

Abstract
GFI1B variants are a rare cause of thrombocytopenia. We report on a male child who was initially diagnosed with immune thrombocytopenia. However, subtle clinical signs led to suspicion for a genetic cause of thrombocytopenia. Gene panel sequencing revealed a rare variant in GFI1B (C168F), which has recently been reported in several families with thrombocytopenia. We demonstrate that this variant significantly alters platelet parameters in population studies. This case highlights how diagnoses of exclusion, such as immune thrombocytopenia, can be confounded by genetic variation. Our understanding of blood disorders will undoubtedly evolve from an increased knowledge of human genetic variation.
Keywords: GFI1B, thrombocytopenia, hematopoiesis, bone marrow failure, immune thrombocytopenia, genetics
1. Introduction
One of the most common causes of low platelet counts in children is immune thrombocytopenia (ITP)1. However, ITP is a diagnosis of exclusion, and distinguishing ITP from other causes of thrombocytopenia in children can be challenging. Consideration of genetic causes of thrombocytopenia is important, as treatment and prognosis may considerably differ depending on the underlying cause. One genetic cause of thrombocytopenia stems from variants in the growth factor-independent 1B (GFI1B) gene, which encodes a master transcription factor critical for hematopoiesis2. We describe the case of a male child who presented with thrombocytopenia and was initially diagnosed with ITP, but who was later found to carry a rare variant in GFI1B that has been associated with macrothrombocytopenia.
2. Results
2.1. Case report
A previously healthy 4-year-old male child of South Asian descent was referred to our hematology clinic after initially presenting to his pediatrician for two weeks of increased bruising, petechiae, and an episode of self-resolving epistaxis that lasted about 30 minutes. An initial complete blood count (CBC) collected at the pediatrician’s office revealed a platelet count of 31 000 cells/μL with other blood parameters being entirely normal. Prior platelet counts were also reportedly normal.
During his initial visit to the hematology clinic, the patient was found to have multiple small bruises on both of his legs, but no petechiae. His physical exam also revealed bilateral hypoplastic thumbs. A complete review of systems was notable for a lifelong history of mild recurrent epistaxis, with each episode typically lasting less than 5 minutes. For two weeks prior to this visit, scattered petechiae primarily along the lower extremities were also noted. The patient otherwise has had no other bleeding, hepatosplenomegaly, or relevant symptoms. The patient did appear to have a viral intercurrent illness with rhinorrhea and cough immediately preceding the onset of symptoms. The patient had a past history of an atrial and ventricular septal defect that resolved without intervention, as well as bilateral thumb hypoplasia for which he was undergoing occupational therapy. He otherwise had no history of medical problems. The patient’s family history was unrevealing for any hematologic abnormalities or clinical bleeding.
A complete blood count obtained during his initial hematology visit showed a platelet count of 43 000 cells/μL with an immature platelet fraction of 10.7%. His hemoglobin (Hb) was 11.0 g/dL with a normal mean corpuscular volume (MCV) of 78.3 fL. The reticulocyte percentage was 1.1%. Lactate dehydrogenase (LDH) was slightly elevated at 329 unit/L (normal range 110-295 unit/L). A Direct Coombs test was negative. The remainder of his blood parameters were within normal limits. A peripheral blood smear showed a reduction in platelet number with numerous enlarged platelets that had normal granularity (Figure 1A).
Figure 1.

Peripheral blood smears of the reported patient with the GFI1B C168F variant. Blood smears were visualized under light microscopy at 100× magnification. Time points correspond to (A) the patient’s initial hematology clinic visit, (B) the 1-month follow-up visit, and (C) the 5-month follow-up visit. A 10 μm scale bar is shown.
Given this patient’s history, well appearance, reassuring physical exam, and isolated macrothrombocytopenia, a presumptive diagnosis of immune thrombocytopenia was made. However, given his congenital anomalies (hypoplastic thumbs and a history of an atrioventricular septal defect noted during infancy), Fanconi Anemia and other bone marrow failure syndromes were also considered3. A chromosome breakage analysis was performed at the initial visit and was negative. As this patient’s Buchanan and Adix bleeding score was grade 1-2, he was sent home without any intervention1.
At his one-month follow-up visit, his bruising and petechiae had decreased substantially. A repeat CBC showed an improved platelet count at 156 000 cells/μL, with an elevated mean platelet volume of 12.7 fL. There was also a drop in hemoglobin levels compared to his prior CBC, from 11.0 to 10.0 g/dL, with an inadequate reticulocyte response of 0.9% (absolute reticulocytes 0.038 M cells/μL). A peripheral blood smear showed anisopoikilocytosis with occasional teardrop cells and macrothrombocytopenia (Figure 1B). A hemoglobin electrophoresis found a mildly elevated fetal hemoglobin (HbF) at 2.4%. Therefore, a bone marrow failure genetic panel (using whole exome sequencing with analysis of genes implicated in congenital bone marrow syndromes) was sent to more fully evaluate this possibility3 (list of genes included in Supporting Information Table S1). No bone marrow failure gene mutations were identified, but a likely pathogenic variant in the GFI1B gene (chr9:135863848 G>T in hg19 coordinates, c.503 G>T in exon 8, causing a C168F change) was reported.
