

Abstract

Enantioselective desymmetrization of meso-epoxides by 2-mercaptobenzothiazoles was realized by using the pentacarboxycyclopentadiene-based chiral Brønsted acid in combination of N-isopropylaniline as amine additive to give up to 90.5:9.5 er of the ring opening products.
Introduction
Catalytic enantioselective desymmetrization of meso-epoxides is an attractive method to prepare chiral alcohols1 because the meso-epoxide substrates are readily available and the 1,2-difunctionalized product structures with two adjacent chiral centers are very useful synthetic building blocks.2 Various nucleophiles have been reported to participate in catalytic enantioselective desymmetrization of meso-epoxides, including amines,3 azides,4 alcohols,5 carboxylic acids,6 thiols,7 and halides.8 The majority of these reactions were catalyzed by Lewis acidic transition metal complexes.1 In 2013, Sun et al. successfully realized the first Brønsted acid-catalyzed enantioselective desymmetrization of meso-epoxides by 2-mercaptobenzothiazoles.7d In that report, 1,1′-bi-2-naphthol (BINOL)-derived chiral phosphoric acid bearing 3,3′-(2,4,6-triisopropyl) substituents (commonly abbreviated as TRIP) was identified to be a good catalyst, and up to 92.5:7.5 er was obtained for the ring opening reaction of cyclohexene oxide.
While the research endeavor on strong Brønsted acid catalysis was dominated by BINOL-based chiral phosphoric acid derivatives9 since the pioneering works from Akiyama et al. and Terada et al.,10 syntheses of this class of catalysts were generally considered lengthy, laborious, and expensive, especially for some of the most successful chiral phosphoric acid-derived catalysts bearing bulky 3,3′-substituents, such as TRIP. Simplifying the preparation of strong Brønsted acid catalysts could be synthetically very useful.11
In 2016, Lambert et al. reported a novel, enantiopure, pentacarboxycyclopentadiene (PCCP)-based strong Brønsted acid catalyst, which could be easily prepared from readily available 1,2,3,4,5-pentacarbomethoxycyclopentadiene and chiral (−)-menthol in one transesterification step.12 Catalytic asymmetric Mukaiyama–Mannich reactions and oxocarbenium aldol reactions were realized in good enantioselectivities (Scheme 1a).12 Recently, Diels–Alder reactions between salicylaldehyde acetals and vinyl ethers catalyzed by a different PCCP catalyst were also reported.13 Other groups have also reported transfer hydrogenations and preparations of chiral aminals using Lambert’s catalyst.14 We believe that the development of PCCP-based chiral strong Brønsted acid catalyst could potentially alleviate some of the problematic issues for using BINOL-based chiral phosphoric acid derivatives. Herein, we report the enantioselective desymmetrization of meso-epoxides by 2-mercaptobenzothiazoles using the PCCP-based chiral Brønsted acid with up to 90.5:9.5 er (Scheme 1b).
Scheme 1. Chiral PCCP-Based Brønsted Acid-Catalyzed Reactions.

Results and Discussion
We used the ring opening reaction of cyclohexene oxide (1a) with 2-mercaptobenzothiazole (2a) with chiral Brønsted acid catalyst 1 as the model for optimization of reaction conditions. Initial trial in dichloromethane (DCM) solvent at room temperature (rt) overnight without the addition of chiral catalyst gave racemic product 3a in 18% background yield (Table 1, entry 1). When 2.5 mol % PCCP-based catalyst derived from chiral (−)-menthol was used, 93% isolated yield and 57:43 er were obtained (entry 2).15 We then screened a number of solvents for this reaction (entries 3–15). The best result (99% yield, 72:28 er) was obtained when chloroform was used as solvent (entry 8). We then tried to enhance the selectivity by lowering the reaction temperature. However, the enantioselectivity decreased to 60.5:39.5 er at 0 °C (entry 16) and 40.5:59.5 er at −44 °C (entry 17), which probably means that the favored transition state for the stereochemistry determining step might be reversed at lower temperatures. Further increasing the reaction temperature to 30 °C led to 99% yield and 70.5:29.5 er (entry 18).
Table 1. Optimization of Reaction Conditions.

| entry | catalyst loading (mol %) | solvent | temp (°C) | yield (%)a | er |
|---|---|---|---|---|---|
| 1 | 0b | CH2Cl2 | 10 | 18 | |
| 2 | 2.5 | CH2Cl2 | 11 | 93 | 57:43 |
| 3 | 5 | toluene | 12 | 97 | 49.4:50.5 |
| 4 | 2.5 | ClCH2CH2Cl | 10 | 74 | 56.5:43.5 |
| 5 | 2.5 | PhCl | 15 | 99 | 52:48 |
| 6 | 2.5 | hexane | 16 | 45 | 42:58 |
| 7 | 2.5 | CCl4 | 25 | 83 | 60:40 |
| 8 | 2.5 | CHCl3 | 22 | 99 | 72:28 |
| 9 | 2.5 | THF | 15 | 6 | 50:50 |
| 10 | 2.5 | dioxane | 16 | 10 | 52:48 |
| 11 | 2.5 | EtOAc | 12 | 30 | 46.5:53.5 |
| 12 | 2.5 | acetone | 16 | 42 | 50.5:49.5 |
| 13 | 2.5 | MeCN | 10 | 21 | 52:48 |
| 14 | 2.5 | DMF | 12 | trace | |
| 15 | 2.5 | EtOH | 16 | 31 | 50.5:49.5 |
| 16 | 2.5 | CHCl3 | 0 | 92 | 60.5:39.5 |
| 17 | 2.5 | CHCl3 | –44 to 5c | 89 | 40.5:59.5 |
| 18 | 2.5 | CHCl3 | 30 | 99 | 70.5:29.5 |
Isolated yield.
Without catalyst.
–44 °C for 13 h then −44 to 5 °C for 9 h.
We then tried a number of other sulfur and nitrogen nucleophiles for the ring opening reaction of cyclohexene oxide (1a), but none of the results seemed promising (see Supporting Information for details). However, the addition of catalytic quantity of amine bases to react with the PCCP-based chiral Brønsted acid catalyst and form corresponding hydrogen-bonding adducts or ammonium salts did have some favorable effects on the enantioselectivity (Table 2). Specifically, triethylamine and diisopropylamine gave poor yields and enantioselectivities (entries 1 and 2), but a number of aniline derivatives and pyridine derivatives (entries 3–12) all gave full conversion to the desired product 3a and in certain cases increased enantioselectivities comparing with the result we obtained using the chiral Brønsted acid catalyst 1 alone (entry 13). When N-isopropylaniline 4a was used (entry 4), we achieved the highest 74:26 er, whereas using half or double amount of 4a did not help to increase the enantioselectivity (entries 11 and 12). Presumably, the aniline- and pyridine-type bases (entries 3–12) could form more tightly associated hydrogen-bonding adducts or ammonium salts with the PCCP-based chiral Brønsted acid catalyst through π–π stacking interactions and thus achieve higher enantioselectivity than triethylamine and diisopropylamine (entries 1 and 2).
