

Abstract
Germline TP53 pathogenic variants are rare but associated with a high risk of cancer; they are often identified in the context of clinically diagnosed Li–Fraumeni syndrome predisposing to a range of young onset cancers including sarcomas and breast cancer. The study aim was to conduct a detailed morphological review and immuno‐phenotyping of breast cancer arising in carriers of a germline TP53 pathogenic variant. We compared breast cancers from five defined groups: (1) TP53 carriers with breast cancer (n = 59), (2) early onset HER2‐amplified breast cancer, no germline pathogenic variant in BRCA1/2 or TP53 (n = 55), (3) BRCA1 pathogenic variant carriers (n = 60); (4) BRCA2 pathogenic variant carriers (n = 61) and (5) young onset breast cancer with no known germline pathogenic variant (n = 98). Pathologists assessed a pre‐agreed set of morphological characteristics using light microscopy. Immunohistochemistry (IHC) for HER2, ER, PR, p53, integrin alpha v beta 6 (αvβ6) integrin, α‐smooth muscle actin (α‐SMA) and pSMAD2/3 was performed on tissue microarrays of invasive carcinoma. We confirmed a previously reported high prevalence of HER2‐amplified, ductal no special type invasive breast carcinoma amongst known TP53 germline pathogenic variant carriers 20 of 36 (56%). Furthermore we observed a high frequency of densely sclerotic tumour stroma in cancers from TP53 carriers (29/36, 80.6%) when compared with non‐carriers, 50.9% (28/55), 34.7% (50/144), 41.4% (65/157), 43.8% (95/217) in groups 2–5 respectively. The majority of germline TP53 gene carrier breast tumours had a high intensity of integrin αvβ6, α‐SMA and pSMAD2/3 expression in the majority of cancer cells. In conclusion, aggressive HER2 positive breast cancers with densely sclerotic stroma are common in germline TP53 carriers. High levels of αvβ6 integrin, α‐SMA and pSMAD2/3 expression suggest that the dense stromal phenotype may be driven by upregulated transforming growth factor beta signalling.
Keywords: breast cancer, TP53 pathogenic variant, germline, stroma
Introduction
The importance of functional wild‐type p53 protein is exemplified by the fact that somatic pathogenic variants in TP53 are detected in more than 50% of all cancer types, particularly more aggressive sub‐types, which constitute 28% of breast cancers 1, 2. Germline TP53 pathogenic variants are rare and predispose to early onset breast cancer which, in combination with other tumour types such as malignant brain tumours and sarcoma, is recognised clinically as a manifestation of Li–Fraumeni syndrome (LFS) 3, 4. TP53 encodes the p53 protein which has been referred to as ‘the guardian of the genome’ due to its crucial function in maintaining cellular homeostasis including cell cycle arrest, apoptosis, angiogenesis, metabolism, DNA damage and senescence in response to a number of genotoxic stressors 5.
It has been recognised for over two decades that breast cancers associated with BRCA1 pathogenic variants are predominantly triple receptor (ER, PR and HER2) negative, high grade invasive ductal carcinomas with high numbers of tumour infiltrating lymphocytes (TILs) 6, 7, 8, 9, 10. More recently we reported a preliminary observation that germline TP53 pathogenic variant carriers more commonly developed HER2 positive invasive breast cancer when compared to sporadic cases 9. Our preliminary observation has been confirmed by others 11, 12, 13. In this study, we describe an extended morphological and immunohistochemical characterisation of a larger cohort of early onset breast cancer patients with a germline TP53 pathogenic variant. We were interested to describe not only the tumour features but also the characteristics of the tumour microenvironment (TME), particularly the stroma and presence or absence of TILs given the growing interest in the therapeutic barriers and opportunities of the TME.
Methods
Patients and samples
Formalin fixed paraffin embedded (FFPE) breast cancer tissue blocks were used for this study.
Group 1: The COPE study (Cohort study of TP53 pathogenic variant carriers in early onset breast cancer) acquired breast cancer tissue blocks from patients with a known germline TP53 pathogenic variant and breast cancer (multicentre research ethics committee approval (09/H0501/85) (Table 1).
Table 1.
Cohorts and recruitment eligibility: patient selection and eligibility for groups 1–5: TP53, BRCA1 and BRCA2 gene carriers, HER2+and YBC with no underlying germline pathogenic variant
| Cohort | COPE | POSH | |||
|---|---|---|---|---|---|
| Total no. of patients | 45 | 2956 | |||
| Group | 1 | 2 | 3 | 4 | 5 |
| Selection criterion | Germline TP53 | No germline pathogenic variant HER2+ | Germline BRCA1 | Germline BRCA2 | No germline pathogenic variant YBC |
| No. of patients | 45 | 55 | 60 | 61 | 98 |
| No. of reads | 45 | 55 | 138 | 157 | 217 |
| Morphological review as part of the COPE study* | ✓ | ✓ | |||
| Morphological review as part of the POSH study** | ✓ | ✓ | ✓ | ||
| TMAs | ✓ | ✓ | |||
COPE morphology review (three breast histopathologists).
POSH morphology review involving 13 breast histopathologists described in Shaw et al 22.
Group 2: Tissue blocks were selected from young breast cancer (YBC) cases participating in the Prospective study of Outcomes in Sporadic versus Hereditary breast cancer (POSH) for comparison 14. POSH cases were selected based on the following criteria – tissue block available, no known pathogenic variant in BRCA1, BRCA2 or TP53, invasive breast cancer reported as HER2 positive with associated ductal carcinoma in situ (DCIS) (group 2, n = 55). Ethical approval for POSH was granted in 2000 by South West MREC (00/6/69).
