

Abstract

A flexible and convergent strategy for the stereoselective total synthesis of bioactive marine natural product cytospolide Q has been developed. The key features of this synthesis include Evans anti-aldol reaction for the installation of C-2 and C-3 stereocenters and cycloetherification via epoxide opening followed by concomitant lactonization for the construction of tetrahydrofuran and γ-butyrolactone scaffolds. This synthetic study also revealed that protected oxygenated functionality (methyl ester or benzyl ether) at C-1 position participated readily in epoxide opening.
Introduction
Cytospolides belong to a family of nonanolides which were isolated by Zhang and co-workers during their bioassay-guided fractionation study of a crude acetone extract of Cytospora sp., an endophytic fungus from an evergreen shrub (Ilex canariensis), collected from Gomera, Spain (1–8, Figure 1).1 Many of the members of this family exhibited cytotoxic effects on different human cancer cell lines. The structures and absolute configurations of these molecules were elucidated using detailed spectroscopic analysis. Architecturally, most of these members bear a 10-membered lactone except, cytospolide Q (8). Technically, cytospolide Q (8) does not belong to the nonanolide family. The proposed biogenetic pathway suggests that cytospolide Q has been conceived through transesterification reaction of cytospolide M (6) which is a tetrahydrofuran (THF)-containing nonanolide.1 Bioactivities and interesting structural features have rendered many of these natural products attractive synthetic targets to the organic community.2 In continuation of our ongoing programs to the chemical synthesis of bioactive natural products,3 especially the nonanolides,3f,3g we envisaged the total synthesis of cytospolide Q. Structurally, it bears a unique 15 carbon skeleton embedded with a THF ring and a γ-butyrolactone moiety. Cytospolide Q showed cytotoxicity against human lungs adenocarcinoma (A549) with an IC50 value of 10.55 μg/mL.1b There is only one report on the total synthesis of cytospolide Q.2g Stark et al. had developed a nice strategy for its synthesis in 21 linear steps by mimicking the biosynthetic pathway. However, the overall yield of their synthesis was low (<1%). Furthermore, a partial epimerization of C-2 methyl center of cytospolide Q was observed when the precursor of cytospolide M was subjected for transesterification process in the presence of base.2g Thus, there is a need to explore other possible synthetic routes for cytospolide Q. In this paper, we report a shorter and flexible synthetic route for the total synthesis of the target molecule.
Figure 1.
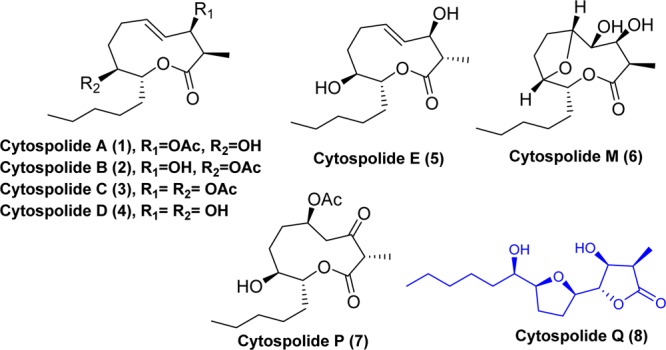
Some representative examples of cytospolides.
Results and Discussion
Retrosynthetic analysis of cytospolide Q (8) is depicted in Scheme 1. The THF ring and the γ-butyrolactone moiety could be installed from suitably protected epoxy ester 9 by a set of cascade reactions: deprotection of the acetonide group followed by concomitant cycloetherification via opening of the epoxide moiety followed by γ-lactonization. The epoxy compound 9 could be constructed from compound 10 using Sharpless asymmetric epoxidation reaction as one of the pivotal steps. Compound 10 could be obtained from aldehyde 11 and sulfone 12 using Julia–Kocienski olefination as one of the key steps.
Scheme 1. Retrosynthetic Analysis of Cytospolide Q (8).

Our synthetic endeavor commenced with the preparation of aldehyde 11 (Scheme 2). Cheap and easily available material, cinnamaldehyde (13), was subjected to Mg-mediated Evans anti-aldol reaction4 using the known oxazolidinone 14(3g,4) in the presence of trimethyl silyl chloride (TMSCl) and Et3N to obtain the corresponding TMS-protected aldol adducts with very good selectivity, favoring the anti-aldol product (dr > 19:1) which were subjected further for TMS deprotection using trifluoro acetic acid (TFA). The crude mixture was purified by silica gel column chromatography to obtain the known compound 15(4a) as a major isomer in 86% overall yield. Next, the free hydroxy group of compound 15 was protected as the corresponding TBS ether using tert-butyldimethylsilyl triflate (TBSOTf)/2,6-lutidine and subsequently was saponified with LiOH·H2O in the presence of H2O2 to afford acid 16 in 65% overall yield. Acid 16 was then treated with an ethereal solution of CH2N2 to obtain ester 17 in an excellent yield (93%). The olefin functionality of ester 17 was finally subjected to ozonolysis to get the required aldehyde 11.
Scheme 2. Synthesis of Aldehyde 11.

Reagent and conditions: (a) 14, MgCl2, Et3N, TMSCl, ethyl acetate (EtOAc), rt then MeOH, TFA, 2 h, 86%; (b) (i) 2,6-lutidine, TBSOTf, CH2Cl2, 0 °C, 30 min, 88%; (ii) LiOH·H2O, H2O2, (THF/H2O, 3:1), 0 °C to rt, 12 h, 74%; (c) diazomethane (CH2N2), Et2O, 0 °C, 93%; (d) O3, CH2Cl2/MeOH (4:1), −78 °C, 30 min, quantitative.
The synthesis of the coupling partner (12) is delineated in Scheme 3. Alcohol 18, prepared from l-arabinose following the literature procedure,5 was treated with I2 in the presence of Ph3P/imidazole to access the corresponding iodo compound. Next, the iodo compound was reacted with allyl magnesium chloride in the presence of CuI and 1,3-dimethyl-3,4,5,6-tetrahydro-2(1H)-pyrimidinone (DMPU) to produce alkene 19 in good overall yield (67%).6 Alkene 19 was then ozonolyzed to obtain the corresponding aldehyde which subsequently was reduced by NaBH4 to yield the known alcohol 20.3g Alcohol 20 was subjected further to Mitsunobu reaction3b,7 in the presence of PTSH (21), di-isopropyl azo dicarboxylate (DIAD), and Ph3P to access sulfide 22 which finally was oxidized to sulfone 12 in 75% overall yield using(NH4)6Mo7O24·4H2O in the presence of 30% aqueous H2O2.3b,8
Scheme 3. Synthesis of Sulfone 12.

Reagents and conditions: (a) (i) Ph3P, imidazole, I2, toluene, 0 °C to rt, 3 h, 83%; (ii) allyl magnesium chloride, CuI, DMPU, THF, −30 °C to rt, 3 h, 81%; (b) (i) O3, CH2Cl2/MeOH (4:1), −78 °C, 30 min; (ii) NaBH4, EtOH, 0 °C to rt, 1 h, 77% over two steps; (c) 21, DIAD, Ph3P, THF, 0 °C to rt, 3.5 h, 88%; (d) (NH4)6Mo7O24·4H2O, 30% aq H2O2, EtOH, 0 °C to rt, 24 h, 85%.