Table 2. Optimization of Amine Additivesa.

Further enhancement of enantioselectivity was achieved by using chiral Brønsted acid catalyst 2, which was similarly synthesized using Lambert’s protocol with readily available (−)-8-phenylmenthol as the chiral source. Under the optimized reaction conditions (see Table S2 in the Supporting Information for details on the temperature optimization for catalyst 2), 99% yield and 89.5:10.5 er were obtained for product 3a (Table 3, entry 1). The increased steric bulkiness of catalyst 2 might contribute to its better performance. We then surveyed a number of substituted 2-mercaptobenzothiazoles, and modest-to-good enantioselectivities were obtained (entries 2–10). Up to 99% yield and 90.5:9.5 er of product 3c were achieved using the 5-trifluoromethoxy-2-mercaptobenzothiazole (2c) as nucleophile for this ring opening reaction with cyclohexene oxide (1a). 2-Mercaptobenzothiazole nucleophiles with electron-donating or electron-neutral substituents generally gave better results (entries 1–8). The low yield for product 3g was probably due to the poor solubility of substrate 2g under the reaction conditions (entry 7). For substrates containing electron-withdrawing substituents, lower yields and er’s were obtained (entries 9–10). When the reaction between substrates 1a and 2a was conducted on 5 mmol scale, the product 3a was also obtained in 99% yield and 89:11 er, and 89% of catalyst 2 was recovered, which showed the same catalytic activity for further reactions.
Table 3. Substrate Scope for 2-Mercaptobenzothiazoles.

| entry | 2 (R) | 3 | yield (%)a | er |
|---|---|---|---|---|
| 1 | 2a (H) | 3a | 99 | 89.5:10.5 |
| 2 | 2b (5-OMe) | 3b | 86 | 86:14 |
| 3 | 2c (5-OCF3) | 3c | 99 | 90.5:9.5 |
| 4 | 2d (5-Me) | 3d | 96 | 88.5:11.5 |
| 5 | 2e (6-Me) | 3e | 99 | 89.5:10.5 |
| 6 | 2f (5-Cl) | 3f | 61 | 90.5:9.5 |
| 7 | 2g (6-Cl) | 3g | 19 | 82.5:17.5 |
| 8 | 2h (5-F) | 3h | 92 | 89.5:10.5 |
| 9 | 2i (5-CN) | 3i | 51 | 87:13 |
| 10 | 2j (5-NO2) | 3j | 32 | 81:19 |
Isolated yield for 0.3 mmol scale reactions.
We then explored the substrate scope for meso-epoxides using 10 mol % catalyst loading for the sake of slightly better enantioselectivity (Table 4). We should also note that the best enantioselectivity for different epoxides were obtained at either 15 or 30 °C but generally decreased at temperatures higher than 30 °C (see Table S3 in the Supporting Information for details). Epoxide ring opening products 3a, 3l, 3m, and 3p were obtained with better enantioselectivities (entries 1, 3, 4, and 7), while the results for 3k, 3n, and 3o were moderate (entries 2, 5, 6). The double epoxide ring opening reaction of substrate 1h with 2.2 equiv 2a gave the desired product 3q with 89:11 er with moderate isolated yield.
Table 4. Substrate Scope for Meso-Epoxides.


Isolated yield for 0.3 mmol scale reactions.
Run at 15 °C.
15 mol % catalyst loading.
Reaction time: 24 h.
2.2 equiv of 2a was used.
Structure of 3q: 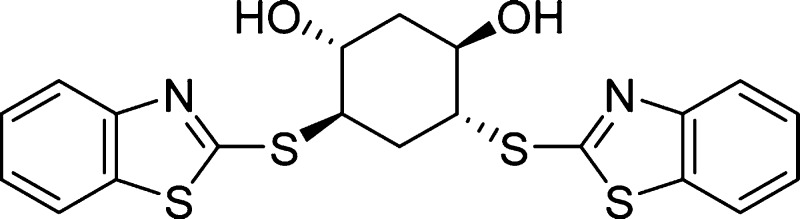 .
.
Conclusions
In conclusion, we identified the (−)-8-phenylmenthol-derived chiral PCCP-based Brønsted acid combined with equal molar amount of amine additive N-isopropylaniline as an efficient catalyst system for the enantioselective desymmetrization of meso-epoxides by 2-mercaptobenzothiazoles. Up to 99% yield and 90.5:9.5 er of the ring opening product were obtained for this reaction, showcasing the potential for further development and application of Lambert’s PCCP-based Brønsted acid catalysts. Other research along this line will be reported in due course.
Experimental Section
General Information
1H and 13C NMR spectra were recorded on a Bruker 400 MHz spectrometer. Thin-layer chromatography was performed on silica gel 60-F254-coated 0.2 mm plates. Flash column chromatography was performed using silica gel (200–300 mesh). All reagents were purchased from commercial suppliers and used without further purification unless otherwise noted. All solvents were purchased from commercial suppliers and purified by standard techniques. The enantiomeric excesses were determined by HPLC analysis, which employed a chiral stationary phase column specified in the individual experiment, by comparing the samples with the appropriate racemic mixtures. The meso-epoxides 1c,161d,171e,171f,6b1g,18 and 1h(19) were prepared according to the reported literature procedures.
The PCCP catalysts 1 and 2 were prepared according to the reported literature procedures.20 Pentacarbomethoxycyclopentadiene (307 mg, 0.86 mmol), (1R,2S,5R)-(−)-8-phenylmenthol (2.00 g, 8.61 mmol, 97%), and N-methylimidazole (0.412 mL, 5.16 mmol) were dissolved in 8.6 mL of toluene (0.1 M) in a two-neck flask. A steady flow of dry N2 was allowed for the removal of methanol. The reaction solution was stirred at 125 °C in an oil bath for 48 h. Upon completion, the reaction was cooled down to rt and concentrated in vacuo. The crude material was purified by silica gel chromatography (0 → 5% MeOH/CH2Cl2), washed with 1 M HCl/CH2Cl2 (3×), and dried with anhydrous MgSO4 to afford catalyst 2.
1,2,3,4,5-Pentacarbo-(−)-menthoxycyclopentadiene (PCCP Catalyst 1)20
Brown solid. 95% yield. 1H NMR (400 MHz, CDCl3): δ 20.30 (s, 1H), 5.05–4.60 (m, 5H), 2.58–0.68 (m, 90H).