H&E‐stained sections for cases in groups 1 and 2 were assessed independently by two pathologists (GM, MS) for a range of morphological features (Table 1); discrepancies were resolved by a third pathologist (AB).
Groups 3–5: Data were available from a previously published morphological review comparing digital and conventional (glass slide) microscopy reading amongst cases from the POSH study 15. This additional dataset was therefore included for comparison of TP53 germline pathogenic variant carriers with other high risk breast cancer susceptibility gene carriers and with young onset non‐carriers. Group 3 comprised known BRCA1 carriers, group 4 known BRCA2 carriers and group 5 YBC with no underlying germline pathogenic variant.
For groups 3–5, cases were randomly distributed between 13 histopathologists who assessed pre‐specified morphological features using conventional (glass slide) microscopy or a digital interface 15. The submitted reports from conventional microscopy were extracted for each reported case. The POSH cases were reported by more than one pathologist (1–3 per case) so, for the purposes of statistical analyses, we treated each report as a separate case. There were 138 pathologists' reports for BRCA1 carriers, 157 for BRCA2 and 217 for early onset non‐carriers.
Morphological assessment
Morphological assessment was performed on 4 μm thick sections cut using a microtome (Leica Biosystems, Milton Keynes, Bucks, UK) from FFPE breast tumours. Sections were mounted on Superfrost Plus™ Adhesion Microscope Slides (ThermoFisher Scientific, Waltham, MA, USA). Slides were stained using H&E on an automated CoverStainer (Dako UK Ltd, Ely, Cambridge, UK).
Tissue microarray construction
H&E sections were reviewed and 1 mm cores of three representative invasive and two representative in situ areas were taken from the corresponding tissue blocks. Cores were inserted into a new recipient paraffin block using a tissue arrayer (Alphelys Minicore(R)3, Langevin, France). 4 μm sections were cut from each TMA using a microtome (Leica).
Immunohistochemistry
To further characterise breast cancer cases with germline TP53 pathogenic variants (group 1), HER2, ER, PR and p53 IHC was performed on TMA sections using an automated system. For HER2, ER and PR IHC, the Roche Diagnostics Limited, West Sussex, UK antibody with the Ventana Benchmark XT platform and the Ultraview‐Universal DAB detection Kit were used. For p53 the Dako antibody was used with Dako PT link for antigen retrieval, a Dako Autostainer Link 48 staining platform and the Envision FLEX detection system. IHC for integrin alpha v beta 6 (αvβ6), α‐smooth muscle actin (α‐SMA) and pSmad2/3 was performed on TMA sections using a method described previously 16. For COPE cases with HER2 positive results, in situ analysis was conducted on whole tumour sections: dual hapten, dual chromagen in situ hybridisation (DDISH) for HER2/Chromosome 17 using the Roche Ventana INFORM kit performed on the Ventana Ultra platform as per manufacturer recommendations. All antibodies were optimised using national diagnostic standards (UK NEQAS central office, Sheffield, S Yorks, UK).
A further available dataset comprising ER, PR, HER2 and p53 scores from 25 TMAs (1260 cases) from the POSH cohort provided the typical frequency of these markers in YBCs.
Scoring and statistical analysis
HER2 was scored as between 0 and 3 according guidelines used in routine clinical practice based on staining intensity, completeness of membrane expression and the proportion of cells stained 17; a score of either 2+ on IHC and a subsequent positive DDISH test or 3+ on IHC alone are considered positive. ER and PR were evaluated using the Allred scoring system where positive was defined as a score of ≥3 18. p53 was scored using a semi‐quantitative modified McCarthy ‘H’ score; the modified scoring system gave a maximum score of 7 based on the proportion of cancer cells staining positive (1 = <25%, 2 = 25–50%, 3 = 50–75%, 4 = >75%) and the strength of staining intensity (1 = weak, 2 = moderate, 3 = strong) 19, 20. αvβ6 and α‐SMA were scored based on the strength of staining intensity (1 = weak, 2 = moderate, 3 = strong) as described by Marsh and colleagues for αvβ6 scoring 16. pSMAD2/3 was scored using the same method as p53. Scoring was manual (two pathologists reaching a consensus via simultaneous viewing of each core).
For pSMAD2/3, the digital pathology Halo Image Analysis software was trained and used. Each core was checked for tissue content, a classifier (evaluation of tissue and background in the image) and a mark‐up (showing individual cell by cell scoring). Cores were excluded where they were incomplete, contained normal tissue or where the software was unable to score the material.
Summary statistics were used to describe the morphological characteristics of the cases. IBM SPSS Statistics program (IBM United Kingdom Limited, Portsmouth, Hampshire, UK) was used for Pearson Chi‐square, Fisher's exact and Wilcoxon signed rank tests. Pearson Chi‐square was used when the number of unpaired cases in each group was >5, Fisher's exact test was used when the number of unpaired cases in each group was <5 and the Wilcoxon signed rank test when testing statistical significance in paired data sets with a skewed distribution of data.
Results
The COPE study recruited samples from 59 patients with a confirmed germline TP53 pathogenic variant. Fourteen cases had insufficient tumour for the study and were excluded. Of the remaining 45 cases, 36 contained invasive carcinoma and the other 9 contained DCIS only. A total of 32 of 36 (88.9%) of invasive carcinoma cases also had areas of DCIS. A total of 41 of 45 (91.1%) of all cases contained DCIS with 40 of 41 (97.6%) of those cases scored as high grade.
Morphological assessment was included for 60 young onset cases with a germline BRCA1 pathogenic variant (group 3), 61 young onset cases with a germline BRCA2 pathogenic variant (group 4) and 98 young onset breast cancer cases (group 5) with no identifiable germline high risk pathogenic variant (BRCA1, BRCA2 or TP53) 21.
