

Abstract

The worldwide use of the broad-spectrum antimicrobial trimethoprim (TMP) has induced the rise of TMP-resistant microorganisms. In addition to resistance-causing mutations of the microbial chromosomal dihydrofolate reductase (Dfr), the evolutionarily and structurally unrelated type II Dfrs (DfrBs) have been identified in TMP-resistant microorganisms. DfrBs are intrinsically TMP-resistant and allow bacterial proliferation when the microbial chromosomal Dfr is TMP-inhibited, making these enzymes important targets for inhibitor development. Furthermore, DfrBs occur in multiresistance plasmids, potentially accelerating their dissemination. We previously reported symmetrical bisbenzimidazoles that are the first selective inhibitors of the only well-characterized DfrB, DfrB1. Here, their diversification provides a new series of inhibitors (Ki = 1.7–12.0 μM). Our results reveal two prominent features: terminal carboxylates and inhibitor length allow the establishment of essential interactions with DfrB1. Two crystal structures demonstrate the simultaneous binding of two inhibitor molecules in the symmetrical active site. Observations of those dimeric inhibitors inspired the design of monomeric analogues, binding in a single copy yet offering similar inhibition potency (Ki = 1.1 and 7.4 μM). Inhibition of a second member of the DfrB family, DfrB4, suggests the generality of these inhibitors. These results provide key insights into inhibition of the highly TMP-resistant DfrBs, opening avenues to downstream development of antibiotics for combatting this emergent source of resistance.
Introduction
Trimethoprim (TMP) is a broad-spectrum diaminopyrimidine antimicrobial agent (Scheme 1).1−3 The target of TMP is the enzyme dihydrofolate reductase (Dfr). Dfrs are ubiquitous and have long been primary targets for the treatment of proliferative disorders such as cancer and infectious diseases because they are essential for the synthesis of DNA precursors.4 Dfrs catalyze the reduction of dihydrofolate (DHF) to tetrahydrofolate with the reducing cofactor NADPH.5 TMP specifically inhibits the bacterial chromosomal Dfr (known as DfrA); moderate TMP resistance can result from its mutations.6
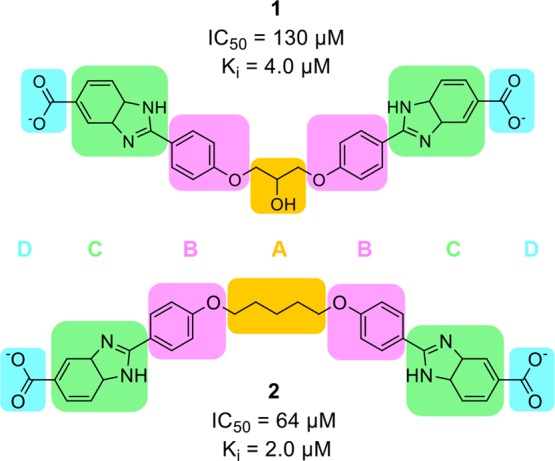
Scheme 1. Previously Reported Symmetrical Bisbenzimidazole-Type Inhibitors of DfrB1, 1 and 2,20 Have the General Structure (A) Central Core; (B) Phenoxy Moieties; (C) Benzimidazole Moieties; and (D) Terminal Carboxylates.
Bacteria may additionally express an additional Dfr: an evolutionarily unrelated type II Dfr (DfrB). DfrB1, also known as R67 DHFR, is the best studied among the DfrB family. It is inert to millimolar concentrations of TMP and therefore allows bacterial proliferation in the presence of TMP that inhibits the chromosomal Dfr.7−10 DfrBs share no sequence or structural relationship with the chromosomal Dfrs: they have evolved convergently to catalyze the same reaction (reduction of dihydrofolate with NADPH) yet do not bind TMP. As a result, Gram-negative bacteria harboring a plasmid-borne DfrB are highly resistant to TMP (Figure 1).5,9,11−15 Thus, DfrBs are not targets of TMP and therefore require independent inhibitor discovery.
Figure 1.

Treatment of bacterial infections with the antibiotic TMP selectively inhibits the target, chromosomal Dfr, leading to bacterial cell death. The presence of the evolutionarily unrelated type II Dfr (DfrB) in bacteria allows bacterial proliferation due to its intrinsic resistance to TMP.
As a result of its simple structure and efficacy, the low-cost TMP has been in constant and heavy use as an antibiotic worldwide for decades.1,2,5 The combination of TMP and sulfonamide (SUL) is broadly used against aerobic bacteria and occasionally against protozoa.1 The World Health Organization has classified TMP–SUL as highly important because of its effectiveness in the treatment of human urinary and respiratory tract infections.1,2,5 The TMP–SUL combination is also a part of the primary front-line treatment for infections of known or unknown origin in veterinary applications1 and is used in routine preventive measures, further increasing its distribution. For example, TMP is heavily used in aquaculture of shrimp16 and fish,14,16 where it is disseminated into surrounding waters. It is also widely used in livestock which is of particular concern because resistance to TMP in livestock,17 especially cattle1 and swine,13,15 is known to be transferable to humans.1,18 Consequently, the global incidence of TMP resistance in humans is increasing;19 we recently identified a dfrB gene integrated into a multiresistance cassette in a North American TMP-resistant clinical sample,10 highlighting the need for discovery of DfrB inhibitors.