1,2,3,4,5-Pentacarbo-(−)-8-phenylmenthoxycyclopentadiene (PCCP Catalyst 2)
Brown solid. 85% yield. 1H NMR (400 MHz, CDCl3): δ 20.06 (s, 1H), 7.45–6.80 (m, 25H), 5.40–4.75 (m, 5H), 2.95–0.60 (m, 85H). 13C NMR (101 MHz, CDCl3): δ 170.8, 165.4, 164.6, 163.8, 162.1, 161.9, 161.3, 159.7, 152.2, 151.6, 151.3, 151.1, 150.4, 150.4, 150.2, 149.5, 144.5, 140.3, 138.6, 137.0, 129.3, 128.8, 128.3, 128.0, 128.0, 127.8, 127.7, 127.0, 125.8–124.7, 123.4, 107.6, 79.8, 77.9, 77.7, 77.1, 75.6, 75.3, 60.2, 51.7, 51.1, 50.7, 50.4, 50.2, 50.1, 49.4, 42.4, 41.6, 41.5, 41.3, 41.1, 41.0, 40.7, 40.6, 40.6, 40.5, 40.3, 39.7, 39.4, 35.4, 34.9, 34.7, 34.3, 32.2, 31.8–30.9, 29.3, 28.9, 27.8, 27.4, 27.1, 26.5, 26.4, 22.9, 22.6, 22.3, 22.1, 22.0, 21.9, 21.7, 21.6, 21.4, 14.1. HRMS (ESI-TOF) m/z: [M + H]+ calcd for C90H117O10, 1357.8641; found, 1357.8649.
Typical Procedure for the Synthesis of 2-Mercaptobenzothiazoles (2b–2j)
The synthesis by a modified literature method21 is given as follows: To a solution of potassium ethylxanthate (1.76 g, 11.0 mmol) in dimethylformamide (7.5 mL) was added 2-bromo-5-methoxyaniline (1.00 g, 5.0 mmol). The reaction mixture was stirred for 12 h at 120 °C in an oil bath. After cooling down to rt, the reaction mixture was poured into ice water (200 mL), followed by adding 1 M HCl until pH = 2. Then, the mixture was filtered and the cake was washed by deionized water, dried under infrared, and then recrystallized from chloroform. Then, the solid was dissolved in EtOAc, stirred with 200 mg activated charcoal (powder) for 30 min at rt, and filtered through celite to yield 5-methoxybenzo[d]thiazole-2-thiol (2b) as a white solid (221 mg, 22%).
5-Methoxybenzo[d]thiazole-2-thiol (2b)22
White solid. 22% yield. 1H NMR (400 MHz, DMSO-d6): δ 13.63 (s, 1H), 7.57 (d, J = 8.8 Hz, 1H), 6.92 (dd, J = 8.8, 2.4 Hz, 1H), 6.82 (d, J = 2.4 Hz, 1H), 3.79 (s, 3H).
5-(Trifluoromethoxy)benzo[d]thiazole-2-thiol (2c)
2c was prepared from 2-bromo-5-trifluoromethoxyaniline. Reaction condition: 120 °C, 4 h. Colorless crystal, 57% yield. mp 193–195 °C. 1H NMR (400 MHz, DMSO-d6): δ 13.90 (s, 1H), 7.85–7.80 (m, 1H), 7.34–7.28 (m, 1H), 7.21 (s, 1H). 13C NMR (101 MHz, DMSO-d6): δ 191.9, 147.8, 142.5, 128.9, 123.8, 120.5 (q, J = 263 Hz), 117.5, 105.5. HRMS (ESI-TOF) m/z: [M + H]+ calcd for C8H5F3NOS2, 251.9759; found, 251.9760.
5-Methylbenzo[d]thiazole-2-thiol (2d)23
2d was prepared from 2-chloro-5-methylaniline. Reaction condition: 150 °C, 14 h. White solid. 31% yield.
6-Methylbenzo[d]thiazole-2-thiol (2e)24
2e was prepared from 2-chloro-4-methylaniline. Reaction condition: 150 °C, 12 h. White solid. 40% yield.
5-Chlorobenzo[d]thiazole-2-thiol (2f)22
2f was prepared from 2,5-dichloroaniline. Reaction condition: 120 °C, 12 h. White solid. 56% yield.
6-Chlorobenzo[d]thiazole-2-thiol (2g)22
2g was prepared from 2,4-dichloroaniline. Reaction condition: 120 °C, 4 h. White solid. 74% yield.
5-Fluorobenzo[d]thiazole-2-thiol (2h)25
2h was prepared from 2,5-difluoroaniline. Reaction condition: 120 °C, 11 h. White solid. 55% yield.
2-Mercaptobenzo[d]thiazole-5-carbonitrile (2i)26
2i was prepared from 3-amino-4-chlorobenzonitrile. Reaction condition: 120 °C, 4 h. Yellow solid. 42% yield.
5-Nitrobenzo[d]thiazole-2-thiol (2j)27
2j was prepared from 2-chloro-5-nitroaniline. Reaction condition: 100 °C, 4 h. Yellow solid. 41% yield.
General Procedure for the Synthesis of Racemic Ring Opening Products (rac-3)
To a suspension of 2-mercaptobenzothiazole (0.15 mmol) and meso-epoxide (0.15 mmol) in DCM (2 mL) was added a catalytic amount of TsOH. The reaction mixture was stirred at a 40 °C water bath for 10 min and then concentrated under reduced pressure. The residue was purified by flash column chromatography to give the desired product.
General Procedure for the Asymmetric Synthesis of Ring Opening Products (3)
A flame-dried 10 mL Schlenk tube was charged with 2-mercaptobenzothiazole (0.36 mmol), cyclopentadiene (0.015 mmol), N-isopropylaniline (0.015 mmol), and dry chloroform (1.8 mL). The reaction mixture was brought to 15 °C and then meso-epoxide (0.3 mmol) was added. The reaction mixture was stirred overnight, and then NaHCO3 powder (200 mg) was added to the reaction solution. Then, the reaction mixture was filtered and concentrated under reduced pressure. The residue was purified by flash column chromatography to give the desired product.
(1R,2R)-2-(Benzo[d]thiazol-2-ylthio)cyclohexan-1-ol (3a)7d (90:10 er)
[α]D20 +21.5 (c 1.0, CHCl3). Colorless liquid, 79 mg, 99% yield. HPLC (Daicel CHIRALPAK AD-H column, iPrOH/hexane = 10/90, flow rate = 1.0 mL/min): tR = 14.8 min (9.9%): tR = 17.7 min (90.1%).
Absolute configuration was determined by the comparison of the HPLC retention time in ref (7d).
(1R,2R)-2-((5-Methoxybenzo[d]thiazol-2-yl)thio)cyclohexan-1-ol (3b)7d (86:14 er)
[α]D20 +10.7 (c 1.0, CHCl3). Colorless liquid, 76 mg, 86% yield. HPLC (Daicel CHIRALPAK AD-H column, iPrOH/hexane = 10/90, flow rate = 1.0 mL/min): tR = 17.0 min (13.8%): tR = 21.0 min (86.2%).