The completion of total synthesis of cytospolide Q (8) is described in Scheme 4. Both the coupling partners 11 and 12 were subjected to crucial Julia–Kocienski olefination3b,9 using KHMDS to get the coupled products 10 and 10a as an inseparable mixture (E/Z ≈ 4:1) in good yield. The stereochemistry of the major product was assigned as an E-isomer from the corresponding 1H NMR signals (3JH4–H5 = 15.3 Hz). Several attempts were made to improve the E-selectivity of the required isomer by changing the reaction conditions and the bases such as LiHMDS, NaHMDS, and LDA. Unfortunately, none of them superseded the KHMDS result. Next, the mixture of compounds 10 and 10a was treated with tetra-n-butyl ammonium fluoride (TBAF) to obtain compounds 23 and 23a in 87% yield as an inseparable mixture. This mixture of compounds was then subjected to kinetic asymmetric epoxidation, following the Sharpless protocol.10 The E-isomer underwent epoxidation in faster rate relative to its Z-counterpart to produce the required epoxide 9 in a gratifying yield. Next, we planned for a set of cascade reactions where the acid-catalyzed acetonide deprotection followed by concomitant epoxide opening for cycloetherification followed by γ-lactonization could be achieved in a single step.11 Thus, epoxide 9 was treated initially with camphor sulfonic acid (CSA)/MeOH. The target compound (8) was obtained in 19% yield, whereas compound 24 was formed as a major side product in 49% yield. The spectroscopic data (NMR and HRMS) and optical rotation (observed [α]D27 −1.5 (c 0.08, CHCl3); reported [α]D −4.4 (c 0.04, CHCl3)] of compound 8 were in good agreement with the data reported1b for the isolated cytospolide Q which unambiguously confirmed its total synthesis.
Scheme 4. Completion of the Total Synthesis of Cytospolide Q (8).

Reagents and conditions: (a) KHMDS, THF, 15 min, −78 °C, then 11, 1 h, 61%, based on recovered starting material 12, (E/Z > 4:1); (b) TBAF, THF, 0 °C to rt, 3 h, 87%; (c) Ti(OiPr)4, l-(+)-diisopropyl tartarate (DIPT), tert-butyl hydrogen peroxide (TBHP), molecular sieves (MSs) 4 Å, CH2Cl2, −20 °C, 6 days, 72% based on E-olefin used; (d) CSA, MeOH, rt, 5 h, 19% wrt compound 8 and 49% wrt compound 24; (e) 2,2-DMP, CSA, 0 °C to rt, 3 h, 70%.
The structure of compound 24 was elucidated through 2D-NMR studies. In order to confirm the stereochemistry as well as to understand its formation, compound 24 was reacted further with 2,2-dimethoxy propane (2,2-DMP) in the presence of CSA to result the acetonide-protected compound 25. 13C NMR data of compound 25 showed a signal at δ 101.2 ppm establishing the anti-relationship between the C-3 and C-5 hydroxy centers.12 This also proved that the methyl ester participated in epoxide opening preferably on γ-position over the other possible δ-position to yield a γ-butyrolactone moiety. This fact was supported further by the literature precedents where carboxylic acid13 or ester14 or amide15 was reported to undergo intramolecular epoxide opening to form γ-butyrolactone. At this point, we planned to screen other reagents to test whether the yield of the targeted molecule (8) could be increased further by inhibiting the formation of γ-butyrolactone (24) with variation of acidity of the reaction medium. However, trials with TFA or 80% AcOH–H2O or BF3·Et2O or Zn(NO3)2·6H2O gave almost similar results. It was further observed that in the case of HF-py in THF, the ester carbonyl-assisted epoxide opening took place even before any substantial deprotection of the acetonide group.
All of the above observations led us to believe that the ester moiety at C-1 position facilitated the formation of unwanted side product as a major compound in our case. Next, we planned to replace the ester moiety of compounds 23 and 23a with benzyl ether. Thus, ester functionality of the mixture of compounds 23 and 23a was reduced by LiBH4 to get an inseparable mixture of corresponding diols (Scheme 5). Our initial efforts to benzylate the free primary alcohol moiety of the diols using BnBr in the presence of different bases (NaHMDS, LiHMDS, and NaH) were not efficient because of the formation of substantial amount of di-benzyl-protected compounds. Finally, BnBr in the presence of Ag2O provided the monobenzylated products 26 and 26a in 54% yield as an inseparable mixture.16 The mixture of these compounds was then subjected to Sharpless kinetic asymmetric epoxidation reaction to get the required product 27 in good yield. However, it was highly disappointing to see that compound 27 suffered a similar fate like the epoxy ester (9) under acidic conditions. When compound 27 was treated with CSA in MeOH, the benzyl ether reacted promptly in preference to C-8 hydroxy to form the unrequired THF compound 28 as a major product (54%). The benzyl group was deprotected during the process. The required product 29 was obtained in trace amount which could be detected only by mass spectrometry.
Scheme 5. Alternative Effort toward the Total Synthesis of Cytospolide Q.

Reagents and Conditions: (a) (i) LiBH4, moist Et2O, 0 °C to rt, 1.5 h, 84%; (ii) BnBr, Ag2O, toluene, rt, 24 h, 54%; (b) Ti(OiPr)4, l-(+)-DIPT, TBHP, MS 4 Å, CH2Cl2, −20 °C, 6 days, 70% based on E-olefin used; (c) CSA, MeOH, rt, 1 h, 54% wrt compound 28.
Conclusions
In summary, we have developed a convergent route for the total synthesis of marine natural product cytospolide Q in 10 linear steps from the known compound 18 with an overall yield of 2.8%. This synthetic study also revealed that methyl ester and the benzyl ether at C-1 position participated in epoxide opening reaction faster compared to C-8 hydroxy to form the unrequired γ-butyrolactone- and THF-containing compounds, respectively, as the major products. We are of the opinion that the yield of the targeted molecule could possibly be increased further by masking of C-1 position with an olefinic moiety to prevent any kind of unwanted participation in epoxide opening. This may need stepwise formation of required THF moiety followed by functional group manipulations to install the lactone moiety. Efforts are presently in progress in that direction which will be reported in due course.
Experimental Section
General Experimental Procedures
Reactions, sensitive to moisture, were carried out under argon atmosphere in a dried glassware fitted with a Teflon-coated magnetic stirring bar using anhydrous solvents, unless otherwise noted. Liquids sensitive to air and moisture were added into the reaction mixture via a gastight syringe fitted with a stainless-steel needle or by cannula. The progress of a reaction was monitored by a thin-layer chromatography (TLC, Silica gel 60 F254) plate using UV light followed by charring the chromatography plate using ethanolic solution of anisaldehyde (prepared with 1% AcOH and 3.3% concd H2SO4) as a developing agent. All workup and purification processes were done with reagent-grade distilled solvents, unless otherwise stated. Column chromatographic purifications were executed using silica gel 60–120, 100–200, and 230–400 mesh. Yields mentioned based on chromatographically and spectroscopically homogeneous materials, unless otherwise stated. Optical rotation was recorded only for pure compound using a sodium (589, D line) lamp which was represented as [α]D25 (c = mg/100 mL, solvent). The IR spectrum of the liquid sample was measured using a thin film. A Quadruple-TOF micro-MS system using an electrospray ionization (ESI) technique was used to collect HRMS data. 1H NMR spectra were taken in 300, 400, and 500 MHz spectrometers in appropriate solvents and calibrated using a residual undeuterated solvent as an internal reference. The chemical shifts have been represented in parts per million (ppm) scales. Multiplicities of NMR signals have been reported as s (singlet), d (doublet), t (triplet), q (quartet), br (broad), m (multiplet, for unresolved lines), and so forth. 13C and 2D NMR spectra were measured on 75, 100, and 125 MHz spectrometers.