At his two- and five-month follow-up appointments, the patient remained asymptomatic with no further bleeding symptoms. His platelet levels remained in the low-normal range at 151 000 cells/μL with an MPV of 12.7 fL at his two-month follow-up appointment, and 159 000 cells/μL with an MPV of 12.8 fL at his five-month follow-up appointment. During this time his hemoglobin level recovered to 11.0 g/dL with a persistently elevated HbF of 2.6%. A blood smear at his five-month follow-up appointment confirmed improvement in platelet count (Figure 1C). Given the resolution of his symptoms, no further diagnostic workup or treatment was pursued.
2.2. The C168F variant in GFI1B
Growth factor-independent 1B (GFI1B) encodes a transcription factor that plays a crucial role in hematopoiesis2. In mice, GFI1B is essential for the development of both erythroid and megakaryocytic lineages, as well as for maintenance of hematopoietic stem cells4,5. In humans, mutations in GFI1B may cause bleeding disorders with thrombocytopenia and compromised platelet function, although the observed phenotypes are variable6–8. In addition, common genetic variants in GFI1B have been associated with variation in platelet and other blood cell counts2,9.
Notably, the C168F GFI1B variant identified in our patient has been recently found to segregate in an autosomal dominant manner in three unrelated families with mild to moderate macrothrombocytopenia, but without significant bruising or bleeding symptoms10,11. Comparison of affected and unaffected relatives within each family suggests that the C168F mutation is associated with a decrease in platelet counts without signs of α-granule deficiency, in contrast to other GFI1B mutations that result in changes in platelet granules10,12. Computational models predict that the C168F mutation changes the conformation of the first non-DNA-binding zinc finger domain13. Additionally, in vitro studies suggest that this mutation causes de-repression of other genes through disruption of the protein’s usual transcriptional repressor activity10,14. Furthermore, several additional carriers of the C168F mutation have been reported in the literature to demonstrate elevated platelet sizes. One individual heterozygous for the allele had an MPV of 12.5 fL, and in one family the mean platelet size among carriers was ~30% larger compared to an unaffected family member, as measured by platelet diameter8,15. Moreover, two individuals homozygous for the C168F mutation have been reported to have marked thrombocytopenia and abnormal platelet function, suggesting that this variant is indeed a loss-of-function or hypomorphic allele11,13.
Given these findings, we decided to examine this variant within a large population-based study, the UK Biobank9,16. Within the UK Biobank, this variant has an allele frequency of 0.005487263 in individuals of South Asian ancestry. We assessed the associations between the genotype of this variant and 20 blood parameters in 7,628 individuals of South Asian ancestry using a linear regression with covariates of age, sex, and the top 10 principal components to control for population stratification. Among South Asians, the minor T allele is associated with a lower platelet count (p = 6.76 × 10−13) and a higher mean platelet volume (p = 5.47 × 10−23) (Figure 2). The presence of the GFI1B C168F variant is estimated to confer an average decrease in platelet count of 50 900 cells/μL (standard error (se) = 7070 cells/μL), and an increase in mean platelet volume of 1.29 fL (se = 0.13 fL). Importantly, this effect size would be sufficient to decrease the platelet count to the range that can be observed in individuals with ITP and cause macrothrombocytopenia in carriers.
Figure 2.
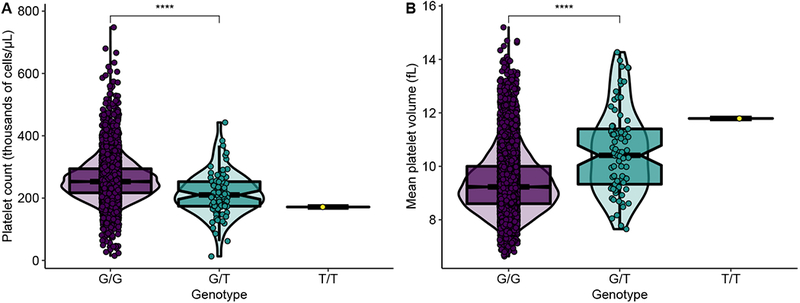
Association between platelet parameters and genotype of GFI1B C168F variant within South Asian population (n = 7628) in the UK Biobank. There is a dose-dependent effect of the C168F variant on (A) platelet count and (B) mean platelet volume. Only one individual was homozygous for the C168F variant in the UK Biobank, so no density plot is shown for the TT genotype.