Morphological review of breast tumours derived in germline TP53 carriers
The five young onset breast cancer cohorts were compared. Descriptive summary statistics are provided in Table 2.
Table 2.
Morphological review comparing subgroups: morphological features in invasive breast tumour groups 1–5
| Morphological feature | Subgroup | |||||
|---|---|---|---|---|---|---|
| Feature | Grading | TP53 | HER2+ | BRCA1 | BRCA2 | YBC |
| Tumour grade | 1 | 2/36 (5.6%) | 1/55 (1.8%) | 7/138 (5.1%) | 13/157 (8.3%) | 35/217 (16.1%) |
| 2 | 16/35 (44.4%) | 26/55 (47.3%) | 33/138 (23.9%) | 80/157 (51.0%) | 86/217 (40.0%) | |
| 3 | 18/36 (50.0%) | 28/55 (50.9%) | 98/138 (71.0%) | 64/157 (40.8%) | 96/217 (44.2%) | |
| Tumour border | Pushing | 0/36 (0.0%) | 3/55 (5.4%) | 80/138 (58.0%) | 63/157 (40.1%) | 94/217 (43.3%) |
| Infiltrative | 36/36 (100.0%) | 52/55 (94.6%) | 58/138 (42.0%) | 93/157 (59.2%) | 122/217 (56.2%) | |
| Missing | 0/36 (0.0%) | 0/55 (0.0%) | 0/138 (0.0%) | 1/157 (0.6%) | 1/217 (0.5%) | |
| Lymphocytic infiltration | Absent | 14/36 (38.9%) | 10/55 (18.2%) | 13/138 (9.4%) | 41/157 (26.1%) | 58/217 (26.7%) |
| Mild | 16/36 (44.4%) | 35/55 (63.6%) | 59/138 (42.8%) | 67/157 (42.7%) | 96/217 (44.2%) | |
| Prominent | 6/36 (16.7%) | 10/55 (18.2%) | 66/138 (47.8%) | 48/157 (30.6%) | 63/217 (29.0%) | |
| Missing | 0/36 (0.0%) | 0/55 (0.0%) | 0/138 (0.0%) | 1/157 (0.6%) | 0/217 (0.0%) | |
| Vascular invasion | Absent | 24/36 (66.7%) | 36/55 (65.4%) | 118/138 (85.5%) | 115/157 (73.2%) | 168/217 (77.4%) |
| Present | 12/36 (33.3%) | 19/55 (34.6%) | 20/138 (14.5%) | 39/157 (24.8%) | 43/217 (19.8%) | |
| Missing | 0/36 (0.0%) | 0/55 (0.0%) | 0/138 (0.0%) | 3/157 (1.9%) | 6/217 (2.8%) | |
| Tumour stroma | Cellular | 1/36 (2.8%) | 6/55 (10.9%) | 26/138 (18.8%) | 23/157 (14.6%) | 33/217 (15.2%) |
| Sclerotic | 29/36 (80.6%) | 28/55 (50.9%) | 50/138 (36.2%) | 65/157 (41.4%) | 95/217 (43.8%) | |
| Desmoplastic | 6/36 (16.7%) | 20/55 (36.4%) | 39/138 (28.3%) | 34/157 (21.7%) | 63/217 (29.0%) | |
| Myxoid | 0/36 (0.0%) | 1/55 (1.8%) | 3/138 (2.2%) | 11/157 (7.0%) | 6/217 (2.8%) | |
| Other | 0/36 (0.0%) | 0/55 (0.0%) | 19/138 (13.8%) | 23/157 (14.6%) | 20/217 (9.2%) | |
| Missing | 0/36 (0.0%) | 0/55 (0.0%) | 1/138 (0.7%) | 1/157 (0.6%) | 0/217 (0.0%) | |
Missing data refers to an unreported feature.
A low level of lymphocytic infiltration was reported in both TP53 carriers and HER2 positive cases (absent/mild lymphocytic infiltration): TP53 30 of 36 (83.3%), HER2 positive 45 of 55 (81.8%). This particularly contrasts with the BRCA1 carriers where lymphocytic infiltration is a well‐recognised feature with a significantly lower proportion reported as having absent or mild lymphocytic infiltrate, 72 of 138 (52.2%). BRCA2 carriers were similar to YBC, non‐carriers with 108 of 157 (68.8%) and 154 of 217 (71.0%) respectively. The tumour border was more often infiltrative in the TP53 cohort, 36 of 36 (100.0%), and amongst HER2 positive cases, 52 of 55 (94.5%), compared to the BRCA1, BRCA2 and YBC cases – 58 of 138 (42.0%), 93 of 157 (59.2%), 122 of 217 (56.2%) respectively. Vascular invasion was more frequently reported as ‘present’ in cases which were TP53 12 of 36 (33.3%) and HER2 positive 19 of 55 (34.5%) compared to BRCA1, BRCA2 and YBC – 20 of 138 (14.5%), 39 of 157 (24.8%) and 43 of 217 (19.8%) respectively. The most striking morphological feature distinguishing the TP53 germline carriers was the presence of a densely sclerotic tumour stroma (Figure 1) reported in 29 of 36 (80.6%) of cases, compared with 28 of 55 (50.9%) of young patients with HER2 positive breast cancer with no germline TP53 pathogenic variant (p = 0.004) and lower still in other groups; 50 of 138 BRCA1 carriers (36.2%, p < 0.001), 65 of 157 BRCA2 carriers (41.4%, p < 0.001) and 95 of 217 in YBC (43.8%, p < 0.001) (Table 3 and Figure 2).
Figure 1.
