sci U S A 111(2) (2014) 681–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Cutilletta AF, Rudnik M, Zak R, Muscle and non-muscle cell RNA polymerase activity during the development of myocardial hypertrophy, J Mol Cell Cardiol 10(8) (1978) 677–87. [DOI] [PubMed] [Google Scholar]
- [18].Sano M, Abdellatif M, Oh H, Xie M, Bagella L, Giordano A, Michael LH, DeMayo FJ, Schneider MD, Activation and function of cyclin T-Cdk9 (positive transcription elongation factor-b) in cardiac muscle-cell hypertrophy, Nat Med 8(11) (2002) 1310–7. [DOI] [PubMed] [Google Scholar]
- [19].Sano M, Wang SC, Shirai M, Scaglia F, Xie M, Sakai S, Tanaka T, Kulkarni PA, Barger PM, Youker KA, Taffet GE, Hamamori Y, Michael LH, Craigen WJ, Schneider MD, Activation of cardiac Cdk9 represses PGC-1 and confers a predisposition to heart failure, EMBO J 23(17) (2004) 3559–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Casafont I, Navascues J, Pena E, Lafarga M, Berciano MT, Nuclear organization and dynamics of transcription sites in rat sensory ganglia neurons detected by incorporation of 5’-fluorouridine into nascent RNA, Neuroscience 140(2) (2006) 453–62. [DOI] [PubMed] [Google Scholar]
- [21].Monte E, Rosa-Garrido M, Karbassi E, Chen H, Lopez R, Rau CD, Wang J, Nelson SF, Wu Y, Stefani E, Lusis AJ, Wang Y, Kurdistani SK, Franklin S, Vondriska TM, Reciprocal Regulation of the Cardiac Epigenome by Chromatin Structural Proteins Hmgb and Ctcf: IMPLICATIONS FOR TRANSCRIPTIONAL REGULATION, J Biol Chem 291(30) (2016) 15428–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Drygin D, Lin A, Bliesath J, Ho CB, O’Brien SE, Proffitt C, Omori M, Haddach M, Schwaebe MK, Siddiqui-Jain A, Streiner N, Quin JE, Sanij E, Bywater MJ, Hannan RD, Ryckman D, Anderes K, Rice WG, Targeting RNA polymerase I with an oral small molecule CX-5461 inhibits ribosomal RNA synthesis and solid tumor growth, Cancer Res 71(4) (2011) 1418–30. [DOI] [PubMed] [Google Scholar]
- [23].Rosa-Garrido M, Chapski DJ, Schmitt AD, Kimball TH, Karbassi E, Monte E, Balderas E, Pellegrini M, Shih TT, Soehalim E, Liem D, Ping P, Galjart NJ, Ren S, Wang Y, Ren B, Vondriska TM, High-Resolution Mapping of Chromatin Conformation in Cardiac Myocytes Reveals Structural Remodeling of the Epigenome in Heart Failure, Circulation 136(17) (2017) 1613–1625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Bienko M, Crosetto N, Teytelman L, Klemm S, Itzkovitz S, van Oudenaarden A, A versatile genome-scale PCR-based pipeline for high-definition DNA FISH, Nat Methods 10(2) (2013) 122–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Therizols P, Illingworth RS, Courilleau C, Boyle S, Wood AJ, Bickmore WA, Chromatin decondensation is sufficient to alter nuclear organization in embryonic stem cells, Science 346(6214) (2014) 1238–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Rajabi M, Kassiotis C, Razeghi P, Taegtmeyer H, Return to the fetal gene program protects the stressed heart: a strong hypothesis, Heart Fail Rev 12(3–4) (2007) 331–43. [DOI] [PubMed] [Google Scholar]
- [27].Hannan RD, Jenkins A, Jenkins AK, Brandenburger Y, Cardiac hypertrophy: a matter of translation, Clin Exp Pharmacol Physiol 30(8) (2003) 517–27. [DOI] [PubMed] [Google Scholar]
- [28].Brandenburger Y, Arthur JF, Woodcock EA, Du XJ, Gao XM, Autelitano DJ, Rothblum LI, Hannan RD, Cardiac hypertrophy in vivo is associated with increased expression of the ribosomal gene transcription factor UBF, FEBS Lett 548(1–3) (2003) 79–84. [DOI] [PubMed] [Google Scholar]
- [29].Goodfellow SJ, Graham EL, Kantidakis T, Marshall L, Coppins BA, Oficjalska-Pham D, Gerard M, Lefebvre O, White RJ, Regulation of RNA polymerase III transcription by Maf1 in mammalian cells, J Mol Biol 378(3) (2008) 481–91. [DOI] [PubMed] [Google Scholar]