(2R,3R,E)-3-((tert-Butyldimethylsilyl)oxy)-2-methyl-5-phenylpent-4-enoic Acid (16)
To an ice-cold solution of the known compound 15 (7 g, 19.15 mmol) in anhydrous CH2Cl2 (50 mL) under argon, 2,6-lutidine (5.25 mL, 47.87 mmol) and TBSOTf (5.28 mL, 23.0 mmol) were added sequentially. The reaction mixture was stirred for 30 min at the same temperature prior to quenching with saturated aqueous NaHCO3 solution (10 mL). The organic layer was separated, and the aqueous layer was extracted with CH2Cl2 (2 × 100 mL). The combined organic extracts were washed with aqueous CuSO4, water, and brine, dried over Na2SO4, filtered, and concentrated in vacuo. Purification of the crude residue by flash column chromatography (SiO2, 100–200 mesh, 5–8% EtOAc in hexane as an eluent) furnished the corresponding TBS-protected compound (8.1 g, 88%) as a yellowish liquid: Rf = 0.50 (10% EtOAc/hexane); [α]D27 −23.9 (c 1.8, CHCl3); 1H NMR (CDCl3, 300 MHz): δ 7.34–7.20 (m, 10H), 6.53 (d, J = 15.9 Hz, 1H), 6.13 (dd, J = 15.9, 8.1 Hz, 1H), 4.71–4.60 (m, 2H), 4.17–4.03 (m, 3H), 3.37 (dd, J = 13.2, 3.0 Hz, 1H), 2.64 (dd, J = 13.2, 10.2 Hz, 1H), 1.09 (d, J = 7.2 Hz, 3H), 0.86 (s, 9H), 0.08 (s, 3H), 0.04 (s, 3H); 13C NMR (CDCl3, 75 MHz): δ 175.3, 153.3, 136.7, 135.7, 132.3, 130.4, 129.5, 129.1, 128.7, 127.8, 127.4, 126.6, 76.0, 66.0, 55.5, 44.7, 38.5, 26.0, 18.2, 14.0, −3.8, −4.4 ppm; IR (neat) νmax: 3450, 2928, 1790, 1694, 1480, 1385, 1244, 1210, 1120, 1004 cm–1; HRMS (ESI) m/z: calcd for C28H37O4NaNSi [M + Na]+, 502.2390; found, 502.2387.
30% aqueous H2O2 (1.7 mL) and LiOH·H2O (923 mg, 22.0 mmol) were added sequentially to an ice-cold solution of the above silylated compound (5.27 g, 11.00 mmol) in THF/water (32 mL, 3:1). The reaction mixture was allowed to attain room temperature and stirred further for 12 h. The reaction mixture was cooled again to 0 °C and neutralized with 1 N HCl. The resultant mixture was extracted with EtOAc (3 × 100 mL), washed with water and brine, dried over Na2SO4, filtered, and concentrated under reduced pressure. The residue was purified by column chromatography (SiO2, 230–400 mesh, 12–24% EtOAc in hexane as an eluent) to get acid 16 (2.61 g, 74%) as a colorless liquid: Rf = 0.30 (20% EtOAc/hexane); [α]D29 −25.4 (c 0.8, CHCl3); 1H NMR (CDCl3, 300 MHz): δ 7.37–7.20 (m, 5H), 6.53 (d, J = 15.9 Hz, 1H), 6.07 (dd, J = 15.9, 7.5 Hz, 1H), 4.42 (m, 1H), 2.64 (m, 1H), 1.20 (d, J = 7.2 Hz, 3H), 0.86 (s, 9H), 0.07 (s, 3H), 0.02 (s, 3H); 13C NMR (CDCl3, 75 MHz): δ 180.6, 136.6, 132.4, 129.7, 128.7, 127.9, 126.7, 76.0, 47.4, 25.9, 18.2, 13.3, −3.8, −4.9 ppm; HRMS (ESI) m/z: calcd for C18H28O3NaSi [M + Na]+, 343.1705; found, 343.1707.
(2R,3R,E)-Methyl 3-((tert-Butyldimethylsilyl)oxy)-2-methyl-5-phenylpent-4-enoate (17)
Acid 16 (5.6 g, 17.47 mmol) was dissolved in Et2O (50 mL) and cooled to 0 °C. A chilled ethereal solution of CH2N2 prepared by dissolving NMU (5.4 g, 52.41 mmol) in 40% KOH was added until the yellow color persisted. The mixture was warmed to room temperature and evaporated under residue pressure. The crude residue was purified by column chromatography (SiO2, 100–200 mesh, 5% EtOAc in hexane as an eluent) to afford the ester 17 (5.44 g, 93%) as slightly viscous liquid: Rf = 0.55 (5% EtOAc/hexane); [α]D27 −34.6 (c 0.9, CHCl3); 1H NMR (CDCl3, 300 MHz): δ 7.39–7.24 (m, 5H), 6.53 (d, J = 15.9 Hz, 1H), 6.06 (dd, J = 15.9, 7.5 Hz, 1H), 4.41 (t, J = 7.5 Hz, 1H), 3.69 (s, 3H), 2.64 (m, 1H), 1.08 (d, J = 6.9 Hz, 3H), 0.87 (s, 9H), 0.06 (s, 3H), 0.02 (s, 3H); 13C NMR (CDCl3, 75 MHz): δ 175.4, 136.8, 132.1, 130.3, 128.7, 127.8, 126.6, 76.3, 51.6, 47.4, 25.8, 18.1, 13.3, −3.8, −4.9 ppm; IR (neat) νmax: 3418, 2920, 2851, 1730, 1440, 1387, 1046, 771, 715 cm–1; HRMS (ESI) m/z: calcd for C19H30O3NaSi [M + Na]+, 357.1862; found, 357.1864.
(4S,5R)-4-(But-3-en-1-yl)-2,2-dimethyl-5-pentyl-1,3-dioxolane (19)
To an ice-cold mixture of Ph3P (5.25 g, 20.0 mmol), imidazole (2.68 g, 40.0 mmol), and alcohol 18 (2.02 g, 10.0 mmol) in toluene (60 mL) under argon, I2 (5.2 g, 20.5 mmol) was added portion-wise. The resultant solution was allowed to attain room temperature and stirred for 3 h. The solvent was evaporated, and the residue was purified by silica gel column chromatography (SiO2, 100–200 mesh, 2–5% EtOAc in hexane as an eluent) to afford the corresponding iodide (2.58 g, 83%) as a colorless oil: Rf = 0.30 (2% EtOAc/hexane); [α]D27 +1.71 (c 2.1, CHCl3); 1H NMR (CDCl3, 300 MHz): δ 4.32 (q, J = 6.9 Hz, 1H), 4.13–4.07 (m, 1H), 3.15 (d, J = 6.9 Hz, 2H), 1.60–1.48 (m, 2H), 1.46 (s, 3H), 1.35–1.25 (m, 9H), 0.90 (t, J = 6.6 Hz, 3H);13C NMR (CDCl3, 75 MHz): δ 108.4, 78.5, 78.1, 31.9, 29.3, 28.7, 26.2, 25.9, 22.6, 14.1, 4.4 ppm; IR (neat) νmax: 2986, 2929, 2858, 1456, 1379, 1245, 1217, 1036, 871, 790, 594, 512 cm–1.
To a solution of the above iodide (1.5 g, 4.8 mmol) and CuI (366 mg, 1.92 mmol) in anhydrous THF (20 mL) and 1,3-dimethyl-3,4,5,6-tetrahydro-2(1H)-pyrimidinone (4.5 mL) at −30 °C under argon, allyl magnesium chloride (2 M in THF, 7.2 mL 14.4 mmol) was added. The solution was stirred at room temperature for 3 h and subsequently was quenched with saturated aqueous NH4Cl solution (10 mL). The resultant mixture was extracted with EtOAc (3 × 60 mL), washed with water and brine, dried over Na2SO4, filtered, and concentrated under reduced pressure. The crude residue was purified by column chromatography (SiO2, 100–200 mesh, 2–5% EtOAc in hexane as an eluent) to give olefin 19 (880 mg, 81%) as a colorless oil: Rf = 0.6 (5% EtOAc/hexane); 1H NMR (300 MHz, CDCl3): δ 5.90–5.77 (m, 1H), 5.03 (d, J = 17.1 Hz, 1H), 4.97 (d, J = 10.2 Hz, 1H), 4.04–4.01 (m, 2H) 2.33–2.23 (m, 1H), 2.14–2.02 (m, 1H) 1.66–1.39 (m, 9H), 1.37–1.30 (m, 10H), 0.88 (t, J = 6.6 Hz, 3H); 13C NMR (75 MHz, CDCl3): δ 138.4, 114.9, 107.5, 78.1, 77.4, 32.0, 30.4, 29.8, 29.2, 28.8, 26.0, 22.7, 14.1; IR (neat) νmax: 2986, 2929, 2858, 1456, 1379, 1245, 1223, 1098, 871, cm–1; HRMS (ESI) m/z: calcd for C14H26O2Na [M + Na]+, 249.1830; found, 249.1833.