(1R,2R)-2-((5-(Trifluoromethoxy)benzo[d]thiazol-2-yl)thio)cyclohexan-1-ol (3c) (90.5:9.5 er)
[α]D20 +7.1 (c 1.0, CHCl3). Colorless solid, 104 mg, 99% yield. mp 37–39 °C. HPLC (Daicel CHIRALPAK AD-H column, iPrOH/hexane = 10/90, flow rate = 1.0 mL/min): tR = 10.3 min (9.6%): tR = 11.1 min (90.4%). 1H NMR (400 MHz, CDCl3): δ 7.74–7.69 (m, 2H), 7.20–7.16 (m, 1H), 4.21 (d, J = 3.2 Hz, 1H), 3.77–3.69 (m, 1H), 3.68–3.60 (m, 1H), 2.30–2.18 (m, 2H), 1.84–1.74 (m, 2H), 1.63–1.51 (m, 1H), 1.51–1.42 (m, 1H), 1.41–1.30 (m, 2H). 13C NMR (101 MHz, CDCl3): δ 170.5, 153.2, 147.9, 133.8, 121.6, 120.5 (q, J = 258.6 Hz), 118.0, 113.9, 74.6, 55.6, 35.6, 32.2, 26.0, 24.0. HRMS (ESI-TOF) m/z: [M + H]+ calcd for C14H15F3NO2S2, 350.0491; found, 350.0499.
(1R,2R)-2-((5-Methylbenzo[d]thiazol-2-yl)thio)cyclohexan-1-ol (3d) (88.5:11.5 er)
[α]D20 +13.2 (c 1.0, CHCl3). Colorless liquid, 81 mg, 96% yield. HPLC (Daicel CHIRALPAK AD-H column, iPrOH/hexane = 10/90, flow rate = 1.0 mL/min): tR = 14.0 min (11.5%): tR = 16.7 min (88.5%). 1H NMR (400 MHz, CDCl3): δ 7.67 (s, 1H), 7.58 (d, J = 8.0 Hz, 1H), 7.12 (d, J = 8.0 Hz, 1H), 4.77 (s, 1H), 3.66–3.58 (m, 2H), 2.44 (s, 3H), 2.26–2.17 (m, 2H), 1.81–1.72 (m, 2H), 1.59–1.39 (m, 2H), 1.38–1.28 (m, 2H). 13C NMR (101 MHz, CDCl3): δ 167.2, 152.9, 136.3, 132.6, 126.2, 121.7, 120.5, 74.9, 55.6, 35.7, 32.2, 26.1, 24.1, 21.4. HRMS (ESI-TOF) m/z: [M + H]+ calcd for C14H18NOS2, 280.0824; found, 280.0828.
(1R,2R)-2-((6-Methylbenzo[d]thiazol-2-yl)thio)cyclohexan-1-ol (3e) (89.5:10.5 er)
[α]D20 +24.4 (c 1.0, CHCl3). Colorless solid, 83 mg, 99% yield. mp 84–86 °C. HPLC (Daicel CHIRALPAK AD-H column, iPrOH/hexane = 10/90, flow rate = 1.0 mL/min): tR = 20.5 min (10.4%): tR = 43.0 min (89.6%). 1H NMR (400 MHz, CDCl3): δ 7.72 (d, J = 8.0 Hz, 1H), 7.49 (s, 1H), 7.19 (d, J = 8.0 Hz, 1H), 4.81 (br s, 1H), 3.66–3.56 (m, 2H), 2.42 (s, 3H), 2.26–2.16 (m, 2H), 1.80–1.71 (m, 2H), 1.58–1.38 (m, 2H), 1.37–1.27 (m, 2H). 13C NMR (101 MHz, CDCl3): δ 165.9, 150.7, 135.8, 134.8, 127.7, 121.1, 120.8, 74.8, 55.6, 35.6, 32.2, 26.1, 24.1, 21.5. HRMS (ESI-TOF) m/z: [M + H]+ calcd for C14H18NOS2, 280.0824; found, 280.0828.
(1R,2R)-2-((5-Chlorobenzo[d]thiazol-2-yl)thio)cyclohexan-1-ol (3f)7d (90.5:9.5 er)
[α]D20 +12.2 (c 1.0, CHCl3). White solid, 55 mg, 61% yield. mp 74–76 °C. HPLC (Daicel CHIRALPAK AD-H column, iPrOH/hexane = 10/90, flow rate = 1.0 mL/min): tR = 15.5 min (9.6%): tR = 17.4 min (90.4%).
(1R,2R)-2-((6-Chlorobenzo[d]thiazol-2-yl)thio)cyclohexan-1-ol (3g) (82.5:17.5 er)
[α]D20 +14.2 (c 1.0, CHCl3). White solid, 17 mg, 19% yield. mp 87–89 °C. HPLC (Daicel CHIRALPAK AD-H column, iPrOH/hexane = 10/90, flow rate = 1.0 mL/min): tR = 19.9 min (17.6%): tR = 29.5 min (82.4%). 1H NMR (400 MHz, CDCl3): δ 7.76–7.71 (m, 1H), 7.70–7.67 (m, 1H), 7.38–7.32 (m, 1H), 4.35 (br s, 1H), 3.71–3.56 (m, 2H), 2.30–2.15 (m, 2H), 1.82–1.73 (m, 2H), 1.61–1.40 (m, 2H), 1.40–1.29 (m, 2H). 13C NMR (101 MHz, CDCl3): δ 168.0, 151.1, 136.7, 130.5, 126.9, 122.1, 120.6, 74.6, 55.7, 35.5, 32.2, 26.0, 24.0. HRMS (ESI-TOF) m/z: [M + H]+ calcd for C13H15ClNOS2, 300.0278; found, 300.0282.
(1R,2R)-2-((5-Fluorobenzo[d]thiazol-2-yl)thio)cyclohexan-1-ol (3h) (89.5:10.5 er)
[α]D20 +14.5 (c 1.0, CHCl3). White solid, 78 mg, 92% yield. mp 72–73 °C. HPLC (Daicel CHIRALPAK AD-H column, iPrOH/hexane = 10/90, flow rate = 1.0 mL/min): tR = 16.2 min (10.4%): tR = 17.4 min (89.6%). 1H NMR (400 MHz, CDCl3): δ 7.58 (dd, J = 8.8, 5.2 Hz, 1H), 7.48 (dd, J = 9.6, 2.4 Hz, 1H), 7.87 (dt, J = 8.8, 2.4 Hz, 1H), 4.33–4.29 (m, 1H), 3.67–3.53 (m, 2H), 2.22–2.11 (m, 2H), 1.77–1.68 (m, 2H), 1.55–1.43 (m, 1H), 1.43–1.32 (m, 1H), 1.32–1.17 (m, 2H). 13C NMR (101 MHz, CDCl3): δ 170.2, 161.9 (d, J = 244.4 Hz), 153.4 (d, J = 12.1 Hz), 130.8, 121.6 (d, J = 10.1 Hz), 113.1 (d, J = 25.3 Hz), 108.0 (d, J = 24.2 Hz), 74.8, 55.7, 35.6, 32.2, 26.1, 24.1. HRMS (ESI-TOF) m/z: [M + H]+ calcd for C13H15FNOS2, 284.0574; found, 284.0580.