- [30].Mitchell-Jordan S, Chen H, Franklin S, Stefani E, Bentolila LA, Vondriska TM, Features of endogenous cardiomyocyte chromatin revealed by super-resolution STED microscopy, J Mol Cell Cardiol 53(4) (2012) 552–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Franklin S, Chen H, Mitchell-Jordan S, Ren S, Wang Y, Vondriska TM, Quantitative analysis of the chromatin proteome in disease reveals remodeling principles and identifies high mobility group protein B2 as a regulator of hypertrophic growth, Mol Cell Proteomics 11(6) (2012) M111 014258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Patterson M, Barske L, Van Handel B, Rau CD, Gan P, Sharma A, Parikh S, Denholtz M, Huang Y, Yamaguchi Y, Shen H, Allayee H, Crump JG, Force TI, Lien CL, Makita T, Lusis AJ, Kumar SR, Sucov HM, Frequency of mononuclear diploid cardiomyocytes underlies natural variation in heart regeneration, Nat Genet 49(9) (2017) 1346–1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Gonzalez-Rosa JM, Sharpe M, Field D, Soonpaa MH, Field LJ, Burns CE, Burns CG, Myocardial Polyploidization Creates a Barrier to Heart Regeneration in Zebrafish, Dev Cell 44(4) (2018) 433–+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Sutherland H, Bickmore WA, Transcription factories: gene expression in unions?, Nat Rev Genet 10(7) (2009) 457–66. [DOI] [PubMed] [Google Scholar]
- [35].Guelen L, Pagie L, Brasset E, Meuleman W, Faza MB, Talhout W, Eussen BH, de Klein A, Wessels L, de Laat W, van Steensel B, Domain organization of human chromosomes revealed by mapping of nuclear lamina interactions, Nature 453(7197) (2008) 948–51. [DOI] [PubMed] [Google Scholar]
- [36].Finlan LE, Sproul D, Thomson I, Boyle S, Kerr E, Perry P, Ylstra B, Chubb JR, Bickmore WA, Recruitment to the nuclear periphery can alter expression of genes in human cells, PLoS Genet 4(3) (2008) e1000039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Cahoy JD, Emery B, Kaushal A, Foo LC, Zamanian JL, Christopherson KS, Xing Y, Lubischer JL, Krieg PA, Krupenko SA, Thompson WJ, Barres BA, A transcriptome database for astrocytes, neurons, and oligodendrocytes: a new resource for understanding brain development and function, J Neurosci 28(1) (2008) 264–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Moller S, Bernardi M, Interactions of the heart and the liver, Eur Heart J 34(36) (2013) 2804–11. [DOI] [PubMed] [Google Scholar]
- [39].Sayed D, He M, Yang Z, Lin L, Abdellatif M, Transcriptional Regulation Patterns Revealed by High Resolution Chromatin Immunoprecipitation during Cardiac Hypertrophy, J Biol Chem 288(4) (2013) 2546–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Ho L, Crabtree GR, Chromatin remodelling during development, Nature 463(7280) (2010) 474–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Papait R, Cattaneo P, Kunderfranco P, Greco C, Carullo P, Guffanti A, Vigano V, Stirparo GG, Latronico MV, Hasenfuss G, Chen J, Condorelli G, Genome-wide analysis of histone marks identifying an epigenetic signature of promoters and enhancers underlying cardiac hypertrophy, Proc Natl Acad Sci U S A 110(50) (2013) 20164–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Nothjunge S, Nuhrenberg TG, Gruning BA, Doppler SA, Preissl S, Schwaderer M, Rommel C, Krane M, Hein L, Gilsbach R, DNA methylation signatures follow preformed chromatin compartments in cardiac myocytes, Nature communications 8(1) (2017) 1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Zullo JM, Demarco IA, Pique-Regi R, Gaffney DJ, Epstein CB, Spooner CJ, Luperchio TR, Bernstein BE, Pritchard JK, Reddy KL, Singh H, DNA sequence-dependent compartmentalization and silencing of chromatin at the nuclear lamina, Cell 149(7) (2012) 1474–87. [DOI] [PubMed] [Google Scholar]