3-((4S,5R)-2,2-Dimethyl-5-pentyl-1,3-dioxolan-4-yl)propan-1-ol (20)
Olefin 19 (2.02 g, 8.9 mmol) was dissolved in a mixture of CH2Cl2 and MeOH (16 mL, 4:1) and was cooled to −78 °C. Ozone was bubbled through the solution until it turned blue in about 30 min. The ozone supply was cut off, and oxygen was then bubbled through the solution until it turned colorless. Me2S (1 mL) was added. The solution was warmed to room temperature and stirred further for 6 h. The solvent was evaporated, and the residue was dissolved in anhydrous EtOH (20 mL) under argon. The solution was cooled to 0 °C, and NaBH4 (371 mg, 9.8 mmol) was added. The solution was allowed to attain room temperature, and stirring was continued further for 1 h. The reaction was then quenched by saturated aqueous NH4Cl solution (5 mL). The mixture was extracted with EtOAc (2 × 60 mL), washed with water and brine, dried over Na2SO4, filtered, and concentrated in vacuo. Purification of the crude product by column chromatography (SiO2, 100–200 mesh, 15–25% EtOAc in hexane as an eluent) resulted alcohol 20 (1.58 g, 77% over two steps) as a colorless oil: Rf = 0.25 (20% EtOAc/hexane); [α]D28 −2.0 (c 4.2, CHCl3); 1H NMR (500 MHz, CDCl3): δ 4.06–4.04 (m, 2H), 3.70–3.66 (m, 2H), 2.20 (bs, 1H), 1.75–1.55 (m, 2H), 1.53–1.36 (m, 7H), 1.33–1.24 (m, 9H), 0.88 (m, 3H); 13C NMR (100 MHz, CDCl3): δ 107.6, 78.3, 78.2, 62.9, 32.0, 29.9, 29.8, 28.6, 26.8, 26.0, 22.7, 14.1 ppm; IR (neat) νmax: 3407, 2933, 1218, 1029 cm–1; HRMS (ESI) m/z: calcd for C13H26O3Na [M + Na]+, 253.1780; found, 253.1782.
5-((3-((4S,5R)-2,2-Dimethyl-5-pentyl-1,3-dioxolan-4-yl)propyl)thio)-1-phenyl-1H-tetrazole (22)
To an ice-cold solution of alcohol 20 (2.02 g, 8.80 mmol) in anhydrous THF (30 mL) under argon, Ph3P (2.76 g, 10.55 mmol) and 5-phenyl-1H-tetrazole (21) (1.88 g, 10.55 mmol) were added sequentially. After stirring for 30 min at the same temperature, DIAD (2.0 mL, 10.55 mmol) was added dropwise. The mixture was warmed slowly to room temperature and stirred for 3 h. Solvent was removed in vacuo, and flash column chromatography (SiO2, 230–400 mesh, 4–8% EtOAc in hexane as an eluent) of the resultant residue afforded pure compound 22 (3.01 g, 88%) as a colored oil: Rf = 0.27 (10% EtOAc/hexane); [α]D27 −8.8 (c 0.6, CHCl3); 1H NMR (CDCl3, 300 MHz): δ 7.59–7.52 (m, 5H), 4.08–4.00 (m, 2H), 3.48–3.43 (m, 2H), 2.10–2.00 (m, 1H), 1.98–1.84 (m, 1H), 1.69–1.46 (m, 4H), 1.45–1.35 (m, 4H), 1.34–1.25 (m, 8H), 0.90 (m, 3H); 13C NMR (CDCl3, 75 MHz): δ 154.4, 133.8, 130.2, 129.9, 124.0, 107.7, 78.1, 33.4, 32.0, 29.7, 28.9, 28.7, 26.2, 26.1, 26.0, 22.6, 14.1 ppm; IR (neat) νmax: 3475, 2983, 2933, 2859, 1597, 1500, 1457, 1379, 1243, 1218, 1071 cm–1; HRMS (ESI) m/z: calcd for C20H30N4O2NaS [M + Na]+, 413.1987; found, 413.1989.
5-((3-((4S,5R)-2,2-Dimethyl-5-pentyl-1,3-dioxolan-4-yl)propyl)sulfonyl)-1-phenyl-1H-tetrazole (12)
To an ice-cold solution of compound 22 (2.77 g, 7.10 mmol) in EtOH (15 mL), (NH4)6Mo7O24·4H2O (435 mg, 0.35 mmol) and 30% H2O2 (8 mL) were added sequentially. The reaction mixture was warmed slowly to room temperature and stirred for 24 h. The solvent was evaporated, and the residue was extracted with EtOAc (2 × 60 mL), washed with water and brine, dried over Na2SO4, and concentrated in vacuo. Flash column chromatography (SiO2, 100–200 mesh, 25% EtOAc in hexane as an eluent) of the crude residue provided sulfone 12 (2.55 g, 85%) as a colorless oil: Rf = 0.57 (20% EtOAc/hexane); [α]D27 −7.5 (c 0.8, CHCl3); 1H NMR (CDCl3, 300 MHz): δ 7.71–7.63 (m, 2H), 7.62–7.57 (m, 3H), 4.08–4.01 (m, 2H), 3.91–3.75 (m, 2H), 2.29–2.12 (m, 1H), 2.10–2.04 (m, 1H), 1.71–1.57 (m, 2H), 1.56–1.47 (m, 2H), 1.40–1.32 (m, 3H), 1.31–1.19 (m, 9H), 0.89 (m, 3H); 13C NMR (CDCl3, 75 MHz): δ 153.5, 133.1, 131.5, 129.8, 125.2, 107.9, 78.0, 56.0, 31.9, 29.6, 28.6, 28.5, 26.1, 25.9, 22.6, 19.7, 14.1 ppm; IR (neat) νmax: 3458, 2984, 2933, 2860, 1498, 1460, 1341, 1218, 1152, 1064 cm–1; HRMS (ESI) m/z: calcd for C20H30N4O4NaS [M + Na]+, 445.1885; found, 445.1886.
(2R,3R)-Methyl 3-((tert-Butyldimethylsilyl)oxy)-7-((4S,5R)-2,2-dimethyl-5-pentyl-1,3-dioxolan-4-yl)-2-methylhept-4-enoate (10,10a)
Ester 17 (2 g, 6.0 mmol) was dissolved in a mixture of solvent of CH2Cl2 and MeOH (15 mL, 4:1) and was cooled to −78 °C. Ozone was bubbled through the solution until the solution turned blue in about 30 min. The ozone supply was cut off, and oxygen was bubbled through the solution until the solution turned colorless. The reaction was quenched by Me2S (1 mL) and warmed to room temperature. Stirring was continued for another 6 h. The solvent was evaporated, and the residue was passed through a pad of silica gel. The solvent was evaporated, and the residue was dried in vacuo for 2 h to remove the relatively volatile benzaldehyde by-product. Aldehyde 11 was obtained as a colorless liquid which was used directly for the next reaction without further purification: Rf = 0.55 (5% EtOAc/hexane); HRMS (ESI) m/z: calcd for C12H24O4SiNa [M + Na]+, 261.1517; found, 261.1519.