To date, few efforts have been made toward the discovery of inhibitors for DfrBs. In the DfrB family, only DfrB1 has been structurally and kinetically characterized.20−24 In contrast to all chromosomal Dfrs (and their plasmid-borne, close variants), including the human Dfr (hDfr), that share a conserved monomeric fold with clearly defined substrate and cofactor binding regions, DfrB1 is a doughnut-shaped homotetrameric enzyme with a single, central active-site tunnel (Figure 2). Each protomer consists of an SH3-like domain that contributes equally to the formation of the active site.7,23,25 The hourglass-shaped active-site tunnel has a central neck that opens into the opposing tunnel mouths (Figure 2). Its calculated volume is greater than twice that of the Escherichia coli DfrA active site, despite catalyzing the same reaction.8 As a result of its symmetry, it can bind two DHF substrate molecules, two NADPH cofactor molecules, or one of each with positive cooperativity.26−28
Figure 2.
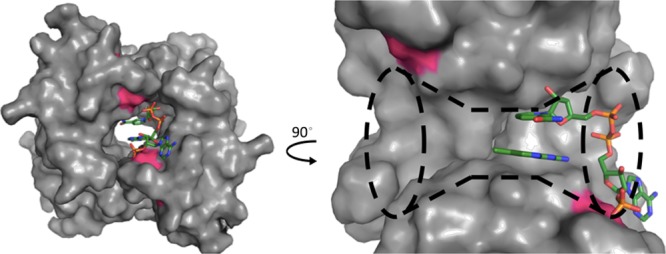
Left: surface representation of the tetrameric DfrB1. The substrates NADPH and DHF, in sticks representation, are bound inside the active-site tunnel (PDB code 2RK1).29 Right: close-up of the tunnel; only two DfrB1 protomers are shown. The pABA-Glu tail of DHF was not resolved as a result of its dynamic behavior;30 simulations indicate that it salt bridges with a Lys32 (pink surface, left opening) at the flared active-site mouth, opposite the Lys32 (pink surface, right opening) where the NADPH 2′-phosphate forms a similar interaction.30
This unusual binding promiscuity has resulted in DfrBs being considered as poorly evolved enzymes and led to the identification of nonselective inhibitors of DfrB1, some among which bind in two copies.4,8,27 2,4-Diaminopyrimidines, including methotrexate (MTX) and aminopterin (AMT), known for decades to be antimicrobial or antineoplastic inhibitors that target chromosomal Dfrs,3,31,32 bind weakly to apo-DfrB1, although no binding was observed with holo-DfrB1.3,9−15,27 Pemetrexed (PMTX), 2-desamino-5,8-dideazafolic acid, 5,8-dideazapterin analogues, and 5-deazafolic acid analogues nonspecifically not only inhibit DfrB1 in the low micromolar range but also inhibit the human Dfr (hDfr) and other mammalian enzymes from the folate pathway with higher affinity, precluding their use as antibiotic agents.4,27,33,34 In addition, the mainly hydrophobic active-site cavity of DfrB1 procures micromolar binding of various symmetrical or asymmetrical polycyclic molecules including novobiocin.4,8,35
We previously reported the first selective inhibitors of DfrB1, obtained by a fragment-based design.20 Among the weakly inhibiting, low molecular weight fragments that were identified, 1H-benzimidazole-5-carboxylic acid was selected to build symmetrical compounds that reflect the symmetry of the DfrB1 active site.20 The resulting symmetrical bisbenzimidazoles offered competitive inhibition that was 100- to 1000-fold more efficient than the initial fragment, with Ki in the low micromolar range (Scheme 1).20 Preliminary crystallographic results and kinetic analyses demonstrated that two molecules of 1 bind simultaneously inside the DfrB1 tunnel.20 Despite having achieved selective inhibition, little insight was obtained into the nature of the 2:1 inhibitor/target interactions.
With the goal of achieving a 1:1 inhibitor/target ratio that is more amenable to downstream antibiotic development, we investigated the contribution to binding of the substructures of 1 and 2: the central core, phenoxy moieties, benzimidazole moieties, and terminal carboxylates (Scheme 1). In addition, the length of the molecule and geometric constraints were explored to determine binding requirements. Two crystal structures resolved with 1 or one among the most effective; new inhibitors confirmed the previously proposed 2:1 inhibitor/target binding stoichiometry and, along with LowModeMD simulations and steady-state kinetic analyses, provided insights into key binding interactions. These findings inspired the design of analogues that were confirmed to bind with 1:1 stoichiometry. We further revealed a broader biological impact by demonstrating that a second member of the DfrB family, identified in a North American TMP-resistant clinical sample,10 is also effectively inhibited by these novel bisbenzimidazole inhibitors.