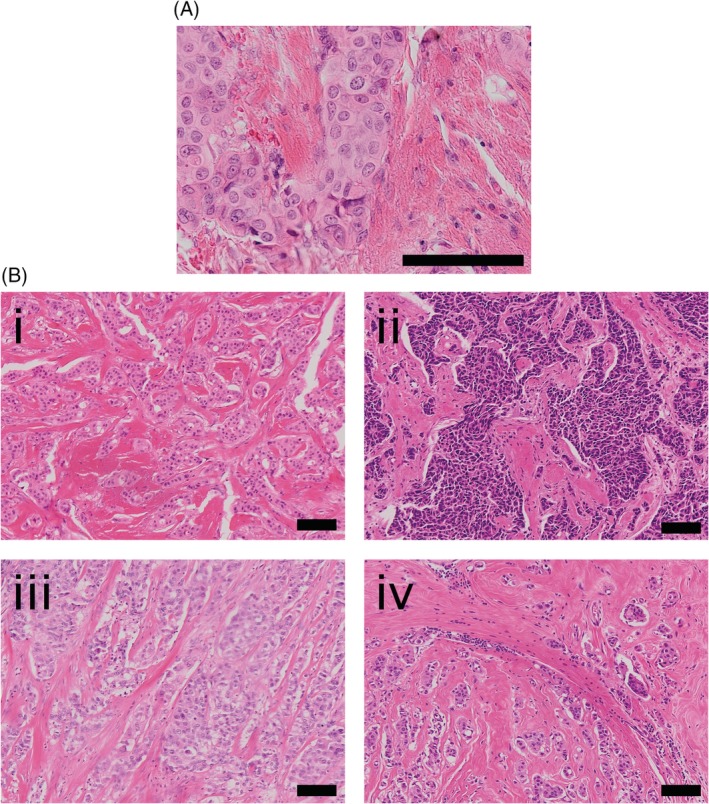
Sclerotic stroma in TP53 carriers. (A) A magnified area of invasive tumour surrounded by sclerotic stroma. (B) (i)–(iv) Four patient samples with invasive carcinoma surrounded by a sclerotic stroma. Images were taken on the Olympus Dotslide at an objective magnification of ×20 (A) or ×10 (B). Scale bars represent 100 μm.
Table 3.
The distribution of stromal types in YBC onset cohorts
| Cohort | Sclerotic – (% of cohort) | Sclerotic + (% of cohort) | Missing data |
|---|---|---|---|
| TP53 | 7/36 (19.4%) | 29/36 (80.6%) | 0/36 (0.0%) |
| HER2+ | 27/55 (49.1%) | 28/55 (50.9%) | 0/55 (0.0%) |
| BRCA1 | 87/138 (63.0%) | 50/138 (36.2%) | 1/138 (0.7%) |
| BRCA2 | 91/157 (58.0%) | 65/157 (41.4%) | 1/157 (0.6%) |
| YBC | 122/217 (56.2%) | 95/217 (43.8%) | 0/217 (0.0%) |
Figure 2.
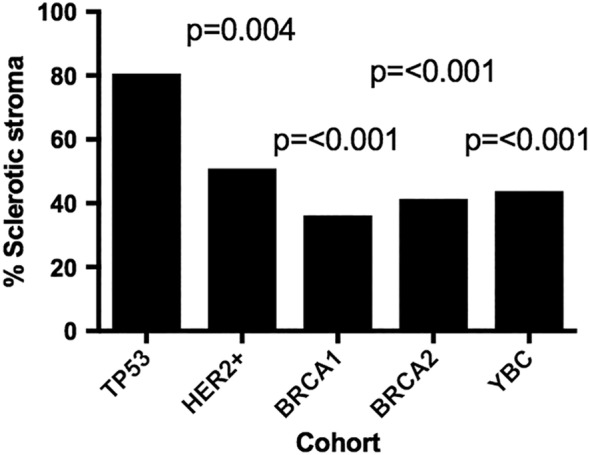
TP53 carriers had a significantly higher proportion of sclerotic tumour stroma. The bar chart shows the frequencies of sclerotic stroma between cohorts. Statistics were performed on TP53 carriers against each of the other groups using the Pearson Chi‐square test. Missing data were excluded.
Receptor status in TP53 carriers
Hormone receptor status was evaluated and compared to available data in young onset breast cancer cohorts between TP53 gene carriers (n = 36) and data available from the POSH cohort (n = 1260). TP53 gene carriers were significantly more likely to be ER+/PR+/HER2+ (p < 0.001) than in the POSH cohort (Table 4).
Table 4.
Receptor status compared with POSH cohort data
| Tumour receptor status | COPE cohort, TP53 gene carriers (%) n = 36 | POSH cohort (%) n = 1260 | P value* |
|---|---|---|---|
| HER2+/ER+/PR+ | 13/36 (36.1%) | 145/1260 (11.5%) | <0.001 |
| HER2+/ER+/PR− | 1/36 (2.8%) | 34/1260 (2.7%) | ns |
| HER2+/ER−/PR+ | 0/36 (0.0%) | 10/1260 (0.8%) | ns |
| HER2+/ER−/PR− | 6/36 (16.7%) | 97/1260 (7.7%) | 0.023 |
| HER2−/ER+/PR+ | 7/36 (19.4%) | 477/1260 (37.9%) | ns |
| HER2−/ER+/PR− | 2/36 (5.6%) | 77/1260 (6.1%) | ns |
| HER2−/ER−/PR+ | 0/36 (0.0%) | 27/1260 (2.1%) | ns |
| HER2−/ER−/PR− | 3/36 (8.3%) | 393/1260 (31.2%) | 0.008 |
| Missing data | 4/36 (11.1%) | 0/1260 (0.0%) |
Summary statistics for receptor status in invasive breast tumours within TP53 carriers and the YBC onset cohort POSH. Missing data refers to an unreported feature.