- [44].Reddy KL, Zullo JM, Bertolino E, Singh H, Transcriptional repression mediated by repositioning of genes to the nuclear lamina, Nature 452(7184) (2008) 243–7. [DOI] [PubMed] [Google Scholar]
- [45].Kind J, Pagie L, Ortabozkoyun H, Boyle S, de Vries SS, Janssen H, Amendola M, Nolen LD, Bickmore WA, van Steensel B, Single-cell dynamics of genome-nuclear lamina interactions, Cell 153(1) (2013) 178–92. [DOI] [PubMed] [Google Scholar]
- [46].Kirby TJ, Patel RM, McClintock TS, Dupont-Versteegden EE, Peterson CA, McCarthy JJ, Myonuclear transcription is responsive to mechanical load and DNA content but uncoupled from cell size during hypertrophy, Mol Biol Cell 27(5) (2016) 788–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Cisse II, Izeddin I, Causse SZ, Boudarene L, Senecal A, Muresan L, Dugast-Darzacq C, Hajj B, Dahan M, Darzacq X, Real-time dynamics of RNA polymerase II clustering in live human cells, Science 341(6146) (2013) 664–7. [DOI] [PubMed] [Google Scholar]
- [48].Cho WK, Jayanth N, Mullen S, Tan TH, Jung YJ, Cisse II, Super-resolution imaging of fluorescently labeled, endogenous RNA Polymerase II in living cells with CRISPR/Cas9-mediated gene editing, Sci Rep 6 (2016) 35949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Cho WK, Jayanth N, English BP, Inoue T, Andrews JO, Conway W, Grimm JB, Spille JH, Lavis LD, Lionnet T, Cisse II, RNA Polymerase II cluster dynamics predict mRNA output in living cells, Elife 5 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Faro-Trindade I, Cook PR, A conserved organization of transcription during embryonic stem cell differentiation and in cells with high C value, Mol Biol Cell 17(7) (2006) 2910–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Sayed D, Yang Z, He M, Pfleger JM, Abdellatif M, Acute targeting of general transcription factor IIB restricts cardiac hypertrophy via selective inhibition of gene transcription, Circ Heart Fail 8(1) (2015) 138–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Fanucchi S, Shibayama Y, Burd S, Weinberg MS, Mhlanga MM, Chromosomal contact permits transcription between coregulated genes, Cell 155(3) (2013) 606–20. [DOI] [PubMed] [Google Scholar]
- [53].Towbin BD, Gonzalez-Aguilera C, Sack R, Gaidatzis D, Kalck V, Meister P, Askjaer P, Gasser SM, Step-wise methylation of histone H3K9 positions heterochromatin at the nuclear periphery, Cell 150(5) (2012) 934–47. [DOI] [PubMed] [Google Scholar]
- [54].Poleshko A, Shah PP, Gupta M, Babu A, Morley MP, Manderfield LJ, Ifkovits JL, Calderon D, Aghajanian H, Sierra-Pagan JE, Sun Z, Wang Q, Li L, Dubois NC, Morrisey EE, Lazar MA, Smith CL, Epstein JA, Jain R, Genome-Nuclear Lamina Interactions Regulate Cardiac Stem Cell Lineage Restriction, Cell 171(3) (2017) 573–587 e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Kehat I, Accornero F, Aronow BJ, Molkentin JD, Modulation of chromatin position and gene expression by HDAC4 interaction with nucleoporins, J Cell Biol 193(1) (2011) 21–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figure 1: Characterization of transcription factory structure and function in cardiomyocytes. A) 5’Fluorouridine (5’FU) is a uracil analogue that can permeate live cells and be incorporated into synthesizing RNA. Sites of 5’FU incorporation can then be detected using immunofluorescence. Representative image of neonatal rat ventricular myocytes treated with 4mM 5’FU for 30min is shown (left). RNase A treatment, post-5’FU