Results and Discussion
Crystal Structures Reveal Two Bound Molecules of the Inhibitor
DfrB1 is an unusual enzyme in that its four protomers assemble to form a single, 24 Å-long active-site tunnel that bisects the tetrameric structure (Figure 2).36 The homotetramer is thus defined by a 222 symmetry where the center of the active site is situated at the intersection of three axes of symmetry; the asymmetric unit contains one-quarter of the active-site pore. In prior work, we identified 1 (Scheme 1) as a competitive inhibitor of DfrB1, with a Ki = 4.0 μM.20 Its Hill coefficient of n = 2.1 and competitive mode of binding were consistent with the simultaneous binding of more than one molecule of 1 within the active-site cavity. Consistent with this, we reported preliminary crystallographic data suggesting that two molecules of 1 lie lengthwise in the tunnel.20
The particular symmetry of DfrB1 poses a challenge in the course of analyzing electron density of ligands crystallized in the active site. It has been previously shown29 that ligand binding typically breaks the crystallographic symmetry of the DfrB1 tunnel, either upon binding the substrates NADPH and DHF (PDB ID: 2RK1(29)) or by binding a ligand that is asymmetric or whose symmetry does not coincide with the crystallographic symmetry axis (PDB ID: 2RK2).29 The electron density observed in the asymmetric unit is the average density over the four symmetric quarters of the active site. This renders interpretation of the electron density difficult, particularly in cases with low ligand occupancy.
To gain further insights into the mode of inhibitor binding, we synthesized 3 that bears an asymmetric central core (Figure 3; Scheme S1; detailed synthetic methods are provided in the Supporting Information). Inhibition (Ki = 3.5 μM) is essentially unchanged from that of 1 (Ki = 4.0 μM) and both are selective, as no inhibition of human Dfr (hDfr) was observed (Table 1). We obtained a crystal structure of DfrB1 in complex with 3, which we report here along with the completed analysis of the crystal structure of 1 (Table S1). In both structures, two molecules of the soaked inhibitor are juxtaposed within the tetrameric active site, in an extended conformation (Figure 3). Only the 1,3-dioxopropan-2-ol or 1,4-dioxobutan-2-ol central cores could be resolved; they lie in the central neck of the tunnel. The quality of the electron density decreased toward the benzimidazole extremities, in the direction of the active-site mouths. This is analogous to the DfrB1:DHF crystal structure, where only the pteridin moiety was resolved in the center of the active site29 as a result of the dynamic behavior of its pABA-Glu tail21,30 and is suggestive of a similar binding mode.
Figure 3.

Crystal structures of DfrB1 (A) with inhibitor 1 PDB ID 6NY0 and (B) with inhibitor 3 PDB ID 6NXZ. Because of the ambiguity of the electron density in the asymmetric unit, we were unable to definitely assign one enantiomer of the racemic central core of 3. We modeled the R-enantiomer in the active site, for further analysis. As the binding modes of 1 and 3 are slightly offset from the center of symmetry, they are observed in two slightly different binding modes when the asymmetric unit is expanded to show the tetramer. The inhibitors were modeled at 0.5 occupancy per asymmetric unit. This approximated two fully occupied binding sites in the tetrameric active-site tunnel.
Table 1. Inhibition Upon Central Core Substitution and Alteration of the Central Core Lengtha.
| DfrB1 | hDfr |
|||
|---|---|---|---|---|
| compound | length (Å)b | IC50 (μM) | Ki (μM)c | IC50 (μM)d |
| 3 | 28.9 | 110 ± 10 | 3.5 ± 1.7 | >500 |
| 4 | 28.9 | 160 ± 50 | 4.9 ± 1.6 | ND |
| 5 | 31.2 | 60 ± 10 | 1.9 ± 0.3 | ND |
| 6 | 33.7 | 180 ± 95 | 5.5 ± 3.0 | ND |
| 7 | 36.1 | 70 ± 30 | 2.1 ± 0.9 | ND |
| 8 | 38.5 | >100d | ND | |
| 9 | 41.0 | >200d | ND | |
| 10 | 28.7 | 90 ± 20 | 2.8 ± 0.8 | >400 |
| 11 | 27.6 | [200 ± 130]e | [6.1 ± 3.9]e | >100 |
Values are given as the average ± standard deviation from the mean of at least triplicates of two independent experiments.
Calculated with Chemdraw 3D.
Ki was calculated from IC50 values.
Inhibitors were tested at the highest possible concentration, considering constraints due to high absorbance or to precipitation.