Sulfone 12 (3.04 g, 7.2 mmol) was dissolved in anhydrous THF (10 mL) under argon and cooled to −78 °C. KHMDS (0.5 M in toluene, 14.0 mL, 7.0 mmol) was added and stirred for 15 min. A solution of the above aldehyde 11 (6.0 mmol) dissolved in anhydrous THF (10 mL) was cannulated into the reaction mixture and stirred for 1 h at the same temperature. The reaction mixture was quenched with saturated aqueous NH4Cl solution (5 mL). The mixture was extracted with EtOAc (2 × 50 mL), washed with water and brine, dried over Na2SO4, and finally concentrated in vacuo. Purification of the resultant residue by flash column chromatography (SiO2, 230–400 mesh, 2–8% EtOAc in hexane as an eluent) provided an inseparable mixture of compounds 10 and 10a (1.66 g, 61% based on recovered sulfone 12) as a colored liquid, 507 mg of sulfone 12 was recovered: Rf = 0.17 (5% EtOAc/hexane); (mixture of isomers) 1H NMR (CDCl3, 300 MHz): δ 5.67–5.57 (m, 1H), 5.34 (dd, J = 15.3, 7.8 Hz, 1H), 4.17 (t, J = 8.1 Hz, 1H), 4.03–4.00 (m, 2H), 3.65 (m, 3H), 2.50 (m, 1H), 2.30–2.25 (m, 1H), 2.22–2.16 (m, 1H), 1.56–1.40 (m, 6H), 1.39–1.27 (m, 10H), 1.00 (d, J = 7.2 Hz, 3H), 0.90–0.87 (m, 3H), 0.83 (m, 9H), 0.01 (s, 3H), 0.00 (s, 3H); 13C NMR (CDCl3, 75 MHz): δ 175.7, 132.9, 131.3, 107.5, 78.1, 76.4, 51.5, 47.3, 32.0, 29.7, 29.5, 28.9, 28.7, 26.1, 26.0, 25.8, 22.7, 18.1, 14.1, 13.4, −3.8, −4.9 ppm; IR (neat) νmax: 3460, 2983, 2936, 2860, 1739, 1458, 1377, 1246, 1218, 1168, 1044 cm–1; HRMS (ESI) m/z: calcd for C25H48O5SiNa [M + Na]+, 479.3169; found, 479.3167.
(2R,3R)-Methyl-6-((4S,5R)-2,2-dimethyl-5-pentyl-1,3-dioxolan-4-yl)-3-hydroxy-2-methylhept-4-enoate (23,23a)
To an ice-cooled solution of esters 10 and 10a (1.54 g, 3.37 mmol) in anhydrous THF (15 mL) under argon, TBAF (1 M solution in THF, 4.0 mL, 4.0 mmol) was added. The reaction mixture was allowed to attain room temperature and stirred for 3 h. The reaction was then quenched with saturated aqueous NH4Cl solution (3 mL) and extracted with EtOAc (2 × 50 mL), washed with water and brine, dried over Na2SO4, and concentrated in vacuo. Flash column chromatography of the resultant residue (SiO2, 100–200 mesh, 15% EtOAc in hexane as an eluent) afforded the corresponding mixture of esters 23 and 23a (1.02 g, 87%) as oil: Rf = 0.37 (20% EtOAc/hexane); (mixture of isomers) 1H NMR (CDCl3, 300 MHz): δ 5.80–5.70 (m, 1H), 5.47 (ddt, J = 15.3, 7.5, 1.2 Hz, 1H), 4.15 (t, J = 7.2 Hz, 1H), 4.06–3.98 (m, 2H), 3.71 (s, 3H), 2.54 (quintet, J = 6.5 Hz, 1H), 2.31–2.23 (m, 1H), 2.20–2.03 (m, 1H), 1.63–1.42 (m, 6H), 1.41–1.25 (m, 10H), 1.14 (d, J = 7.2 Hz, 3H), 0.91–0.83 (m, 3H); 13C NMR (CDCl3, 75 MHz): δ 176.2, 133.6, 130.5, 107.5, 78.1, 77.4, 74.8, 51.9, 45.6, 32.0, 29.7, 29.5, 29.0, 28.7, 26.1, 26.0, 22.7, 14.2, 14.1 ppm; IR (neat) νmax: 3460, 2985, 2939, 2871, 1745, 1460, 1375, 1245, 1219, 1168, 1044 cm–1; HRMS (ESI) m/z: calcd for C19H34O5Na [M + Na]+, 365.2304; found, 365.2306.
(2R,3S)-Methyl-3-((2S,3S)-3-(2-((4S,5R)-2,2-dimethyl-5-pentyl-1,3-dioxolan-4-yl)ethyl)oxiran-2-yl)-3-hydroxy-2-methylpropanoate (9)
To a suspension of activated 4 Å MSs (powdered, 300 mg) in anhydrous CH2Cl2 (20 mL) under argon, Ti(OiPr)4 (1.7 mL, 5.8 mmol) and (+)-DIPT (1.32 mL, 6.3 mmol) were added sequentially at −20 °C. After being stirred for 15 min, the mixture of esters 23 and 23a (1.8 g, 5.3 mmol) dissolved in anhydrous CH2Cl2 (8 mL) was cannulated. After 15 min, TBHP (4.0 M solution in toluene, 5.0 mL, 21 mmol) was added and stirring was continued for 6 days at the same temperature. The reaction mixture was quenched with an aqueous solution of tartaric acid (200 mg in 2 mL of water), diluted with Et2O (20 mL), warmed to room temperature, and stirred further for 1 h. The resultant mixture was extracted with Et2O (2 × 50 mL), washed with water and brine, and concentrated in vacuo. The crude residue was dissolved in Et2O (30 mL) and cooled to 0 °C, and a cooled 10% solution of NaOH in brine (10 mL) was added into it. The mixture was stirred for 2 h at room temperature to complete the hydrolysis of DIPT. Et2O was removed, and the aqueous part was extracted with Et2O (2 × 60 mL), washed with water and brine, dried (Na2SO4), filtered, and concentrated in vacuo. Purification of the crude residue by column chromatography (SiO2, 230–400 mesh, 14–24% EtOAc in hexane as an eluent) afforded required epoxide 9 (1.02 g, 72% based on E-olefin used) as a colorless oil. 825 mg of a diastereomeric mixture (E/Z ≈ 1:1) of compounds 23 and 23a was recovered: Rf = 0.30 (20% EtOAc/hexane); [α]D27 −12.6 (c 0.8, CHCl3); 1H NMR (CDCl3, 500 MHz): δ 4.07–4.04 (m, 1H), 3.72 (s, 3H), 3.66 (q, J = 5.0, 2.0 Hz, 1H), 3.00–2.99 (m, 1H), 2.81 (dd, J = 7.5, 5.0 Hz, 2H), 2.72 (quintet, J = 6.5 Hz, 1H), 1.80–1.72 (m, 1H), 1.69–1.58 (m, 3H), 1.51–1.48 (m, 3H), 1.44–1.35 (m, 3H), 1.32–1.24 (m, 11H), 0.88 (t, J = 6.5 Hz, 3H); 13C NMR (CDCl3, 100 MHz): δ 175.6, 107.7, 78.1, 72.3, 59.2, 55.8, 52.0, 43.1, 32.0, 29.8, 29.7, 28.7, 28.3, 26.1, 26.1, 22.7, 14.1, 13.7 ppm; IR (neat) νmax: 3454, 2984, 2952, 2860, 1738, 1635, 1458, 1378, 1246, 1217, 1170, 1039 cm–1; HRMS (ESI) m/z: calcd for C19H34O6Na [M + Na]+, 381.2253; found, 381.2254.
(2S,2′R,3S,4R,5′S)-3-Hydroxy-5′-((R)-1-hydroxyhexyl)-4-methylhexahydro-[2,2′-bifuran]-5(2H)-one (8)/(3R,4S,5R)-4-Hydroxy-3-methyl-5-((1R,4S,5R)-1,4,5-trihydroxydecyl)dihydrofuran-2(3H)-one (24)
The epoxy ester 9 (102 mg, 0.29 mmol) was dissolved in anhydrous MeOH (2 mL) under argon and cooled to 0 °C, and then CSA (7 mg, 0.029 mmol) was added. The reaction mixture was stirred for 5 h prior to quenching with saturated aqueous NaHCO3 solution (0.5 mL). The solvent was evaporated, and the residue was extracted with CH2Cl2 (3 × 20 mL), washed with brine, dried over Na2SO4, evaporated, and concentrated in vacuo. Purification of the crude residue by column chromatography (SiO2, 230–400 mesh, 3–8% MeOH in CH2Cl2 as an eluent) afforded cytospolide Q (8) (15 mg, 19%) as a colorless oil and compound 24 (42 mg, 49%) as a white amorphous solid.