3. Discussion
ITP is a diagnosis of exclusion. One recent study estimated that nearly 1 in 7 patients who are initially diagnosed with ITP are later found to have another cause for their thrombocytopenia, indicating that a high index of suspicion for other diagnoses should be maintained17. In the pediatric population, genetic causes of thrombocytopenia should be considered, especially if there is a positive family history of thrombocytopenia and/or accompanying laboratory abnormalities that are not fully explained by ITP. For example, isolated thrombocytopenia is sometimes the initial presenting symptom in patients with inherited bone marrow failure syndromes, such as Fanconi Anemia or congenital amegakaryocytic thrombocytopenia (CAMT)18. Familial platelet disorders such as those caused by dominant mutations in ANKRD26 and ETV6 should also be considered, as these diseases confer an increased risk of developing malignancies18.
Our patient was initially thought to have ITP. Several aspects of his history and presentation made ITP a likely diagnosis. First, the patient reportedly had normal platelet levels in the past. Alternate etiologies of thrombocytopenia (e.g. infections, drugs/toxins) were also unlikely given the clinical history obtained. When his platelet levels recovered and symptoms improved after one month without any medical intervention, this provided further support for ITP, a typically transient disease in children19. However, the persistently low-normal level of his platelets, history of recurrent epistaxis, new anemia, borderline abnormal blood smear with occasional teardrop cells, and mildly elevated HbF levels prompted a workup for bone marrow failure syndromes, which revealed a heterozygous missense variant in GFI1B.
The specific GFI1B C168F mutation has only been reported in a handful of individuals to date who all have mild to moderate macrothrombocytopenia10,13. The more severe phenotype reported with this variant in the homozygous state, along with functional assays, suggests that this variant is indeed a loss-of-function or hypomorphic allele10,13. We extended these observations through an analysis of individuals of South Asian ancestry within the UK Biobank cohort, where this variant is associated with a lower platelet count and higher platelet volume to an extent that would be sufficient to cause a macrothrombocytopenia.
Our patient appears to have a low baseline platelet count, which is likely explained by the GFI1B C168F variant. It is possible that he may have had ITP on top of his preexisting thrombocytopenia, which could have further reduced his platelet count. Alternatively, he may have had suppression of thrombopoiesis from another cause, such as an intercurrent viral illness, in the setting of the GFI1B variant lowering baseline platelet counts and altering normal thrombopoiesis. This combination of events would explain the sudden dip in his platelet count and subsequent recovery to a low-normal platelet count. The increased HbF observed in this patient is interesting, and it would be valuable to examine the extent to which this allele may impact fetal hemoglobin gene regulation and erythropoiesis in future studies20,21. The relevance of the GFI1B mutation to our patient’s thumb and cardiac abnormalities remains unclear; there have been no reported associations between the GFI1B variant and these phenotypes.
In summary, our case demonstrates that persistent thrombocytopenia in the presence of other hematologic alterations should prompt a workup for potential genetic causes of thrombocytopenia. ITP is a diagnosis of exclusion, and can sometimes exacerbate or mask another cause of thrombocytopenia. More broadly, this case illustrates how our understanding of diagnoses of exclusion may be revised as we uncover germline genetic variation with large effects on hematopoiesis. Indeed, as we observe in this case, leveraging the power of population genetics can generate further insights into the effects of variants of uncertain significance and help us to refine diagnoses.
Supplementary Material
Supporting Information Table S1. The list of genes tested in our bone marrow failure exome sequencing gene panel. Genes with an asterisk either did not undergo deletion/duplication analysis or had limited coverage.
Acknowledgments
We thank members of the Sankaran laboratory and Dr. Rachael Grace for helpful discussions and comments. This work was supported by National Institutes of Health (NIH) grants R01 DK103794 and R33 HL120791, as well as the New York Stem Cell Foundation (to V.G.S.). A.N.C. and E.L.B. are supported by the Howard Hughes Medical Institute Research Fellows Program. V.G.S. is a New York Stem Cell Foundation-Robertson Investigator. This research was conducted by using the UK Biobank resource under projects 11898 and 31063.
Abbreviations
- GFI1B
Growth factor-independent 1B
- ITP
Immune thrombocytopenia
- CBC
Complete blood count
- Hb
Hemoglobin
- MCV
Mean corpuscular volume
- LDH
Lactate dehydrogenase
- HbF
Fetal hemoglobin
- CAMT
Congenital amegakaryocytic thrombocytopenia
- ANKRD26
Ankyrin Repeat Domain 26
- ETV6
ETS Variant 6
- se
Standard error
Footnotes
Conflicts of Interest Statement
The authors declare no competing interests.