Extrapolated value, as the compound was insoluble above 100 μM in 10% DMSO ND, not determined.
In our initial analysis of the crystal structure of 1 in complex of DfrB1,20 the data were not sufficient for us to confidently conclude that the density could be definitively modeled as 1. We observe very similar electron density in the complex with the chemically similar 3; this density was visible only when inhibitor soaking was performed. Given these results, combined with prior reports of ligand-bound DfrB1 structures,29 we are now confident in modeling 1 and 3 in these crystal structures.
In Silico Reconstruction of the Network of Interactions that Describe Inhibitor Binding
To model the missing portion of the bound inhibitors, we built the cocrystallized inhibitor molecules outward from the central core coordinates and performed LowModeMD (Molecular Operating Environment). The crystal structure with 3 (PDB ID 6NXZ) was selected as a template for building all other inhibitors, reserving the crystal structure with 1 to validate the models. Throughout the hundreds of simulated poses obtained for the full-length modeled inhibitors, the two molecules of 3 consistently adopted a helical conformation along the length of the active-site tunnel (Figure 4). During most of the simulation, the terminal carboxylates formed electrostatic bonds with the ε-amino of either of the two Lys32 at each tunnel mouth and established H-bonds with the backbone of the nearby Gly35 and Ala36 (Figures 4; S1; Tables S2 and S3). We refer to this predominant bonding pattern as the “Lys32 network”. On some occasions, the terminal carboxylates of 3 also H-bonded with the hydroxyl of Ser34. Alternatively, the terminal carboxylates interacted with the hydroxyl of two out of the three following residues: Tyr46, Thr48, and Thr51, which we will refer to as the “YTT cluster” (Figure 4). This confirms the dynamic binding of the inhibitor extremities, which is consistent with the lack of clear electron density in the complexed crystal structures. The benzimidazole moieties of 3 established multiple interactions with Val66, Gln67, Ile68, and Tyr69: the amine H-bonded with the backbone carbonyl of Val66, and the phenyl and imidazole rings formed intermittent π-hydrogen bonds or edge-to-face aromatic interactions with the Val66 backbone amine and Tyr69, respectively (Figure 4; Tables S2 and S3). The benzimidazole moiety, phenoxy moiety, and central core made multiple H-bonds with Gln67 and Ile68. Occasionally, the benzenes from the phenoxy moieties of 3 formed π-hydrogen bonds with the backbone amine of Ile68. Finally, the central core hydroxyl established H-bonding interactions with the side-chain amide of Gln67, as observed in the cocrystal structure.
Figure 4.

In silico reconstruction of 3 in the active site of DfrB1 based on the crystal structure PDB ID 6NXZ. Interactions of DfrB1 with 3 are highlighted.
Geometry of Inhibitor Binding is Orthogonal to that of the Bound Substrates
DfrB is a dimer of dimers: monomers assemble into dimers by one set of contacts, and dimers assemble into a 222 symmetric homotetramer by a distinct set of contacts, resulting in two distinct types of dimer interfaces. The symmetry dictates that the ligand-binding surface provided in quadrants I/II (see Figure 5) is equivalent to that created in quadrants III/IV and that these are distinct from the ligand-binding surface provided in quadrants I/IV and II/III (themselves equivalent). During the catalytic cycle, positive cooperativity favors binding of DHF onto the NADPH-bound DfrB1 (holo-DfrB1) to form the productive (DfrB1·NADPH·DHF) complex. NADPH binding (quadrants I and II) is thus followed by the binding of DHF (quadrants III and IV) according to an ordered mechanism of the ternary complex formation (Figure 5).37 Naturally, the active-site symmetry allows binding in the opposite quadrants (NADPH in quadrants III/IV and DHF in I/II). The active site can also accommodate the binding of two DHF or two NADPH molecules,27,28,30,37 illustrating its promiscuity; binding occurs in the same mode (quadrants I/II and III/IV).
Figure 5.

Substrate-binding surfaces of DfrB1 in quadrants I/II and III/IV, identical to each other, are occupied by the substrates NADPH and DHF (PDB ID 2RK1;28 left panel). The distinct inhibitor-binding surfaces in quadrants I/IV and II/III, identical to each other, are occupied by the central core of a pair of 3 molecules (PDB ID 6NXZ; right panel). As each of the inhibitor-binding surfaces partially occludes both of the substrate-binding surfaces, neither substrate-binding site is accessible with the inhibitor bound surface. This provides strong evidence that binding of DHF is incompatible with binding of one molecule of 3 (overlay, center panel).