2-(((1R,2R)-2-Hydroxycyclohexyl)thio)benzo[d]thiazole-5-carbonitrile (3i) (87:13 er)
[α]D20 +4.2 (c 1.5, CHCl3). White solid, 44 mg, 51% yield. mp 114–116 °C. HPLC (Daicel CHIRALPAK AD-H column, iPrOH/hexane = 20/80, flow rate = 1.0 mL/min): tR = 22.2 min (13.0%): tR = 24.3 min (87.0%). 1H NMR (400 MHz, CDCl3): δ 8.11 (s, 1H), 7.83 (d, J = 8.4 Hz, 1H), 7.53 (d, J = 8.4 Hz, 1H), 3.84–3.75 (m, 1H), 3.73–3.70 (m, 1H), 3.69–3.61 (m, 1H), 2.33–2.18 (m, 2H), 1.86–1.76 (m, 2H), 1.66–1.54 (m, 1H), 1.54–1.44 (m, 1H), 1.44–1.33 (m, 2H). 13C NMR (101 MHz, CDCl3): δ 171.0, 152.3, 140.4, 127.0, 125.1, 122.0, 118.6, 109.9, 74.3, 55.7, 35.5, 32.2, 26.0, 24.0. HRMS (ESI-TOF) m/z: [M + H]+ calcd for C14H15N2OS2, 291.0620; found, 291.0624.
(1R,2R)-2-((5-Nitrobenzo[d]thiazol-2-yl)thio)cyclohexan-1-ol (3j) (81:19 er)
[α]D20 +1.7 (c 1.0, CHCl3). White solid, 30 mg, 32% yield. mp 116–118 °C. HPLC (Daicel CHIRALPAK AD-H column, iPrOH/hexane = 30/70, flow rate = 1.0 mL/min): tR = 12.4 min (19.2%): tR = 17.4 min (80.8%). 1H NMR (400 MHz, CDCl3): δ 8.66 (d, J = 2.4 Hz, 1H), 8.17 (dd, J = 8.8, 2.4 Hz, 1H), 7.85 (d, J = 8.8 Hz, 1H), 3.86–3.79 (m, 1H), 3.70–3.62 (m, 2H), 2.34–2.27 (m, 1H), 2.26–2.19 (m, 1H), 1.86–1.77 (m, 2H), 1.67–1.56 (m, 1H), 1.55–1.45 (m, 1H), 1.44–1.33 (m, 2H). 13C NMR (101 MHz, CDCl3): δ 172.0, 152.5, 146.8, 142.0, 121.3, 119.1, 116.6, 74.4, 55.8, 35.6, 32.2, 26.0, 24.1. HRMS (ESI-TOF) m/z: [M + H]+ calcd for C13H15N2O3S2, 311.0519; found, 311.0518.
(1R,2R)-2-(Benzo[d]thiazol-2-ylthio)cyclopentan-1-ol (3k) (81.5:18.5 er)
[α]D20 +66.5 (c 1.0, CHCl3). Colorless liquid, 55 mg, 73% yield. HPLC (Daicel CHIRALPAK AD-H column, iPrOH/hexane = 10/90, flow rate = 1.0 mL/min): tR = 10.7 min (18.4%): tR = 14.2 min (81.6%). 1H NMR (400 MHz, CDCl3): δ 7.81 (d, J = 8.4 Hz, 1H), 7.73 (d, J = 8.0 Hz, 1H), 7.44–7.38 (m, 1H), 7.33–7.27 (m, 1H), 5.34 (br s, 1H), 4.38 (dd, J = 12.0, 8.0 Hz, 1H), 3.90 (dd, J = 16.0, 8.0 Hz, 1H), 2.37–2.26 (m, 1H), 2.20–2.09 (m, 1H), 1.94–1.75 (m, 3H), 1.67–1.56 (m, 1H). 13C NMR (101 MHz, CDCl3): δ 168.7, 152.3, 135.0, 126.3, 124.6, 121.0, 121.0, 81.5, 54.2, 34.3, 30.9, 23.3. HRMS (ESI-TOF) m/z: [M + H]+ calcd for C12H14NOS2, 252.0511; found, 252.0516.
(1R,2R)-2-(Benzo[d]thiazol-2-ylthio)cycloheptan-1-ol (3l) (87:13 er)
[α]D20 +27.3 (c 1.0, CHCl3). Colorless liquid, 62 mg, 74% yield. HPLC (Daicel CHIRALPAK AD-H column, iPrOH/hexane = 10/90, flow rate = 1.0 mL/min): tR = 13.1 min (13.1%): tR = 16.4 min (86.9%). 1H NMR (400 MHz, CDCl3): δ 7.84 (d, J = 8.4 Hz, 1H), 7.74 (dd, J = 8.0, 0.4 Hz, 1H), 7.44–7.38 (m, 1H), 7.33–7.27 (m, 1H), 5.03 (d, J = 2.0 Hz, 1H), 4.10 (ddd, J = 6.4, 3.6, 2.4 Hz, 1H), 3.96 (ddd, J = 8.8, 6.8, 3.2 Hz, 1H), 2.18–2.10 (m, 1H), 1.96–1.49 (m, 9H). 13C NMR (101 MHz, CDCl3): δ 168.0, 152.5, 135.3, 126.2, 124.5, 121.2, 121.0, 77.9, 58.3, 34.8, 31.8, 29.0, 27.1, 23.1. HRMS (ESI-TOF) m/z: [M + H]+ calcd for C14H18NOS2, 280.0824; found, 280.0827.
(1R,6R)-6-(Benzo[d]thiazol-2-ylthio)cyclohex-3-en-1-ol (3m) (86.5:13.5 er)
[α]D20 +40.5 (c 1.0, CHCl3). Colorless solid, 63 mg, 80% yield. mp 59–61 °C. HPLC (Daicel CHIRALPAK AD-H column, iPrOH/hexane = 10/90, flow rate = 1.0 mL/min): tR = 11.3 min (13.7%): tR = 19.0 min (86.3%). 1H NMR (400 MHz, CDCl3): δ 7.86 (d, J = 8.0 Hz, 1H), 7.73 (d, J = 8.0 Hz, 1H), 7.44–7.38 (m, 1H), 7.33–7.28 (m, 1H), 5.70–5.64 (m, 1H), 5.63–5.57 (m, 1H), 4.32 (br s, 1H), 4.03–3.93 (m, 2H), 2.79–2.61 (m, 2H), 2.40–2.23 (m, 2H). 13C NMR (101 MHz, CDCl3): δ 167.0, 152.5, 135.5, 126.3, 125.3, 124.7, 124.7, 121.6, 121.0, 70.8, 51.3, 34.8, 31.8. HRMS (ESI-TOF) m/z: [M + H]+ calcd for C13H14NOS2, 264.0511; found, 264.0518.