incorporation, shows absence of 5’FU signal, demonstrating specificity of labeling to RNA (right). Scale bar: 5μm. B) RNA polymerase I activity quenches 5’FU signal into the nucleolus while treatment with CX-5461 allows for selective labeling of transcriptional activity in the nucleoplasm, which is associated with sites of active RNA polymerase II (phosphorylated at serine 2) occupancy, indicated by co-localization (yellow). Scale bar = 5µm. C) The distribution of nucleoli number does not differ with hypertrophic stress. D) NRVMs were treated with CX-5461, an RNA polymerase I inhibitor (2µM, 15min), prior to the addition of 5’FU. Thus, 5’FU labeling is reflective of RNA polymerase II and III activity. Note the absence of signal in the nucleolus, marked by nucleolin. Scale bar = 5µm. E) CX-5461 treatment efficiency in inhibiting RNA polymerase I was confirmed by measuring ribosomal RNA expression, ITS (premature) and 18S (mature) at 15min (before addition of 5’FU) and 45min (time point at which cells were fixed for immunostaining). 15min: n=4; 45min: n=6. Error bars represent SEM. F) NRVMs were treated with 10µM phenylephrine for 48hr, after which they were fixed and labeled with phalloidin and DAPI for cell and nuclear size measurements. Scale bar = 25µm. G) Quantifications of cell and nuclear sizes show significant increases in cell (60% increase) and total nuclear (8% increase) areas. The ratio of total nuclear area to cell area was significantly reduced in the hypertrophic cardiomyocytes, highlighting greater growth of the cell versus nucleus (nuclear occupancy is reduced to 8% of the cell from 12%). The increase in nuclear size does not affect nuclear shape, indicated by circularity (a perfect circle has circularity value of 1). Control: n=259 cells (279 nuclei); Phenylephrine: n=255 cells (279 nuclei). H) Nuclei number after phenylephrine treatment is unaltered. Control: n=323 cells, Phenylephrine: n=372 cells. I) NRVMs treated with isoproterenol (1µM for 48hr) show minimal hypertrophy response. Scale bar = 25µm. J) Phenylephrine treatment induces robust pathological gene expression. K) The relationship between mean 5’FU intensity and cell area were compared. There was no significant correlation between nascent transcript levels and cell size (control: r=0.177, p=0.133; phenylephrine: r=0.083, p=0.515). Control: n=73 cells; Phenylephrine: n=64 cells. * p<0.05; ** p<0.001 [Mann-Whitney; for nucleoli number distribution, Chi-squared].
Supplemental Figure 2: Super-resolution mapping of cardiac transcription factories. A) NRVMs treated with phenylephrine were labeled for active RNA polymerase II, phosphorylated at serine 2, and 5’fluorouridine incorporation (2µM CX-546, 4mM 5’FU) and imaged with STED microscopy. Left image shows the confocal image of a control nucleus with its corresponding super-resolution image on the right. Boxed regions highlight the level of detail that can be resolved with the STED microscope. Scale bar = 2µm (1µm for magnification). B) Nuclear area increases with hypertrophy in the cells used for these analyses. C) RNA polymerase II clusters are mapped and color-coded based on mean cluster intensity. Analyses examining the intensity ranges (measuring the minimum and maximum intensities per nucleus) demonstrate no difference with hypertrophic agonist treatment. Control: n=10 nuclei; Phenylephrine: n=11 nuclei. Scale bar = 5µm. D) Adult cardiomyocyte transcription factories were analyzed in situ from heart tissue sections. Cardiac nuclei were distinguished from non-cardiomyocyte nuclei using desmin labeling. Scale bar = 50µm (5µm for magnification). ** p<0.001 [Mann-Whitney].