The crystal structure of DfrB1·3 demonstrates that 3 binds with a geometry orthogonal to that of the substrates: quadrants I/IV and II/III are occupied by either of the bound inhibitor molecules (Figure 5). As a result of this orthogonal binding mode, binding of an inhibitor molecule is incompatible with binding of NADPH or DHF because of steric hindrance (Figure 5). Despite the orthogonal binding geometry of the substrates and inhibitor and despite their structural differences, it is remarkable that the substrates and 3 share key binding residues: Lys32, Ala36, Tyr46, Thr51, Val66, Gln67, Ile68, and Tyr69. Among these, Lys32, Gln67, lle68, and Tyr69 are functional residues, justifying the inhibitory effect of 3.
In Silico Reconstruction of the Network of Interactions with Inhibitors 1 and 2
We used the model of DfrB1·3 as a template to predict binding interactions with 1 and 2. One molecule of 3 was replaced by a first molecule of the query inhibitor and a first LowModeMD simulation was performed. Substitution of the second molecule of 3 by the second molecule of 1 or 2 was then followed by a second LowModeMD simulation. A root-mean-square deviation (rmsd) of 2.4 ± 0.6 Å between the eight atoms constituting the 1,3-dioxopropan-2-ol core of 1 in the crystal structure (PDB ID 6NY0) and the ensemble of nearly 3000 simulated conformers obtained validated the model.
The interaction pattern observed for 1 and 2 throughout the hundreds of simulated poses was similar to that observed for 3 (summarized in Tables S2 and S3; Figure S1). Again, the terminal carboxylates formed frequent electrostatic and H-bonds with the Lys32 network and less frequently H-bonded with the YTT cluster, particularly for 2. The benzimidazoles again established bonds with Val66, Ile68, and Tyr69 but lost the H-bond with the Gln67 side chain. The phenoxy oxygens of 1 bind the Gln67 γ-amide as well as the Ile68 backbone-NH while those of 2 are bound instead to the Val66 backbone-NH. In addition to π-hydrogen bonding with the Ile68 backbone-NH, the phenoxy benzenes of 1 formed a novel edge-to-face interaction with Tyr69. The central core of 3 is bound similarly to 1, but the pentyl central core of 2 lacks that H-bonding capacity. These models indicate that the four segments generally contribute to inhibitor binding, yet some variation is tolerated without compromising affinity.
Does Inhibitor Length Matter?
The bound crystal structures and simulations indicate that inhibitors 1–3 bind lengthwise within the active-site tunnel in a helical conformation and their carboxylates interact alternatively with the Lys32 network or the YTT cluster at both tunnel mouths. The location of these binding hotspots suggests that inhibitor length may play a role in allowing establishment of contacts. To verify the effect of central core length on affinity, central cores from 2 to 12 methylene units in length (4 to 9) were synthesized and assayed (Figure 6, Table 1, and Scheme S1). Inhibition was observed in the low micromolar range for compounds with central cores of up to eight methylene units (4 to 7) (≤36.1 Å; Table 1), but none was observed above this length (8 and 9; Table 1).
Figure 6.

Inhibitors 4–7 and 10 and 11. Central core substitution relative to 1 and 2 is shown.
As with 2, the alkyl central cores of 4 to 7 cannot establish electrostatic interactions. LowModeMD simulations with 4 to 7 showed that, as the length of the central core increased, the terminal carboxylates are bound to the Lys32 network more frequently than to the YTT cluster (Table S2). Interactions of their benzimidazole moieties were comparable to those observed with 3 (Tables S2 and S3). The phenoxy oxygens and phenyls of 4 and 6 formed interactions with the same amino acids as 3. In addition, the phenoxy oxygens and phenyls of 5 are bound to Val66 and Ile68 backbone-NHs. In contrast, the (CH2)8 central core of 7 prevented interactions of the phenoxy ring with DfrB1. The sole remaining interaction was the π-hydrogen bond with the Ile68 backbone-NH. Increasing hydrophobicity of the longest central cores (>(CH2)8, or >36 Å) reduced solubility, limiting the concentrations that could be assayed; nonetheless, it was possible to determine that they procured no inhibition at concentrations where shorter analogues clearly did. As a result, 36 Å was considered the maximal length for the design of further bisbenzimidazole-type compounds.
Central Core Substitution
The H-bonding established between the hydroxylated central core of 1 and 3 and the active site suggested that the central core was a target for diversification. Substitution with a rigid (trans-2-butene; 10) or a bulky (phenyl; 11) moiety (Table 1; Scheme S1) procured inhibition similar to 1 (Table 1). Consistent with the good affinity of 2 and 4–7, which all bear an alkyl central core, this demonstrates that H-bonding in the central core is not a key to affinity. Selectivity was maintained, as 10 and 11 did not inhibit hDfr.
LowModeMD simulations with 10 showed no new interactions with its rigidified central core. However, the core modified interactions of the benzimidazole moieties, which no longer interacted with the Val66 backbone but established a π-hydrogen bond with the Ile68 backbone instead (Tables S2 and S3). The terminal carboxylates of 10 still H-bonded principally with the Lys32 network and alternatively with the YTT cluster, with the exception of Thr48.