(2R,3R)-3-(Benzo[d]thiazol-2-ylthio)-1,2,3,4-tetrahydronaphthalen-2-ol (3n) (81.5:18.5 er)
[α]D20 −3.2 (c 1.0, CHCl3). Colorless liquid, 87 mg, 93% yield. HPLC (Daicel CHIRALPAK AD-H column, iPrOH/hexane = 10/90, flow rate = 1.0 mL/min): tR = 15.0 min (18.3%): tR = 18.2 min (81.7%). 1H NMR (400 MHz, CDCl3): δ 7.87 (d, J = 8.0 Hz, 1H), 7.76 (d, J = 8.0 Hz, 1H), 7.46–7.40 (m, 1H), 7.36–7.30 (m, 1H), 7.21–7.13 (m, 3H), 7.12–7.08 (m, 1H), 4.57 (br s, 1H), 4.25–4.11 (m, 2H), 3.45 (dd, J = 16.4, 5.6 Hz, 1H), 3.34 (dd, J = 16.0, 5.6 Hz, 1H), 3.08 (dd, J = 16.4, 10.4 Hz, 1H), 3.00 (dd, J = 16.0, 8.4 Hz, 1H). 13C NMR (101 MHz, CDCl3): δ 166.7, 152.5, 135.5, 134.3, 133.9, 129.0, 128.0, 126.7, 126.4, 126.3, 124.8, 121.6, 121.1, 71.7, 51.9, 38.2, 34.8. HRMS (ESI-TOF) m/z: [M + H]+ calcd for C17H16NOS2, 314.0668; found, 314.0669.
(4R,5R)-5-(Benzo[d]thiazol-2-ylthio)octan-4-ol (3o) (68:32 er)
[α]D20 +11.4 (c 1.0, CHCl3). Colorless liquid, 55 mg, 62% yield. HPLC (Daicel CHIRALPAK AD-H column, iPrOH/hexane = 10/90, flow rate = 1.0 mL/min): tR = 5.8 min (31.9%): tR = 7.8 min (68.1%). 1H NMR (400 MHz, CDCl3): δ 7.84 (d, J = 8.0 Hz, 1H), 7.73 (d, J = 8.0 Hz, 1H), 7.43–7.38 (m, 1H), 7.33–7.27 (m, 1H), 4.13 (s, 1H), 3.89 (s, 1H), 3.73–3.66 (m, 1H), 1.97–1.79 (m, 2H), 1.69–1.39 (m, 6H), 0.94 (dt, J = 7.2, 2.4 Hz, 6H). 13C NMR (101 MHz, CDCl3): δ 166.7, 152.7, 135.6, 126.2, 124.5, 121.4, 121.0, 73.4, 56.4, 38.3, 34.0, 20.7, 19.2, 14.1, 13.8. HRMS (ESI-TOF) m/z: [M + H]+ calcd for C15H22NOS2, 296.1137; found, 296.1143.
(1R,2R)-2-(Benzo[d]thiazol-2-ylthio)-1,2-diphenylethan-1-ol (3p) (85:15 er)
[α]D20 −338.7 (c 1.5, CHCl3). Colorless liquid, 98 mg, 90% yield. HPLC (Daicel CHIRALPAK AS-H column, iPrOH/hexane = 10/90, flow rate = 1.0 mL/min): tR = 9.0 min (15.2%): tR = 10.6 min (84.8%). 1H NMR (400 MHz, CDCl3): δ 7.94 (d, J = 8.0 Hz, 1H), 7.73 (d, J = 7.6 Hz, 1H), 7.48–7.42 (m, 1H), 7.35–7.30 (m, 1H), 7.25–7.17 (m, 10H), 5.29–5.26 (m, 1H), 5.22–5.16 (m, 2H). 13C NMR (101 MHz, CDCl3): δ 167.7, 152.4, 141.7, 137.9, 135.6, 128.6, 128.4, 128.1, 128.0, 127.8, 126.8, 126.3, 124.7, 121.6, 121.1, 78.8, 61.6. HRMS (ESI-TOF) m/z: [M + H]+ calcd for C21H18NOS2, 364.0824; found, 364.0829.
(1R,3R,4R,6R)-4,6-Bis(benzo[d]thiazol-2-ylthio)cyclohexane-1,3-diol (3q) (89:11 er)
[α]D20 −191.3 (c 1.0, CHCl3). Colorless solid, 65 mg, 49% yield. mp 41–42 °C. HPLC (Daicel CHIRALPAK AS-H column, iPrOH/hexane = 20/80, flow rate = 1.0 mL/min): tR = 8.5 min (88.9%): tR = 13.9 min (11.1%). 1H NMR (400 MHz, CDCl3): δ 7.83 (d, J = 8.4 Hz, 2H), 7.70 (d, J = 8.0 Hz, 2H), 7.40–7.34 (m, 2H), 7.30–7.24 (m, 2H), 4.32 (dd, J = 12.0, 6.0 Hz, 2H), 4.24 (br s, 2H), 4.16 (dd, J = 12.0, 6.0 Hz, 2H), 2.59 (t, J = 6.0 Hz, 2H), 2.19 (t, J = 5.6 Hz, 2H). 13C NMR (101 MHz, CDCl3): δ 165.7, 152.6, 135.3, 126.3, 124.7, 121.6, 121.0, 69.6, 50.4, 38.0, 31.3. HRMS (ESI-TOF) m/z: [M + H]+ calcd for C20H19N2O2S4, 447.0324; found, 447.0320.
Acknowledgments
This research was supported in part by National Natural Science Foundation of China (21402005).
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsomega.8b01207.