Supplemental Figure 3: Properties of RNA polymerase II factories in hypertrophic and failing cardiomyocytes. A) The normalized distance to the nuclear envelope shows no change in neonatal cardiomyocytes (normalized to radial distance to account for differences in nuclear size). B) The average of intensity of the top 20% intensity ranked clusters does not differ. When normalizing peripheral distance, there is a decrease in distance from the periphery though not significant. C) The positioning across the nucleus of the most intense clusters shows no change in distribution after hypertrophic stress (left). Enrichment was determined by comparing fraction of the most intense clusters to the fraction of total clusters for each bin (right). D) 5’Fluorouridine intensity is significantly reduced in hypertrophic myocytes. E) The average spacing between nearest neighboring 5’fluorouridine puncta does not change. F) The normalized distance of 5’fluorouridine clusters to the nuclear periphery decreases. G) Cluster number was normalized to nuclear area and indicated no difference with pressure overload in adult mouse cardiac nuclei. The nuclei that were analyzed had similar areas. Nucleoplasmic regions were determined based on presence of RNA polymerase II signal. Regions devoid of signal were regions associated with nucleoli or characteristic cardiomyocyte heterochromatin chromocenters. The ratio of nucleoplasm to nuclear area does not change. H) Cluster distances to the periphery were normalized to area and are not different between groups. I) The mean intensity of the highly ranked clusters significantly increases with TAC stress. The bottom 20%-ranked clusters have increases in mean intensity without changes to spatial organization in the nucleus (distance to nuclear envelope, p=0.689). J) To normalize nuclear distances, the difference between maximum and minimum distances to the nuclear periphery was used to generate 5 bins. Scale bar = 3µm. K) The top 20% most intense clusters were examined. The distribution of intensity shows that these clusters are found across the nucleus without preferential localization as indicated by the comparable mean intensity at each bin across the nucleus (p<0.001, two-way ANOVA). L) Examination of cluster enrichment (fraction of top intense clusters divided by fraction of total clusters per bin) suggests that there is a reduction in highly intense clusters from the nuclear periphery (right). Sham: n=21 nuclei; TAC: n=20 nuclei. * p<0.05; ** p<0.001 [Mann-Whitney; for cluster distribution, Chi-squared]. Error bars represent SEM.
Supplemental Figure 4: Validation of cardiac-specific changes in gene expression. A) RNA expression levels of four genes to be further analyzed by DNA FISH are validated in isolated adult cardiomyocytes. These genes were chosen to represent four categories of expression responses due to stress (activation, silencing, remain active and remain silent) and serve as models to understand genomic organization in cardiomyocytes. Nppa and Atp2a2 expression (normalized to Gapdh) occur in a cardiac-specific manner; the liver expression is shown for comparison (i). Nefl expression, a neuronal gene, serves as a negative control in both heart and liver tissues (normalized to Gapdh expression) (ii). Changes in expression of Nppa and Atp2a2 after TAC are shown (iii), along with raw expression of Gapdh (iv). Error bars represent SEM. B) The changes in expression on the level of RNA reflect the changes in protein abundance. Quantifications of western blots are shown on right. Molecular weights for ANF ~17kDa, SERCA2 ~110kDa, NEFL ~68kDa, GAPDH ~37kDa.
Supplemental Figure 5: Distribution of DNA FISH loci in liver nuclei is unaffected after pressure - overload stress. DNA FISH labeling in liver tissue from mice with heart failure was used to assess tissue-specificity of genomic architecture. Furthermore, based on DAPI patterns, liver nuclei have different nuclear shape and architecture. The distributions of Atp2a2 (A), Nppa (B), Gapdh (C) and Nefl (D) are not affected by TAC surgery. Quantitation is compiled from different labeling experiments from 2 different mice per treatment. Atp2a2: Sham n=78 nuclei/95 loci, TAC n=107/138; Nppa: Sham n=102/119, TAC n=102/125; Gapdh: Sham n=98/116, TAC n=112/119; Nefl: Sham n=99/111, TAC n=104/122. Scale bar = 5µm. P values were calculated using Chi-squared.
Supplemental Figure 6: RNA polymerase II occupancy confirms relationship between gene expression and association with transcription factories. The coordinates of DNA FISH primers were converted from mm10 to mm9 (using liftOver tool) to compare to published RNA polymerase II ChIP - seq (active RNA polymerase II, phosphorylated at serine 2) performed from whole heart lysates of sham and TAC mice (4 days after surgery) [39]. The mapped signal from ChIP-seq (sham in blue, TAC in red) at the regions targeted by DNA FISH is shown on left. Each red bar represents location of 200bp fragments that constitute DNA FISH probe tiling genomic loci; dotted boxes mark gene bodies. Fragment density indicates the number of mapped fragments per bin. ChIP -PCR was performed on isolated cardiomyocytes pooled from 6–7mice/group that were in heart failure (~6 weeks after TAC; right). IPs were normalized to input and fold enrichment over IgG is shown (n=3 technical replicates, data is representative of 4 independent IPs). Locations of the primers used for promoter and gene body regions are indicated by arrows. These results show increased enrichment of RNA polymerase II at the Nppa gene body, decreased enrichment at Atp2a2 and Gapdh genes and no enrichment at the Nefl gene. Error bars represent SEM. P values were calculated using two-tailed t-test.