Data for Compound 8
[α]D27 −1.5 (c 0.08, CHCl3); Rf = 0.57 (10% MeOH/CH2Cl2); 1H NMR (CDCl3, 300 MHz): δ 4.51 (dd, J = 6.9, 3.6 Hz, 1H), 4.12 (dd, J = 8.1, 3.6 Hz, 1H), 3.97–3.85 (m, 2H), 3.84–3.80 (m, 1H), 2.80 (quintet, J = 7.5 Hz, 1H), 2.12–2.06 (m, 1H), 2.03–1.92 (m, 2H), 1.86–1.79 (m, 1H), 1.51–1.47 (m, 1H), 1.39–1.25 (m, 10H), 0.90 (t, J = 6.5 Hz, 3H); 13C NMR (CDCl3, 75 MHz): δ 178.0, 85.3, 83.5, 78.6, 71.6, 71.2, 39.3, 33.2, 31.9, 29.0, 25.7, 23.2, 22.7, 14.1, 8.9 ppm; 1H NMR (CDCl3, 500 MHz): δ 4.52 (dd, J = 7.1, 3.5 Hz, 1H), 4.12 (dd, J = 8.0, 3.6 Hz, 1H), 3.96–3.88 (m, 2H), 3.84–3.79 (m, 1H), 2.81 (quintet, J = 7.5 Hz, 1H), 2.13–2.04 (m, 1H), 1.99–1.91 (m, 2H), 1.88–1.80 (m, 1H), 1.53–1.48 (m, 6H), 1.40–1.24 (m, 9H), 0.91–0.88 (m, 3H); 13C NMR (CDCl3, 125 MHz): δ 177.8, 85.5, 83.6, 78.7, 71.8, 71.4, 39.4, 33.3, 31.9, 29.1, 25.7, 23.5, 22.7, 14.1, 8.9 ppm; IR (neat) νmax: 3431, 3353, 2927, 2857, 1764, 1658, 1474, 1361, 1175, 1046 cm–1; HRMS (ESI) m/z: calcd for C15H26O5Na [M + Na]+, 309.1678; found, 309.1676.
Data for Compound 24
Rf = 0.20 (5% MeOH/CH2Cl2); [α]D27 −7.7 (c 0.9, MeOH); 1H NMR (CD3OD, 300 MHz): δ 4.45 (dd, J = 4.5, 3.0 Hz, 1H), 4.08 (dd, J = 8.4, 3.0 Hz, 1H), 3.93 (td, J = 8.4, 3.0 Hz, 1H), 3.43–3.38 (m, 2H), 2.80 (dq, J = 7.2, 4.8 Hz, 1H), 1.87–1.83 (m, 2H), 1.81–1.75 (m, 1H), 1.72–1.57 (m, 3H), 1.35–1.25 (m, 6H), 1.81 (d, J = 6.9 Hz, 3H), 0.90 (t, J = 6.6 Hz, 1H); 13C NMR (CD3OD, 75 MHz): δ 175.6, 107.7, 78.1, 72.3, 59.2, 55.8, 52.0, 43.1, 32.0, 29.8, 29.7, 28.7, 28.3, 26.1, 26.1, 22.7, 14.1, 13.7 ppm; IR (KBr) νmax: 3436, 3297, 2943, 2856, 1764, 1473, 1322, 1007 cm–1; HRMS (ESI) m/z: calcd for C15H28O6Na [M + Na]+, 327.1783; found, 327.1781.
(4S,4aS,7R,7aS)-4-(2-((4S,5R)-2,2-Dimethyl-5-pentyl-1,3-dioxolan-4-yl)ethyl)-2,2,7-trimethyltetrahydro-6H-furo[3,2-d][1,3]dioxin-6-one (25)
To an ice-cold solution of compound 24 (25 mg, 0.08 mmol) in anhydrous CH2Cl2 (2 mL) under argon, 2,2-DMP (0.5 mL) and CSA (3 mg, 0.0012 mmol) were added sequentially and stirred for 3 h. The reaction was quenched by Et3N (0.5 mL). The resultant mixture was concentrated in vacuo, and the residue was directly purified by flash column chromatography (SiO2, 100–200 mesh, 10% EtOAc in hexane as an eluent) to yield compound 25 (22 mg, 70%) as a colorless oil: Rf = 0.30 (20% EtOAc/hexane); [α]D27 −16.5 (c 0.7, CHCl3); 1H NMR (CDCl3, 300 MHz): δ 4.34 (dd, J = 6.5, 4.0 Hz, 1H), 4.20 (dd, J = 7.0, 4.0 Hz, 1H), 4.05–4.01 (m, 2H), 3.62–3.58 (m, 1H), 2.69 (quintet, J = 6.5 Hz, 1H), 1.85–1.75 (m, 2H), 1.74–1.66 (m, 1H), 1.55–1.47 (m, 2H), 1.45–1.40 (m, 3H), 1.38–1.29 (m, 16H), 1.24 (d, J = 7.0 Hz, 3H), 0.89 (t, J = 6.6 Hz, 3H); 13C NMR (CDCl3, 75 MHz): δ 178.4, 107.6, 101.2, 82.3, 78.1, 70.7, 68.5, 38.5, 32.0, 30.3, 29.6, 28.7, 26.1, 26.1, 25.4, 24.0, 23.9, 22.7, 14.1, 8.2 ppm; IR (neat) νmax: 3443, 3288, 2956, 2824, 1771, 1453, 1356, 1007 cm–1; HRMS (ESI) m/z: calcd for C21H36O6Na [M + Na]+, 407.2410; found, 407.2411.
(2S,3R,E)-1-(Benzyloxy)-7-((4S,5R)-2,2-dimethyl-5-pentyl-1,3-dioxolan-4-yl)-2-methylhept-4-en-3-ol (26,26a)
The mixture of esters 23 and 23a (349 mg, 1.02 mmol) was dissolved in moist Et2O (5 mL) and treated with LiBH4 (46 mg, 2.11 mmol) at 0 °C. The reaction was stirred for 1.5 h prior to quench with saturated aqueous solution of NH4Cl (1 mL). The reaction mixture was extracted with EtOAc (2 × 30 mL), washed with water and brine, dried over Na2SO4, concentrated in vacuo, and purification of the crude residue by column chromatography (SiO2, 100–200 mesh, EtOAc in hexane as eluent) resulted the corresponding inseparable mixture of diols (269 mg, 84%) as a colorless liquid which was taken forwarded to the next step without further characterizations.
To a solution of the above diols (508 mg, 1.62 mmol) in anhydrous toluene (5 mL) under argon, Ag2O (503 mg, 2.17 mmol) and BnBr (0.19 mL, 1.62 mmol) were added sequentially. The reaction mixture was stirred for 24 h at room temperature in the absence of light. After completion of the reaction, the solvent was evaporated and the residue was passed through a pad of Celite. The pad was repeatedly washed with EtOAc, and the eluent was concentrated in vacuo. Flash column chromatography (SiO2, 230–400 mesh, 8–15% EtOAc in hexane as an eluent) of the crude residue yielded an inseparable mixture of primary benzyl ethers 26 and 26a (353 mg, 54%) as liquid: Rf = 0.41 (20% EtOAc/hexane); (mixture of isomers) 1H NMR (CDCl3, 300 MHz): δ 7.37–7.27 (m, 5H), 5.73–5.62 (m, 1H), 5.48 (dd, J = 15.3, 7.2 Hz, 1H), 4.53–4.49 (m, 2H), 4.04–3.94 (m, 3H), 3.59 (dd, J = 9.1, 4.5 Hz, 1H), 3.46 (dd, J = 9.1, 6.8 Hz, 1H), 2.30–2.20 (m, 1H), 2.17–1.99 (m, 1H), 1.93–1.85 (m, 1H), 1.63–1.53 (m, 2H), 1.51–1.45 (m, 1H), 1.44–1.25 (m, 13H), 0.89–0.78 (m, 6H); 13C NMR (CDCl3, 75 MHz): δ 137.9, 132.2, 131.9, 128.6, 127.9, 127.8, 108.6, 78.1, 74.9, 74.8, 73.5, 68.1, 39.0, 32.3, 29.8, 29.6, 29.0, 28.7, 26.1, 22.7, 14.2, 14.1 ppm; IR (neat) νmax: 3405, 2938, 2868, 1724, 1448, 1218, 1031 cm–1; HRMS (ESI) m/z: calcd for C25H40O4Na [M + Na]+, 427.2824; found, 427.2822.