Experimental details of the condition optimization; 1H NMR and 13C NMR spectra; and HPLC trace for products (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- a Jacobsen E. N. Asymmetric Catalysis of Epoxide Ring-Opening Reactions. Acc. Chem. Res. 2000, 33, 421–431. 10.1021/ar960061v. [DOI] [PubMed] [Google Scholar]; b Meninno S.; Lattanzi A. Organocatalytic Asymmetric Reactions of Epoxides: Recent Progress. Chem.—Eur. J. 2016, 22, 3632–3642. 10.1002/chem.201504226. [DOI] [PubMed] [Google Scholar]; c Schneider C. Synthesis of 1,2-Difunctionalized Fine Chemicals Through Catalytic, Enantioselective Ring-Opening Reactions of Epoxides. Synthesis 2006, 3919–3944. 10.1055/s-2006-950348. [DOI] [Google Scholar]; d Wang P.-A. Organocatalyzed enantioselective desymmetrization of aziridines and epoxides. Beilstein J. Org. Chem. 2013, 9, 1677–1695. 10.3762/bjoc.9.192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Heravi M. M.; Lashaki T. B.; Poorahmad N. Applications of Sharpless asymmetric epoxidation in total synthesis. Tetrahedron: Asymmetry 2015, 26, 405–495. 10.1016/j.tetasy.2015.03.006. [DOI] [Google Scholar]; b Zhu Y.; Wang Q.; Cornwall R. G.; Shi Y. Organocatalytic Asymmetric Epoxidation and Aziridination of Olefins and Their Synthetic Applications. Chem. Rev. 2014, 114, 8199–8256. 10.1021/cr500064w. [DOI] [PubMed] [Google Scholar]
- a Chimni S. S.; Kumar V.; Bala N. Design of Peptidyl Thiourea Derivatives as Organocatalysts for the Asymmetric Ring Opening ofmeso-Stilbene Oxides. Asian J. Org. Chem. 2014, 3, 700–705. 10.1002/ajoc.201402040. [DOI] [Google Scholar]; b Kumar M.; Kureshy R. I.; Saravanan S.; Verma S.; Jakhar A.; Khan N.-u. H.; Abdi S. H. R.; Bajaj H. C. Unravelling a new class of chiral organocatalyst for asymmetric ring-opening reaction of meso epoxides with anilines. Org. Lett. 2014, 16, 2798–2801. 10.1021/ol500699c. [DOI] [PubMed] [Google Scholar]; c Regati S.; He Y.; Thimmaiah M.; Li P.; Xiang S.; Chen B.; Zhao J. C.-G. Enantioselective ring-opening of meso-epoxides by aromatic amines catalyzed by a homochiral metal-organic framework. Chem. Commun. 2013, 49, 9836–9838. 10.1039/c3cc45988h. [DOI] [PubMed] [Google Scholar]
- Martinez L. E.; Leighton J. L.; Carsten D. H.; Jacobsen E. N. Highly Enantioselective Ring Opening of Epoxides Catalyzed by (salen)Cr(III) Complexes. J. Am. Chem. Soc. 1995, 117, 5897–5898. 10.1021/ja00126a048. [DOI] [Google Scholar]
- a Matsunaga S.; Das J.; Roels J.; Vogl E. M.; Yamamoto N.; Iida T.; Yamaguchi K.; Shibasaki M. Catalytic Enantioselective meso-Epoxide Ring Opening Reaction with Phenolic Oxygen Nucleophile Promoted by Gallium Heterobimetallic Multifunctional Complexes. J. Am. Chem. Soc. 2000, 122, 2252–2260. 10.1021/ja993650f. [DOI] [Google Scholar]; b Schneider C.; Sreekanth A. R.; Mai E. Scandium-Bipyridine-Catalyzed Enantioselective Addition of Alcohols and Amines tomeso-Epoxides. Angew. Chem., Int. Ed. 2004, 43, 5691–5694. 10.1002/anie.200460786. [DOI] [PubMed] [Google Scholar]
- a Jacobsen E. N.; Kakiuchi F.; Konsler R. G.; Larrow J. F.; Tokunaga M. Enantioselective catalytic ring opening of epoxides with carboxylic acids. Tetrahedron Lett. 1997, 38, 773–776. 10.1016/s0040-4039(96)02414-8. [DOI] [Google Scholar]; b Monaco M. R.; Prévost S.; List B. Organocatalytic asymmetric hydrolysis of epoxides. Angew. Chem., Int. Ed. 2014, 53, 8142–8145. 10.1002/anie.201400170. [DOI] [PubMed] [Google Scholar]
- a Iida T.; Yamamoto N.; Sasai H.; Shibasaki M. New Asymmetric Reactions Using a Gallium Complex: A Highly Enantioselective Ring Opening of Epoxides with Thiols Catalyzed by a Gallium·Lithium·Bis(binaphthoxide) Complex. J. Am. Chem. Soc. 1997, 119, 4783–4784. 10.1021/ja9702576. [DOI] [Google Scholar]; b Ingle G.; Mormino M. G.; Antilla J. C. Lithium BINOL Phosphate Catalyzed Desymmetrization of meso-Epoxides with Aromatic Thiols. Org. Lett. 2014, 16, 5548–5551. 10.1021/ol502527q. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Monaco M. R.; Prévost S.; List B. Catalytic asymmetric synthesis of thiols. J. Am. Chem. Soc. 2014, 136, 16982–16985. 10.1021/ja510069w. [DOI] [PubMed] [Google Scholar]; d Wang Z.; Law W. K.; Sun J. Chiral phosphoric acid catalyzed enantioselective desymmetrization of meso-epoxides by thiols. Org. Lett. 2013, 15, 5964–5966. 10.1021/ol402797v. [DOI] [PubMed] [Google Scholar]
- a Broghammer F.; Brodbeck D.; Junge T.; Peters R. Cooperative Lewis acid-onium salt catalysis as tool for the desymmetrization of meso-epoxides. Chem. Commun. 2017, 53, 1156–1159. 10.1039/c6cc09774j. [DOI] [PubMed] [Google Scholar]; b Denmark S. E.; Barsanti P. A.; Wong K.-T.; Stavenger R. A. Enantioselective Ring Opening of Epoxides with Silicon Tetrachloride in the Presence of a Chiral Lewis Base. J. Org. Chem. 1998, 63, 2428–2429. 10.1021/jo9801420. [DOI] [PubMed] [Google Scholar]; c Gnanamani E.; Someshwar N.; Sanjeevi J.; Ramanathan C. R. Conformationally Rigid Chiral Bicyclic Skeleton-Tethered BipyridineN,N′-Dioxide as Organocatalyst: Asymmetric Ring Opening ofmeso-Epoxides. Adv. Synth. Catal. 2014, 356, 2219–2223. 10.1002/adsc.201400029. [DOI] [Google Scholar]; d Kalow J. A.; Doyle A. G. Enantioselective Ring Opening of Epoxides by Fluoride Anion Promoted by a Cooperative Dual-Catalyst System. J. Am. Chem. Soc. 2010, 132, 3268–3269. 10.1021/ja100161d. [DOI] [PubMed] [Google Scholar]