The rigid phenyl central core of 11 established π-hydrogen bonds with the backbone-NHs of Val66 and Ile68; the former also H-bonded with the phenoxy oxygen, rather than the benzimidazole seen with 3, because of a slight reorganization. Our results demonstrate that rigidification of the central core is tolerated and suggests that further chemical diversification of the core is possible.
Contribution of the Benzimidazole Moieties and Conformational Freedom of the Carboxylates
In our previous report,20 inhibition was lost when the benzimidazole moieties were removed from 2. Loss of inhibition may result from the removal of key functional groups or from shortening the molecule. To verify this, analogues lacking benzimidazole moieties and of central core length varying from (CH2)6 to (CH2)12 (12 to 15)38 were assayed (Figure 7; Table 2). Only 14 demonstrated detectable inhibition of DfrB1. The length of 14 (26.9 Å; Ki = 8.8 μM) is slightly less than that of the shortest bisbenzimidazole inhibitor of Table 1, 4 (28.9 Å; Ki = 4.9 μM), clearly illustrating that removal of the benzimidazoles does not necessarily abolish binding. Longer analogues lacking benzimidazoles could not be assayed because of insufficient solubility. No inhibition was observed with further analogues lacking benzimidazoles where additional length was built in between the phenoxy groups and terminal carboxylates (16 and 17) (Figure 7; Table 2; Scheme S2). Our results are consistent with the benzimidazole moieties contributing to binding, as modeled by LowModeMD of 4, yet demonstrate that they are not critical components of DfrB1 inhibitors (Tables S2 and S3).
Figure 7.

Inhibitors 12–18. Central core length variation and benzimidazole moiety substitution are specified. Inhibitor length was calculated with ChemDraw 3D.
Table 2. Inhibition with Analogues Lacking Benzimidazole Moietiesa.
| DfrB1 |
||
|---|---|---|
| compound | IC50 (μM) | Ki (μM)b |
| 12 | >500c | |
| 13 | ≥500c | |
| 14 | 285 ± 80 | 8.8 ± 2.6 |
| 15 | >250c | |
| 16 | >500c | |
| 17 | >200c | |
| 18 | >200c | |
Values are given as the average ± standard deviation from the mean of at least triplicates of two independent experiments.
Ki was calculated from IC50 values.
Inhibitors were tested at the highest possible concentration, considering constraints due to high absorbance or to precipitation.
Substitution of the benzimidazoles with 2-naphthoic acid (18) (Figure 7; Table 2; Scheme S2) abolished inhibition. Benzimidazole had been identified as a weak inhibitor of DfrB1 through a fragment-based design, as had 2-naphthoic acid.20 Our result demonstrates that in the context of these disubstituted symmetrical molecules, their contribution to binding is not equivalent. The structural analogy of benzimidazole to the adenine moiety of NADPH could be thought to form the basis for its interactions with DfrB1; the adenine moiety is involved in binding, as its removal decreases the affinity of NADPH.27 However, the adenine moiety in the holoenzyme (PDB ID 2RK1) is solvent exposed, establishing no interactions with DfrB1. Notwithstanding that ambiguity, our observations establish that the benzimidazole moieties are significant contributors to DfrB1 inhibition. LowModeMD performed with 14 found interactions similar to those determined above, although the terminal carboxylates formed H-bonds more frequently with the YTT cluster than with the Lys32 network (Tables S2 and S3). In this sole case, no H-bonding was observed between the terminal carboxylates and Gly35 backbone-NH. Finally, the phenoxy moieties made π-hydrogen bonds with the Ser65 β-methylene, Val66 backbone amine, and aromatic C–H of Tyr69, differing somewhat from other inhibitors.
Substitution of the Phenoxy Moieties
We investigated the impact of the phenoxy moieties on binding by modulating their geometric constraints relative to the benzimidazole moieties. The p-orientation of 1 was modified to a m-orientation in 19 (Figure 8; Scheme S3). Despite the m-configuration reflecting the geometry of the flared edges of the active-site tunnel, inhibition decreased (3-fold greater Ki) (Table 3). Simulations suggested the conservation of the intertwined helical conformation of the 2 bound molecules of 19. However, the m-configuration abrogated H-bonding of the benzimidazole amines with the Val66 carbonyl (Tables S2 and S3). Instead, the benzimidazole amines H-bonded with Ile68. The benzimidazole rings interacted through π-hydrogen bonds with the β-carbon hydrogen of Ser65 and the Val66 backbone-NH. Nearly all binding contacts of the phenoxy moieties were abolished; only the phenoxy oxygen H-bond with the Ile68 backbone-NH was maintained. The altered bonding may be reflected in the loss of affinity of 19 relative to 1.
Figure 8.

Inhibitors 19, 20b, and 21. The central core substitution and benzimidazole orientation are shown.