Fisher's exact test.
Stromal markers: αvβ6 and α‐SMA in TP53 carriers
A high number of TP53 carriers showed strong expression of p53 protein (≥5+: 69.4%, 25/36). Additionally, moderate to high expression of the stromal marker integrin αvβ6 was confirmed in 21 of 36 (58.3%) of the invasive tumours of TP53 carriers in this study. High expression of α‐SMA (88.9%, 32/36) and pSMAD2/3 (proportion 3/4: 30/36; 83.3%, intensity 2/3: 29/36; 80.6%) were confirmed in a high proportion of tumours (Figure 3).
Figure 3.
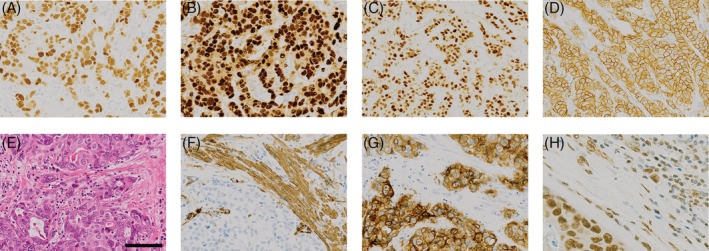
Examples of typical IHC for breast tumours arising in a mutant TP53 background. Tumours are typically ER (A), PR (B) and HER2 (D) positive, show strong nuclear p53 staining (C) and are positive for markers of activated TGFβ signalling (F, αSMA; G, integrin αvβ6; H, pSMAD2/3). A corresponding H&E stain is shown in (E).
We explored whether the type of germline TP53 pathogenic variant present amongst the COPE cases with invasive tumour altered the tumour phenotype. Comparing missense (n = 24), with truncating (n = 12) variants, excluding missing cases, a missense pathogenic variant did appear more likely to be associated with a sclerotic stroma (21/24, 87.5%) than a truncating pathogenic variant (8/12, 67.7%) although this was not statistically significant. HER2 overexpression was slightly less frequent with germline missense (12/24, 50.0%) versus truncating (8/12, 66.7%) variants but again the difference was not statistically significant.
Finally, we looked at the additional morphological data for POSH cases with all four IHC markers (Table 5). Amongst the 1260 young cancers from which TMA IHC scores were available, aberrant p53 staining was present in 302. Of these, 65 also had morphology evaluated in the previous study 22. Sclerotic stroma was reported in 26 of 65 (40.0%) of cases; 3 of 26 (11.5%) were also HER2‐positive but none were ER/PR/HER2 positive. Desmoplastic stroma was reported in 21 of 65 (32.3%) of cases; 5 of 21 (23.8%) of these were also HER2+. Cellular stroma was reported in 6 of 65 (9.2%) cases, all of which were triple negative.
Table 5.
Expression of p53 and stromal markers in TP53 gene carrier breast cancers
| Stain | Scoring | TP53 gene carriers | |
|---|---|---|---|
| p53 (0–7) | 0–1 | 0/36 (0.0%) | |
| 2–4 | 7/36 (19.4%) | ||
| 5+ | 25/36 (69.4%) | ||
| Missing data | 4/36 (11.1%) | ||
| Integrin αvβ6 | Absent/low | 11/36 (30.6%) | |
| Moderate/high | 21/36 (58.3%) | ||
| Missing data | 4/36 (11.1%) | ||
| α‐SMA | Absent/low | 1/36 (2.8%) | |
| Moderate/high | 32/36 (88.9%) | ||
| Missing data | 3/36 (8.3%) | ||
| pSMAD2/3 | Staining proportion | 1 | 1/36 (2.8%) |
| 2 | 0/36 (0.0%) | ||
| 3 | 7/36 (19.4%) | ||
| 4 | 23/36 (63.9%) | ||
| Staining intensity | 1 | 2/36 (5.6%) | |
| 2 | 20/36 (55.6%) | ||
| 3 | 9/36 (25.0%) | ||
| Missing data | 5/36 (13.9%) | ||
Amongst POSH cases 286 of 1260 (20.3%) were HER2 amplified and of these just over half (145) were ER/PR/HER2 positive (Table 4). We had information about stromal morphology in 121 of 1260 cases with complete IHC; 42 of 121 (34.7%) were HER2 positive, only 10 of these were also p53 positive. The stroma in these 10 cases was reported as desmoplastic (5), sclerotic (3), myxoid (2) and other (1). Only two cases were positive for ER/PR/HER2 and p53.
Discussion
Patients with a germline TP53 pathogenic variant typically develop high grade (2 or 3), HER2 positive, infiltrating ductal carcinoma, confirming our previously reported observation and subsequent reports from other groups 11, 12, 13. The frequency of triple positivity (HER2, ER and PR) also appears significantly higher in these patients than young onset cases in general. A number of other morphological features are similar between the cancers in germline TP53 pathogenic variants and the HER2 positive early onset cases from the POSH study. However, the high frequency of breast tumours with densely sclerotic tumour stroma is a novel observation in patients with a germline TP53 pathogenic variant. In comparison to sporadic HER2 amplified young onset breast cancers evaluated using the same methodology, the frequency of densely sclerotic stroma was striking. In comparison, the frequency of sclerotic stroma in sporadic HER2 positive cases was more similar to the proportion reported in other young onset cases; although we recognise that the data from the other young onset groups was taken from a previously reported study so may be less directly comparable. Amongst sporadic HER2 amplified tumours with abnormal p53 expression, there was no obvious excess of sclerotic stroma reported, although numbers were relatively small.