(1S,2S)-3-(Benzyloxy)-1-((2S,3S)-3-(2-((4S,5R)-2,2-dimethyl-5-pentyl-1,3-dioxolan-4-yl)ethyl)oxiran-2-yl)-2-methylpropan-1-ol (27)
To a suspension of activated 4 Å MSs (powdered, 20 mg) in anhydrous CH2Cl2 (3 mL) under argon at −20 °C, Ti(OiPr)4 (0.11 mL, 0.37 mmol) and (+)-DIPT (0.08 mL, 0.40 mmol) were added sequentially. After being stirred for 15 min at the same temperature, the mixture of compounds 26 and 26a (115 mg, 0.34 mmol), dissolved in anhydrous CH2Cl2 (3 mL), was cannulated into the reaction mixture. After 15 min, TBHP (4.0 M solution in toluene, 0.40 mL, 1.3 mmol) was added and stirring was continued for 6 days at the same temperature. The reaction mixture was quenched with a solution of tartaric acid in water (100 mg in 1 mL), diluted with Et2O (10 mL), warmed to the room temperature, and stirred further for 1 h. The organic layer was separated, and the aqueous part was extracted Et2O (2 × 20 mL), washed with water and brine, and concentrated in vacuo. The residue was dissolved in Et2O (5 mL) and cooled to 0 °C, and a cooled 10% solution of NaOH in brine (2 mL) was added into it. The resultant mixture was stirred for 2 h at room temperature to effect the hydrolysis of DIPT. The organic layer was separated, and the aqueous part was extracted Et2O (2 × 20 mL), washed with water and brine, dried (Na2SO4), filtered, and concentrated in vacuo. Purification of crude residue by column chromatography (SiO2, 230–400 mesh, 12–20% EtOAc in hexane as an eluent) afforded epoxide 27 (63 mg, 70% based on E-olefin used) as a colorless oil. 45 mg of a diastereomeric mixture (E/Z ≈ 1:1) of compounds 26 and 26a was recovered: Rf = 0.33 (20% EtOAc/hexane); 1H NMR (CDCl3, 300 MHz): δ 7.37–7.28 (m, 5H), 4.52 (m, 2H), 4.12–3.96 (m, 2H), 3.63 (dd, J = 9.0, 4.5 Hz, 1H), 3.52 (dd, J = 9.0, 6.3 Hz, 1H), 3.48–3.44 (m, 1H), 3.15 (d, J = 1.8 Hz, 1H), 3.00–2.96 (m, 1H), 2.80 (dd, J = 5.4, 2.1 Hz, 1H), 2.09–2.01 (m, 1H), 1.73–1.59 (m, 3H), 1.54–1.45 (m, 3H), 1.45–1.39 (m, 3H), 1.37–1.25 (m, 9H), 1.05 (d, J = 7.2 Hz, 3H), 0.91–0.87 (m, 3H); 13C NMR (CDCl3, 75 MHz): δ 137.9, 128.5, 127.9, 127.7, 107.6, 78.1, 77.3, 74.6, 74.0, 73.6, 59.8, 55.6, 37.6, 32.0, 29.7, 28.7, 28.3, 26.1, 26.0, 22.6, 14.1, 13.5 ppm; IR (neat) νmax: 3471, 2929, 1454, 1245, 1217, 1095 cm–1; HRMS (ESI) m/z: calcd for C25H40O5Na [M + Na]+, 443.2773; found, 443.2774.
(1S,4S,5R)-1-((2S,3S,4S)-3-Hydroxy-4-methyltetrahydrofuran-2-yl)decane-1,4,5-triol (28)
The epoxy ester (27) (40 mg, 0.095 mmol) was dissolved in MeOH (3 mL), and CSA (2.3 mg, 0.001 mmol) was added. The reaction mixture was stirred for 1 h. The reaction was quenched with Et3N (0.5 mL). The resultant mixture was concentrated under reduced pressure, and the resultant residue was purified by column chromatography (SiO2, 100–200 mesh, 2–7% MeOH in CH2Cl2 as an eluent) to afford compound 28 (15 mg, 54%) as liquid and trace amount of compound 29 confirmed by HRMS only.
Data for Compound 28
Rf = 0.30 (10% MeOH/CH2Cl2); 1H NMR (CDCl3, 300 MHz): δ 4.23 (bs, 1H), 3.96 (t, J = 7.8 Hz, 1H), 3.89–3.86 (m, 1H), 3.80 (bs, 1H), 3.70–3.67 (m, 1H), 3.66–3.50 (m, 2H), 2.95 (bs, 1H), 2.32–2.29 (m, 1H), 2.10–1.98 (m, 1H), 1.93–1.89 (m, 1H), 1.74–1.57 (m, 3H), 1.51–1.25 (m, 7H), 1.06 (d, J = 6.9 Hz, 3H), 0.93–0.87 (m, 3H); 13C NMR (CDCl3, 75 MHz): δ 85.5, 75.2, 75.0, 74.3, 73.0, 71.9, 39.7, 32.0, 31.1, 29.8, 28.2, 25.9, 22.7, 14.1, 9.9 ppm; HRMS (ESI) m/z: calcd for C15H30O5Na [M + Na]+, 313.1991; found, 313.1990.
HRMS Data for Compound 29
HRMS (ESI) m/z: calcd for C22H36O5Na [M + Na]+, 403.2460; found, 403.2462.
Acknowledgments
S.C. and G.H.M. thank IACS and Council of Scientific and Industrial Research, New Delhi for their research fellowships. The financial support from Council of Scientific and Industrial Research (project no. 02(0119)/13/EMR-II) and Science and Engineering Research Board (project no. EMR/2016/000988), DST, India, to carry out this work is gratefully acknowledged.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsomega.8b00937.