Table 3. Inhibition Upon Substitution of the Phenoxy Moietiesa.
| DfrB1 |
hDfr | ||
|---|---|---|---|
| compound | IC50 (μM) | Ki (μM)b | IC50 (μM)c |
| 19 | 400 ± 100 | 12 ± 3 | >500 |
| 20b | 80 ± 20 | 2.6 ± 0.7 | >400 |
| 21 | 60 ± 20 | 1.7 ± 0.6 | >400 |
Values are given as the average ± standard deviation from the mean of at least triplicates of two independent experiments.
Ki was calculated from IC50 values.
Inhibitors were tested at the highest possible concentration, considering constraints due to high absorbance.
Addition of halogens may enhance affinity with a target.39,40 Bromination of the phenoxy ring was undertaken in an attempt to complement the large hydrophobic active site of DfrB1. A slight improvement of affinity was observed upon bromination of 3 (Ki = 3.5 μM) to yield 20b (Ki = 2.6 μM) (Figure 8; Table 3; Scheme S4). Furthermore, bromination of an analogue of 2 with a shorter central core to yield 21 (Figure 8; Scheme S5) also resulted in good affinity (Ki = 1.7 μM; Table 3). LowModeMD simulations of 20b and 21 suggest a H-bonding pattern similar to those of 2 and 3 (Tables S2 and S3), where the H-bonds involving the phenoxy oxygens were substituted by halogen bonding. The phenoxy segment of 20b formed electrostatic interactions between the bromine electropositive σ-hole and oxygen lone pair electrons of the Val66 carbonyl and Gln67 side-chain carbonyl. The bromine electropositive σ-hole in 21 created electrostatic interactions with the oxygen lone pair electrons of the Val66 and Ile68 carbonyls and Ser65 hydroxyl. Although those newly formed interactions did not significantly improve affinity, our results demonstrate the tolerance of DfrB1 to brominated inhibitors.
Substituting the Terminal Carboxylates
All simulations presented above predicted that electrostatic interactions of the terminal carboxylates with the Lys32 network were predominant (Tables S2 and S3). In an effort to design compounds that would obviate potential issues associated with carboxylates within pharmacophores,7 we explored alternative terminal substituents by converting these acids into a series of amides (22 to 30; Figure 9, Schemes S4 and S5). The introduction of neutral moieties at this position, with or without H-bonding substituents, was an effort to gain insights into the structural requirements at the termini. Their poor solubility precluded testing at concentrations greater than 50–100 μM, as indicated (Table S4). No inhibition was observed, supporting the hypothesis that the terminal carboxylates play a crucial role in binding to DfrB1. Despite their shortcomings within pharmacophores, they prove to be difficult to substitute in this context.
Figure 9.

Compounds 22–24 and 25–30. The terminal carboxylates were substituted by a set of amides. None inhibit DfrB1.
Reducing Entropic Cost by Binding One Molecule of Inhibitor per Active Site
Enzyme kinetics with 1(20) and crystallography with 3 demonstrated that two molecules of inhibitor simultaneously occupy the DfrB1 active-site tunnel. In an attempt to reduce entropic cost of binding a dimer of bisbenzimidazole inhibitors, we designed a tetrabenzimidazole analogue 31 (Table 4; Scheme S6). It is expected to display similar binding enthalpy and should reduce entropic cost by binding as a single copy. The inhibition potency of 31 was slightly improved (Ki 2- to 12-fold lower than the best inhibitors). A Hill coefficient of 1.1 ± 0.4 was determined, confirming the desired 1:1 binding stoichiometry (Figure 10A). Its methyl ester analogue, 32, did not inhibit, reinforcing the essential nature of the terminal carboxylates (Table S5).
Table 4. Inhibition with 1:1 Target/Ligand Stoichiometrya.

Values are given as the average ± standard deviation from the mean of at least triplicates of two independent experiments.
Ki was calculated from IC50 values.
Figure 10.

Inhibition of DfrB1 with monomeric inhibitor 31. (A) Inhibition, with Hill plot (inset). Values are given as the mean ± standard deviation for triplicate results. The Hill coefficient is 1.1 ± 0.4. (B) In silico reconstruction of 31 in the active site of DfrB1 based on the crystal structure PDB ID 6NXZ. Top: multiple poses adopted by 31 in the active site of DfrB1; only two DfrB1 protomers are shown in the 90° view. Bottom: a representative pose of 31 from a 90° view.
Repeated attempts at crystallization of DfrB1 with 31 were unsuccessful. LowModeMD was executed with 31 (Figure 10B; Tables S2 and S3). Electrostatic interactions, hydrogen, and π-hydrogen bonding were all similar to those observed with 3, apart from the congested quaternary central core. Only one weak H-bond was modeled between a core methylene of 31 and the γ-amide oxygen of Gln67.