- a Parmar D.; Sugiono E.; Raja S.; Rueping M. Complete Field Guide to Asymmetric BINOL-Phosphate Derived Brønsted Acid and Metal Catalysis: History and Classification by Mode of Activation; Brønsted Acidity, Hydrogen Bonding, Ion Pairing, and Metal Phosphates. Chem. Rev. 2014, 114, 9047–9153. 10.1021/cr5001496. [DOI] [PubMed] [Google Scholar]; b Parmar D.; Sugiono E.; Raja S.; Rueping M. Addition and Correction to Complete Field Guide to Asymmetric BINOL-Phosphate Derived Brønsted Acid and Metal Catalysis: History and Classification by Mode of Activation; Brønsted Acidity, Hydrogen Bonding, Ion Pairing, and Metal Phosphates. Chem. Rev. 2017, 117, 10608–10620. 10.1021/acs.chemrev.7b00197. [DOI] [PubMed] [Google Scholar]
- a Akiyama T.; Itoh J.; Yokota K.; Fuchibe K. Enantioselective Mannich-Type Reaction Catalyzed by a Chiral Brønsted Acid. Angew. Chem., Int. Ed. 2004, 43, 1566–1568. 10.1002/anie.200353240. [DOI] [PubMed] [Google Scholar]; b Uraguchi D.; Terada M. Chiral Brønsted Acid-Catalyzed Direct Mannich Reactions via Electrophilic Activation. J. Am. Chem. Soc. 2004, 126, 5356–5357. 10.1021/ja0491533. [DOI] [PubMed] [Google Scholar]
- Hatano M.; Maki T.; Moriyama K.; Arinobe M.; Ishihara K. Pyridinium 1,1′-Binaphthyl-2,2′-disulfonates as Highly Effective Chiral Brønsted Acid–Base Combined Salt Catalysts for Enantioselective Mannich-Type Reaction. J. Am. Chem. Soc. 2008, 130, 16858–16860. 10.1021/ja806875c. [DOI] [PubMed] [Google Scholar]
- Gheewala C. D.; Collins B. E.; Lambert T. H. An aromatic ion platform for enantioselective Bronsted acid catalysis. Science 2016, 351, 961–965. 10.1126/science.aad0591. [DOI] [PubMed] [Google Scholar]
- Gheewala C. D.; Hirschi J. S.; Lee W.-H.; Paley D. W.; Vetticatt M. J.; Lambert T. H. Asymmetric Induction via a Helically Chiral Anion: Enantioselective Pentacarboxycyclopentadiene Brønsted Acid-Catalyzed Inverse-Electron-Demand Diels-Alder Cycloaddition of Oxocarbenium Ions. J. Am. Chem. Soc. 2018, 140, 3523–3527. 10.1021/jacs.8b00260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Qiao X.; El-Shahat M.; Ullah B.; Bao Z.; Xing H.; Xiao L.; Ren Q.; Zhang Z. Cyclopentadiene-based Brønsted acid as a new generation of organocatalyst for transfer hydrogenation of 2-substituted quinoline derivatives. Tetrahedron Lett. 2017, 58, 2050–2053. 10.1016/j.tetlet.2017.04.038. [DOI] [Google Scholar]; b Sui Y.; Cui P.; Liu S.; Zhou Y.; Du P.; Zhou H. Highly Enantioselective Synthesis of Cyclic Aminals with a Cyclopentadiene-Based Chiral Carboxylic Acid. Eur. J. Org. Chem. 2018, 215–218. 10.1002/ejoc.201701561. [DOI] [Google Scholar]; c Zhao X.; Xiao J.; Tang W. Enantioselective Reduction of 3-Substituted Quinolines with a Cyclopentadiene-Based Chiral Bronsted Acid. Synthesis 2017, 49, 3157–3164. 10.1055/s-0036-1589012. [DOI] [Google Scholar]
- The absolute configuration was assigned according to Sun’s literature report.
- Garcia A.; Otte D. A. L.; Salamant W. A.; Sanzone J. R.; Woerpel K. A. Acceleration of Acetal Hydrolysis by Remote Alkoxy Groups: Evidence for Electrostatic Effects on the Formation of Oxocarbenium Ions. Angew. Chem., Int. Ed. 2015, 54, 3061–3064. 10.1002/anie.201410043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y.; Wang M.; Ren W.-M.; Xu Y.-C.; Lu X.-B. Crystalline Hetero-Stereocomplexed Polycarbonates Produced from Amorphous Opposite Enantiomers Having Different Chemical Structures. Angew. Chem., Int. Ed. 2015, 127, 7148–7152. 10.1002/ange.201501417. [DOI] [PubMed] [Google Scholar]
- Robinson M. W. C.; Davies A. M.; Buckle R.; Mabbett I.; Taylor S. H.; Graham A. E. Epoxide ring-opening and Meinwald rearrangement reactions of epoxides catalyzed by mesoporous aluminosilicates. Org. Biomol. Chem. 2009, 7, 2559–2564. 10.1039/b900719a. [DOI] [PubMed] [Google Scholar]
- Rudolph J.; Reddy K. L.; Chiang J. P.; Sharpless K. B. Highly Efficient Epoxidation of Olefins Using Aqueous H2O2and Catalytic Methyltrioxorhenium/Pyridine: Pyridine-Mediated Ligand Acceleration. J. Am. Chem. Soc. 1997, 119, 6189–6190. 10.1021/ja970623l. [DOI] [Google Scholar]
- Gheewala C. D.; Radtke M. A.; Hui J.; Hon A. B.; Lambert T. H. Methods for the Synthesis of Functionalized Pentacarboxycyclopentadienes. Org. Lett. 2017, 19, 4227–4230. 10.1021/acs.orglett.7b01867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaudhuri N. C. Convenient Strategies for the Preparation of Modified 2(3H)-Benzothiazolethiones. Synth. Commun. 1996, 26, 3783–3790. 10.1080/00397919608003794. [DOI] [Google Scholar]
- Huang W.; Tan Y.; Ding M.-W.; Yang G.-F. Improved Synthesis of 2-(3H)Benzothiazolethiones under Microwave Irradiation. Synth. Commun. 2007, 37, 369–376. 10.1080/00397910601038665. [DOI] [Google Scholar]
- Easmon J.; Heinisch G.; Hofmann J.; Langer T.; Grunicke H. H.; Fink J.; Pürstinger G. Thiazolyl and benzothiazolyl hydrazones derived from α-(N)-acetylpyridines and diazines: synthesis, antiproliferative activity and CoMFA studies. Eur. J. Med. Chem. 1997, 32, 397–408. 10.1016/s0223-5234(97)81677-7. [DOI] [Google Scholar]
- Dang P.; Zeng W.; Liang Y. Copper-catalyzed three-component synthesis of benzothiazolethiones from o-iodoanilines, isocyanide, and potassium sulfide. Org. Lett. 2015, 17, 34–37. 10.1021/ol503186w. [DOI] [PubMed] [Google Scholar]
- Zhu L.; Zhang M. Ortho-Selective Nucleophilic Aromatic Substitution Reactions of Polyhaloanilines with Potassium/SodiumO-Ethyl Xanthate: A Convenient Access to Halogenated 2(3H)-Benzothiazolethiones. J. Org. Chem. 2004, 69, 7371–7374. 10.1021/jo049056s. [DOI] [PubMed] [Google Scholar]
- Chen J.-l.Therapeutic modulation of PPAR (gamma) activity. World Patent WO2005086904, Sept 22, 2005.
- Zhu L.; Zhang M.; Dai M. A convenient synthesis of 2-mercapto and 2-chlorobenzothiazoles. J. Heterocycl. Chem. 2005, 42, 727–730. 10.1002/jhet.5570420440. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.