In sporadic breast cancers, somatic TP53 mutations are most frequent in triple negative and HER2 positive tumours, less frequent in ER positive or HER2 negative cancers. Loss of function in the tumour tissue occurs through inactivation or loss of both alleles. Patients with breast cancer arising on a background of an inherited TP53 pathogenic variant have already developed the first hit along the molecular pathway to carcinoma. It is possible that very early loss of TP53 function through somatic mutation in patients with an inherited TP53 pathogenic variant may be the reason for the more frequent development of the sclerotic stroma.
We explored this further by comparing the type of inherited variant. Pathogenic missense variants in TP53 often disrupt the function of the wild type protein (dominant negative effect); this leads to loss of p53 function, even before the evolving cancer cell develops loss or amplification of genomic material, and often leads to a more severe Li–Fraumeni phenotype 23, 24, 25, 26, 27, 28. We hypothesise that a germline pathogenic missense variant would be more likely to be associated with a dense stromal reaction if this early loss of function was the underlying driving factor. Although numbers of cases were quite small, we did observe a higher proportion of cases with dense sclerotic stroma amongst patients with missense pathogenic variants, lending some support to this hypothesis.
There is increasing evidence to suggest that the intricate tumour‐stromal interactions in the TME are essential to driving tumour progression 16, 22, 23, 24, 25, 26. This complex system involves various cell types, including fibroblasts, immune cells and endothelial cells. The novel finding from this study was that breast tumours derived from germline TP53 carriers typically presented with an associated sclerotic tumour stroma. Myofibroblasts are the cellular component of the microenvironment that deposit the rich collagen layer in a morphologically sclerotic stroma 27, 28, 29, 30. Myofibroblasts are characterised by α‐SMA expression 31. Tumours containing a high proportion of cancer‐associated fibroblasts (CAFs) positive for α‐SMA, have been associated with a poorer prognosis as a result of increased migration, invasion, proliferation, angiogenesis and inhibition of infiltrating lymphocytes 16, 22, 23, 24, 25. In TP53 carriers, high expression of α‐SMA in the surrounding stroma was confirmed in 88.9% of cases, suggesting that CAFs are playing a key role.
A key pathway through which CAFs undergo transformation and activate tumour‐promoting processes is via transforming growth factor beta (TGFβ) signalling. One of the key mechanisms by which TGFβ is activated is through the expression of integrin αvβ6 on the cell surface of tumour cells 27. Integrin αvβ6 expression was confirmed in 58.3% of invasive TP53 carrier tumours in this study, compared to only 15–16% of cases noted in a 2000 patient cohort reported elsewhere 32. PhosphoSMAD2/3 proteins are activated through a phosphorylation cascade, which forms the basis of initiated TGFβ signalling. Activated pSMAD2/3 proteins migrate from the cytoplasm to the nucleus and initiate transcription and the downstream deposition of collagen. Confirmation of TGFβ signalling was evidenced by high levels of pSMAD2/3 expression in breast cancers from TP53 carriers.
Patients with inherited TP53 pathogenic variants develop a broad range of tumours including sarcomas and childhood onset adrenocortical carcinomas; an estimated 50% of women develop breast cancer, usually at young ages. Treatment with chemotherapy and radiotherapy is thought to increase the risk of DNA damage‐induced late toxic effects, further increasing the risk of developing second malignancies. The invasive tumour characteristics in this group are typically associated with poor outcomes, including a low level of TILs in 83% of TP53 carriers, with an infiltrative tumour border in 100% and vascular invasion in 33.3%. A potential mechanism by which TILs could be being blocked from migrating towards the tumour is the deposition of a collagen barrier by the CAFs 33, 34, 35, 36, 37, 38. Furthermore, CAFs positive for α‐SMA have previously been reported to increase migration, invasion, proliferation and angiogenesis 26, 39, 40.
Work by other groups has previously implicated p53 in the upregulation of collagen 41. Loss of p53 function was shown to upregulate TGFβ signalling and as a consequence lead to the transcriptional activation of COL1A2 and collagen synthesis 41. Murine models heterozygous for loss of TP53 function (p53+/−) developed an extensive proliferative stromal reaction that was positive for α‐SMA and S100A4, a fibroblast marker 42. Finally, multiple groups have previously suggested that, through indirect cellular contact, tumour cells can inhibit wild‐type p53 activation and stimulate immunosuppression 43, 44.
In summary, we have observed that breast tumours arising in germline TP53 pathogenic variant carriers are highly likely to be high grade, HER2 positive, ER/PR positive tumours with an associated dense sclerotic tumour stroma. The early inactivation of wild‐type p53 may be one of the important mechanisms leading to the activation of TGFβ via integrin αvβ6 and the development of breast cancers with many adverse prognostic characteristics. Given the increased risk of late toxicity from cytotoxic treatments (chemotherapy and radiotherapy) in germline TP53 gene carriers, treatment with targeted agents including anti‐HER2 therapies such as trastuzumab (Herceptin) or antibodies targeting the TGFβ signalling pathway may be safer treatment options for breast cancer patients found to carry a germline TP53 pathogenic variant.
Author contributions statement
DME conceived and designed the study. KP, GM, MS, ES, KW, GT, AB and DME planned and executed the study. ES, LJ, LH, DGE, JB, AH and NP were responsible for sample acquisition. MS and GM conducted morphology reporting. GT, AB, GM and KM scored IHC data. KP, LH and LJ were responsible for TMA curation. All authors reviewed the manuscript, contributed to revisions and approved the final manuscript for submission.