References
- 1.Grace RF, Neunert C. Second-line therapies in immune thrombocytopenia. Hematology. 2016;2016(1):698–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Polfus LM, Khajuria RK, Schick UM, et al. Whole-Exome Sequencing Identifies Loci Associated with Blood Cell Traits and Reveals a Role for Alternative GFI1B Splice Variants in Human Hematopoiesis. Am J Hum Genet. 2016;99(2):481–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Shimamura A, Alter BP. Pathophysiology and management of inherited bone marrow failure syndromes. Blood Rev. 2010;24(3):101–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Qin J, Ramaswamy S, Amzallag A, et al. Distinct, strict requirements for Gfi-1b in adult bone marrow red cell and platelet generation. J Exp Med. 2014;211(5):909–927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Saleque S, Cameron S, Orkin SH. The zinc-finger proto-oncogene Gfi-1b is essential for development of the erythroid and megakaryocytic lineages. Genes Dev. 2002;16(3):301–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rabbolini DJ, Morel-Kopp M-C, Ward CM, Stevenson WS. GFI1B variants associated with thrombocytopenia. Platelets. 2017;28(5):525–527. [DOI] [PubMed] [Google Scholar]
- 7.Schulze H, Schlagenhauf A, Manukjan G, et al. Recessive grey platelet-like syndrome with unaffected erythropoiesis in the absence of the splice isoform GFI1B-p37. Haematologica. 2017;102(9):375–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kitamura K, Okuno Y, Yoshida K, et al. Functional characterization of a novel GFI1B mutation causing congenital macrothrombocytopenia. J Thromb Haemost. 2016;14(7):1462–1469. [DOI] [PubMed] [Google Scholar]
- 9.Ulirsch JC, Lareau CA, Bao EL, et al. Interrogation of human hematopoiesis at single-cell and single-variant resolution. Nat Genet. 2019: 10.1038/s41588-019-0362-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rabbolini DJ, Morel-Kopp MC, Chen Q, et al. Thrombocytopenia and CD34 expression is decoupled from α-granule deficiency with mutation of the first growth factor-independent 1B zinc finger. J Thromb Haemost. 2017;15(11):2245–2258. [DOI] [PubMed] [Google Scholar]
- 11.Favier R, Bergevoet SM, McKinney HL, et al. Inherited missense variants that affect GFI1B function do not necessarily cause bleeding diatheses. Haematologica. 2018:haematol.2018.207712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Marneth AE, Van Heerde WL, Hebeda KM, et al. Platelet CD34 expression and a/d-granule abnormalities in GFI1B- and RUNX1-related familial bleeding disorders. Blood. 2017;129(12):1733–1736. [DOI] [PubMed] [Google Scholar]
- 13.Chen L, Kostadima M, Martens JHA, et al. Transcriptional diversity during lineage commitment of human blood progenitors. Science (80- ). 2014;345(6204):1251033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zweidler-Mckay PA, Grimes HL, Flubacher MM, Tsichlis PN. Gfi-1 encodes a nuclear zinc finger protein that binds DNA and functions as a transcriptional repressor. Mol Cell Biol. 1996;16(8):4024–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Uchiyama Y, Ogawa Y, Kunishima S, et al. A novel GFI1B mutation at the first zinc finger domain causes congenital macrothrombocytopenia. Br J Haematol. 2018;181(6):843–847. [DOI] [PubMed] [Google Scholar]
- 16.Bycroft C, Elliott LT, Young A, et al. The UK Biobank resource with deep phenotyping and genomic data. Nature. 2018;562(7726):203–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Arnold DM, Nazy I, Clare R, et al. Misdiagnosis of primary immune thrombocytopenia and frequency of bleeding: lessons from the McMaster ITP Registry. Blood Adv. 2017;1(25):1–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Noris P, Pecci A. Hereditary thrombocytopenias: A growing list of disorders. Hematology. 2017;2017(1):385–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.George JN. Definition, diagnosis and treatment of immune thrombocytopenic purpura. Haematologica. 2009;94(6):759–762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Basak A, Hancarova M, Ulirsch JC, et al. BCL11A deletions result in fetal hemoglobin persistence and neurodevelopmental alterations. J Clin Invest. 2015;125(6):2363–2368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sankaran VG, Orkin SH. The switch from fetal to adult hemoglobin. Cold Spring Harb Perspect Med. 2013;3(1):a011643. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information Table S1. The list of genes tested in our bone marrow failure exome sequencing gene panel. Genes with an asterisk either did not undergo deletion/duplication analysis or had limited coverage.