Copies of NMR (1H and 13C) and HRMS of representative compounds and 2D-NMR data (COSY, NOESY, and HSQC) of compounds 24 (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- a Lu S.; Kurtán T.; Yang G.; Sun P.; Mándi A.; Krohn K.; Draeger S.; Schulz B.; Yi Y.; Li L.; Zhang W. Cytospolides A-E, new nonanolides from an endophytic fungus, cytospora sp. Eur. J. Org. Chem. 2011, 5452–5459. 10.1002/ejoc.201100675. [DOI] [PubMed] [Google Scholar]; b Lu S.; Sun P.; Li T.; Kurtán T.; Mándi A.; Antus S.; Krohn K.; Draeger S.; Schulz B.; Yi Y.; Li L.; Zhang W. Bioactive nonanolide derivatives isolated from the endophytic fungus cytospora sp. J. Org. Chem. 2011, 76, 9699–9710. 10.1021/jo201755v. [DOI] [PubMed] [Google Scholar]
- a Yadav J. S.; Pandurangam T.; Kumar A. S.; Reddy P. A. N.; Prasad A. R.; Reddy B. V. S.; Rajendraprasad K.; Kunwar A. C. The first stereoselective total synthesis of the Z-isomer of cytospolide E. Tetrahedron Lett. 2012, 53, 6048–6050. 10.1016/j.tetlet.2012.08.108. [DOI] [Google Scholar]; b Rej R. K.; Nanda S. Chemoenzymatic asymmetric total synthesis of nonanolide (Z)-cytospolides D, E and their Stereoisomers. Eur. J. Org. Chem. 2014, 2014, 860–871. 10.1002/ejoc.201301365. [DOI] [Google Scholar]; c Vadhadiya P. M.; Rout J. K.; Ramana C. V. Studies toward the total synthesis of cytospolide E. Tetrahedron 2015, 71, 9088–9094. 10.1016/j.tet.2015.10.018. [DOI] [Google Scholar]; d Kamal A.; Balakrishna M.; Reddy P. V.; Rahim A. First total synthesis of the E- and Z-isomers of cytospolide-D. Tetrahedron: Asymmetry 2014, 25, 148–155. 10.1016/j.tetasy.2013.12.004. [DOI] [Google Scholar]; e Rej R. K.; Kumar R.; Nanda S. Asymmetric synthesis of cytospolides C and D through successful exploration of stereoselective Julia–Kocienski olefination and Suzuki reaction followed by macrolactonization. Tetrahedron 2015, 71, 3185–3194. 10.1016/j.tet.2015.04.014. [DOI] [Google Scholar]; f Rao R. N.; Raju B. C. Stereoselective approach for total synthesis of Z-isomer of cytospolide E. Indian J. Chem., Sect. B: Org. Chem. Incl. Med. Chem. 2017, 56, 423–430. [Google Scholar]; g Ehrlich G.; Stark C. B. W. Total synthesis of cytospolide D and its biomimetic conversion to cytospolides M, O, and Q. Org. Lett. 2016, 18, 4802–4805. 10.1021/acs.orglett.6b02193. [DOI] [PubMed] [Google Scholar]
- a Guchhait S.; Chatterjee S.; Ampapathi R. S.; Goswami R. K. Total synthesis of reported structure of baulamycin A and its congeners. J. Org. Chem. 2017, 82, 2414–2435. 10.1021/acs.joc.6b02838. [DOI] [PubMed] [Google Scholar]; b Das S.; Kuilya T. K.; Goswami R. K. Asymmetric total synthesis of bioactive natural lipid mycalol. J. Org. Chem. 2015, 80, 6467–6489. 10.1021/acs.joc.5b00972. [DOI] [PubMed] [Google Scholar]; c Das S.; Paul D.; Goswami R. K. Stereoselective total synthesis of bioactive marine natural product biselyngbyolide B. Org. Lett. 2016, 18, 1908–1911. 10.1021/acs.orglett.6b00713. [DOI] [PubMed] [Google Scholar]; d Das S.; Goswami R. K. Stereoselective total synthesis of marine cyclodepsipeptide calcaripeptides A-C. J. Org. Chem. 2014, 79, 9778–9791. 10.1021/jo5019798. [DOI] [PubMed] [Google Scholar]; e Kuilya T. K.; Goswami R. K. Stereoselective total synthesis of carolacton. Org. Lett. 2017, 19, 2366–2369. 10.1021/acs.orglett.7b00903. [DOI] [PubMed] [Google Scholar]; f Chatterjee S.; Guchhait S.; Goswami R. K. Stereoselective total synthesis of cytospolide P. J. Org. Chem. 2014, 79, 7689–7695. 10.1021/jo501184t. [DOI] [PubMed] [Google Scholar]; g Chatterjee S.; Kuilya T. K.; Goswami R. K. Studies directed toward the stereoselective synthesis of cytospolide E. ACS Omega 2018, 3, 1041–1059. 10.1021/acsomega.7b01893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Evans D. A.; Tedrow J. S.; Shaw J. T.; Downey C. W. Diastereoselective magnesium halide-catalyzed anti-aldol reactions of chiral N-acyloxazolidinones. J. Am. Chem. Soc. 2002, 124, 392–393. 10.1021/ja0119548. [DOI] [PubMed] [Google Scholar]; b Evans D. A.; Downey C. W.; Shaw J. T.; Tedrow J. S. Magnesium halide-catalyzed anti-aldol reactions of chiral N-acylthiazolidinethiones. Org. Lett. 2002, 4, 1127–1130. 10.1021/ol025553o. [DOI] [PubMed] [Google Scholar]
- Suman P.; Raju B. C. Carbohydrate-based first stereoselective total synthesis of bioactive cytospolide P. Org. Biomol. Chem. 2014, 12, 3358–3361. 10.1039/c4ob00323c. [DOI] [PubMed] [Google Scholar]
- Liu G.; Wang Z. Total synthesis of koninginin D, B and E. Synthesis 2001, 119–127. 10.1055/s-2001-9741. [DOI] [Google Scholar]
- Candy M.; Audran G.; Bienaymé H.; Bressy C.; Pons J.-M. Total synthesis of (+)-crocacin C using hidden symmetry. J. Org. Chem. 2010, 75, 1354–1359. 10.1021/jo902582w. [DOI] [PubMed] [Google Scholar]
- a Cichowicz N. R.; Nagorny P. Synthesis of conjugated polyenes via sequential condensation of sulfonylphosphonates and aldehydes. Org. Lett. 2012, 14, 1058–1061. 10.1021/ol203431e. [DOI] [PubMed] [Google Scholar]; b Crimmins M. T.; Haley M. W.; O’Bryan E. A. Formal synthesis of (+)-sorangicin A. Org. Lett. 2011, 13, 4712–4715. 10.1021/ol201920j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blakemore P. R. The modified Julia olefination: alkene synthesis via the condensation of metallated heteroarylalkylsulfones with carbonyl compounds. J. Chem. Soc., Perkin Trans. 1 2002, 2563–2585. , and the references cited therein 10.1039/b208078h. [DOI] [Google Scholar]
- Gao Y.; Hanson R. M.; Klunder J. M.; Ko S. Y.; Masamunne H.; Sharpless K. B. Catalytic asymmetric epoxidation and kinetic resolution: modified procedures including in situ derivatization. J. Am. Chem. Soc. 1987, 109, 5765–5780. 10.1021/ja00253a032. [DOI] [Google Scholar]
- Hoye T. R.; Tan L. Total synthesis of the potent antitumor, bis-tetrahydrofuranyl annonaceous acetogenins (+)-asimicin and (+)-bullatacin. Tetrahedron Lett. 1995, 36, 1981–1984. 10.1016/0040-4039(95)00207-s. [DOI] [Google Scholar]
- a Skalitzky D. J.; Rychnovsky S. D. Stereochemistry of alternating polyol chains: 13C NMR analysis of 1,3-diol acetonides. Tetrahedron Lett. 1990, 31, 945–948. 10.1016/s0040-4039(00)94399-5. [DOI] [Google Scholar]; b Rychnovsky S. D.; Rogers B. N.; Richardson T. I. Configurational assignment of polyene macrolide antibiotics using the [13C] acetonide analysis. Acc. Chem. Res. 1998, 31, 9–17. 10.1021/ar960223n. [DOI] [Google Scholar]
- Botubol J. M.; Macías-Sánchez A. J.; Collado I. G.; Hernández-Galán R. Stereoselective synthesis and absolute configuration determination of xylariolide A. Eur. J. Org. Chem. 2013, 2013, 2420–2427. 10.1002/ejoc.201201526. [DOI] [Google Scholar]
- Urones J. G.; Marcos I. S.; Basabe P.; Sexmero M. J.; Dibz D.; Garrido N. M.; Prieto J. E. S. Formation of orthoesters in the sharpless asymmetric epoxidation: hemisynthesis of labdanes. Tetrahedron 1990, 46, 2495–2502. 10.1016/s0040-4020(01)82030-5. [DOI] [Google Scholar]
- Davies I. R.; Cheeseman M.; Green R.; Mahon M. F.; Merritt A.; Bull S. D. Efficient asymmetric synthesis of chiral hydroxy-γ-butyrolactones. Org. Lett. 2009, 11, 2896–2899. 10.1021/ol9008365. [DOI] [PubMed] [Google Scholar]
- Bouzide A.; Sauvé G. Highly selective silver (I) oxide mediated monoprotection of symmetrical diols. Tetrahedron Lett. 1997, 38, 5945–5948. 10.1016/s0040-4039(97)01328-2. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.