Acknowledgements
We would like to acknowledge funding for this project from The Pathological Society of Great Britain and Ireland. Data from the POSH study were generated with funding from Cancer Research UK and Breast Cancer Now (BCN). DGE is supported through the National Institute for Health Research Manchester Biomedical Research Centre (IS‐BRC‐1215‐20007). We thank Maria‐Antoinette Lopez for technical support with immunohistochemistry and Dave Johnson for technical support with images. We thank Tom Maishman for advice regarding statistical analyses. The following investigators helped identify and collect cases for COPE: Dr Ramūnas Janavičius, Dr Carole Brewer, Dr Helen Hanson, Dr Jackie Cook, Dr Julian Adlard, Dr Kai Ren Ong, Dr Marc Tischkowitz, Dr Zosia Miedzybrodzka, Ms Cheryl Berlin, Professor Ros Eeles, Dr Gabriella Pichert, Ms Caroline Langman, Dr Alex Murray, Professor Eamonn Sheridan, Dr Rosemarie Davidson, Dr Mariella D'Alessandro and Dr Lynn Greenhalgh.
Conflict of interest statement: DME has provided consultancy to Astra Zeneca in the recent past around BRCA and drug response but there are no other relevant conflicts of interest amongst the authors.
References
- 1. Silwal‐Pandit L, Vollan HK, Chin SF, et al. TP53 mutation spectrum in breast cancer is subtype specific and has distinct prognostic relevance. Clin Cancer Res 2014; 20: 3569–3580. [DOI] [PubMed] [Google Scholar]
- 2. Toledo F, Wahl GM. Regulating the p53 pathway: in vitro hypotheses, in vivo veritas. Nat Rev Cancer 2006; 6: 909–923. [DOI] [PubMed] [Google Scholar]
- 3. Li FP, Fraumeni JF Jr. Rhabdomyosarcoma in children: epidemiologic study and identification of a familial cancer syndrome. J Natl Cancer Inst 1969; 43: 1365–1373. [PubMed] [Google Scholar]
- 4. Malkin D, Li FP, Strong LC, et al. Germ line p53 mutations in a familial syndrome of breast cancer, sarcomas, and other neoplasms. Science 1990; 250: 1233–1238. [DOI] [PubMed] [Google Scholar]
- 5. Lane DP. Cancer. p53, guardian of the genome. Nature 1992; 358: 15–16. [DOI] [PubMed] [Google Scholar]
- 6. Atchley DP, Albarracin CT, Lopez A, et al. Clinical and pathologic characteristics of patients with BRCA‐positive and BRCA‐negative breast cancer. J Clin Oncol 2008; 26: 4282–4288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Rakha EA, Reis‐Filho JS, Ellis IO. Basal‐like breast cancer: a critical review. J Clin Oncol 2008; 26: 2568–2581. [DOI] [PubMed] [Google Scholar]
- 8. Lee E, McKean‐Cowdin R, Ma H, et al. Characteristics of triple‐negative breast cancer in patients with a BRCA1 mutation: results from a population‐based study of young women. J Clin Oncol 2011; 29: 4373–4380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Pathology of familial breast cancer: differences between breast cancers in carriers of BRCA1 or BRCA2 mutations and sporadic cases. Breast Cancer Linkage Consortium. Lancet 1997; 349: 1505–1510. [PubMed] [Google Scholar]
- 10. Lakhani SR, Van De Vijver MJ, Jacquemier J, et al. The pathology of familial breast cancer: predictive value of immunohistochemical markers estrogen receptor, progesterone receptor, HER‐2, and p53 in patients with mutations in BRCA1 and BRCA2 . J Clin Oncol 2002; 20: 2310–2318. [DOI] [PubMed] [Google Scholar]
- 11. Wilson JR, Bateman AC, Hanson H, et al. A novel HER2‐positive breast cancer phenotype arising from germline TP53 mutations. J Med Genet 2010; 47: 771–774. [DOI] [PubMed] [Google Scholar]
- 12. Masciari S, Dillon DA, Rath M, et al. Breast cancer phenotype in women with TP53 germline mutations: a Li–Fraumeni syndrome consortium effort. Breast Cancer Res Treat 2012; 133: 1125–1130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Melhem‐Bertrandt A, Bojadzieva J, Ready KJ, et al. Early onset HER2‐positive breast cancer is associated with germline TP53 mutations. Cancer 2012; 118: 908–913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Eccles D, Gerty S, Simmonds P, et al. Prospective study of outcomes in sporadic versus hereditary breast cancer (POSH): study protocol. BMC Cancer 2007; 7: 160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Copson ER, Maishman TC, Tapper WJ, et al. Germline BRCA mutation and outcome in young‐onset breast cancer (POSH): a prospective cohort study. Lancet Oncol 2018; 19: 169–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Marsh D, Suchak K, Moutasim KA, et al. Stromal features are predictive of disease mortality in oral cancer patients. J Pathol 2011; 223: 470–481. [DOI] [PubMed] [Google Scholar]
- 17. Rakha EA, Pinder SE, Bartlett JM, et al. Updated UK recommendations for HER2 assessment in breast cancer. J Clin Pathol 2015; 68: 93–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Harvey JM, Clark GM, Osborne CK, et al. Estrogen receptor status by immunohistochemistry is superior to the ligand‐binding assay for predicting response to adjuvant endocrine therapy in breast cancer. J Clin Oncol 1999; 17: 1474–1481. [DOI] [PubMed] [Google Scholar]
- 19. Lawson J, Robinson‐Vyas RJ, McQuillan JP, et al. Crowdsourcing for translational research: analysis of biomarker expression using cancer microarrays. Br J Cancer 2017; 116: 237–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. McCarty KS Jr, Szabo E, Flowers JL, et al. Use of a monoclonal anti‐estrogen receptor antibody in the immunohistochemical evaluation of human tumors. Cancer Res 1986; 46: 4244s–4248s. [PubMed] [Google Scholar]
- 21. Shaw EC, Hanby AM, Wheeler K, et al. Observer agreement comparing the use of virtual slides with glass slides in the pathology review component of the POSH breast cancer cohort study. J Clin Pathol 2012; 65: 403–408. [DOI] [PubMed] [Google Scholar]
- 22. Underwood TJ, Hayden AL, Derouet M, et al. Cancer‐associated fibroblasts predict poor outcome and promote periostin‐dependent invasion in oesophageal adenocarcinoma. J Pathol 2015; 235: 466–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. De Monte L, Reni M, Tassi E, et al. Intratumor T helper type 2 cell infiltrate correlates with cancer‐associated fibroblast thymic stromal lymphopoietin production and reduced survival in pancreatic cancer. J Exp Med 2011; 208: 469–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Surowiak P, Murawa D, Materna V, et al. Occurrence of stromal myofibroblasts in the invasive ductal breast cancer tissue is an unfavourable prognostic factor. Anticancer Res 2007; 27: 2917–2924. [PubMed] [Google Scholar]
- 25. Tsujino T, Seshimo I, Yamamoto H, et al. Stromal myofibroblasts predict disease recurrence for colorectal cancer. Clin Cancer Res 2007; 13: 2082–2090. [DOI] [PubMed] [Google Scholar]
- 26. Vong S, Kalluri R. The role of stromal myofibroblast and extracellular matrix in tumor angiogenesis. Genes Cancer 2011; 2: 1139–1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Munger JS, Huang X, Kawakatsu H, et al. The integrin alpha v beta 6 binds and activates latent TGF beta 1: a mechanism for regulating pulmonary inflammation and fibrosis. Cell 1999; 96: 319–328. [DOI] [PubMed] [Google Scholar]
- 28. Lygoe KA, Norman JT, Marshall JF, et al. AlphaV integrins play an important role in myofibroblast differentiation. Wound Repair Regen 2004; 12: 461–470. [DOI] [PubMed] [Google Scholar]
- 29. Serini G, Gabbiani G. Mechanisms of myofibroblast activity and phenotypic modulation. Exp Cell Res 1999; 250: 273–283. [DOI] [PubMed] [Google Scholar]
- 30. Ohtani H, Sasano N. Stromal cell changes in human colorectal adenomas and carcinomas. An ultrastructural study of fibroblasts, myofibroblasts, and smooth muscle cells. Virchows Arch 1983; 401: 209–222. [DOI] [PubMed] [Google Scholar]
- 31. Desmouliere A, Geinoz A, Gabbiani F, et al. Transforming growth factor‐beta 1 induces alpha‐smooth muscle actin expression in granulation tissue myofibroblasts and in quiescent and growing cultured fibroblasts. J Cell Biol 1993; 122: 103–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Moore KM, Thomas GJ, Duffy SW, et al. Therapeutic targeting of integrin αvβ6 in breast cancer. J Natl Cancer Inst 2014; 106: dju169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Huo CW, Chew G, Hill P, et al. High mammographic density is associated with an increase in stromal collagen and immune cells within the mammary epithelium. Breast Cancer Res 2015; 17: 79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Garcia‐Mendoza MG, Inman DR, Ponik SM, et al. Neutrophils drive accelerated tumor progression in the collagen‐dense mammary tumor microenvironment. Breast Cancer Res 2016; 18: 49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Liotta LA. Tumor invasion and metastases‐‐role of the extracellular matrix: Rhoads memorial award lecture. Cancer Res 1986; 46: 1–7. [PubMed] [Google Scholar]
- 36. Salmon H, Franciszkiewicz K, Damotte D, et al. Matrix architecture defines the preferential localization and migration of T cells into the stroma of human lung tumors. J Clin Invest 2012; 122: 899–910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Klingberg F, Hinz B, White ES. The myofibroblast matrix: implications for tissue repair and fibrosis. J Pathol 2013; 229: 298–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Torres S, Bartolome RA, Mendes M, et al. Proteome profiling of cancer‐associated fibroblasts identifies novel proinflammatory signatures and prognostic markers for colorectal cancer. Clin Cancer Res 2013; 19: 6006–6019. [DOI] [PubMed] [Google Scholar]
- 39. Massarelli G, Tanda F, Bosincu L, et al. Myofibroblasts in the epithelial‐stromal junction of basal cell carcinoma. Appl Pathol 1983; 1: 25–30. [PubMed] [Google Scholar]
- 40. Lewis MP, Lygoe KA, Nystrom ML, et al. Tumour‐derived TGF‐beta1 modulates myofibroblast differentiation and promotes HGF/SF‐dependent invasion of squamous carcinoma cells. Br J Cancer 2004; 90: 822–832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Ghosh AK, Bhattacharyya S, Varga J. The tumor suppressor p53 abrogates Smad‐dependent collagen gene induction in mesenchymal cells. J Biol Chem 2004; 279: 47455–47463. [DOI] [PubMed] [Google Scholar]
- 42. Hill R, Song Y, Cardiff RD, et al. Selective evolution of stromal mesenchyme with p53 loss in response to epithelial tumorigenesis. Cell 2005; 123: 1001–1011. [DOI] [PubMed] [Google Scholar]
- 43. Bar J, Feniger‐Barish R, Lukashchuk N, et al. Cancer cells suppress p53 in adjacent fibroblasts. Oncogene 2009; 28: 933–936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Guo G, Marrero L, Rodriguez P, et al. Trp53 inactivation in the tumor microenvironment promotes tumor progression by expanding the immunosuppressive lymphoid‐like stromal network. Cancer Res 2013; 73: 1668–1675. [DOI] [PMC free article] [PubMed] [Google Scholar]