A second strategy to improve entropic cost albeit with lower molecular weight than 31 involved nonsymmetrical phenoxybenzimidazole-based compounds 34–38, 39a, and 40–48 (Figure 11; Tables S6 and S7; Schemes S5, S7 and S8). They include the benzimidazole and phenoxy moieties that procure affinity (Tables 2 and 3) and take advantage of the tolerance to bromination and to modification of the central core moiety (Table 1). However, none inhibited DfrB1 at the highest concentration where they were soluble (30–2500 μM; Tables S6 and S7).
Figure 11.
Representative nonsymmetrical compounds 37, 40, 43, and 48. The terminal carboxylates, phenoxy moiety, and core were substituted. Four compounds among the 15 synthesized and assayed are shown. None among the 15 nonsymmetrical compounds inhibited DfrB1 (Tables S6 and S7).
Pursuing efforts to improve the entropic cost with a lower molecular weight, we designed a V-shaped analogue of 11 based on an o-substituted central phenyl core (33) (Scheme S1). This configuration imitates half of 31 and was designed to fill half of the active-site tunnel and competitively prevent DfrB1 activity. Binding a single molecule of 33 should reduce the entropic cost yet it would suffer from lower binding enthalpy due to loss of contacts. Upon verification, V-shaped 33 lost little affinity (Ki = 7.4 μM, 2-fold weaker than 1) which was surprisingly similar to the affinity of its linear, p-substituted analogue, 11 (Ki = 6.1 μM). The Hill coefficient of 1.1 ± 0.1 confirmed the desired 1:1 binding stoichiometry of 33 (Figure 12). Inhibitor 33 was selective, displaying no inhibition of hDfr (Table 4).
Figure 12.
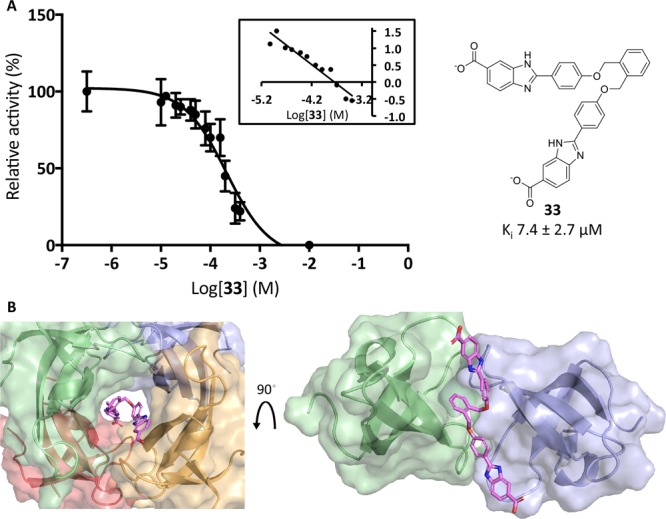
(A) Inhibition of DfrB1 with 33. Values are given as the mean ± standard deviation for triplicate results. Inset: Hill plot of panel (A). The Hill coefficient is 1.1 ± 0.1. (B) In silico reconstruction of 33 in the active site of DfrB1 (left) and from a top view (right).
The Dixon plot analysis of the inhibition of DfrB1 with 33 at varying NADPH concentrations and saturating DHF demonstrated that binding of 33 is competitive with respect to NADPH binding (Figure S2). In contrast, the Dixon analysis upon varying DHF concentrations with saturating NADPH did not reflect a classical form of inhibition (Figure S2). Uncompetitive inhibition is expected to produce parallel lines with a nonzero slope, whereas noncompetitive inhibition gives intersecting lines with an x-intercept of −Ki,41 neither of which corresponds to the observed pattern of nearly horizontal lines when varying DHF. This demonstrates that saturating NADPH precluded binding of subsaturating concentrations of 33. The preferential binding of NADPH on the apo-DfrB1 (Kd1 = 2.5 μM) relative to DHF (Kd1 = 120 μM) justifies the greater impact of NADPH than DHF on binding of 33.37
LowModeMD simulations are consistent with one molecule of 33 preventing binding of NADPH or DHF to DfrB1 by occupying the active-site tunnel (Figure 12; Tables S2 and S3). As a result of its o-substituted central core, V-shaped 33 adopted an elongated, heavily twisted conformation, precluding simultaneous binding of two molecules of 33 (Figure S3). Because a single molecule of 33 was bound, the frequency of contacts predicted between 33 and DfrB1 is lower than other inhibitors: all other simulations included four terminal carboxylates, either because two molecules were bound simultaneously or a single, 4-armed molecule (31) was bound (Tables S2 and S3). Nonetheless, the Ki of 33 (7.4 μM) remains on the same order of magnitude as the best inhibitors (1.1 to 2.6 μM), demonstrating that potency is essentially maintained. The nature of the interactions was the same as that for 3 except near their central cores because the phenyl core of 33 established π-hydrogen bonds with the alkyl side chains of Gln67 and the Tyr69 hydroxyl. These weak interactions were not sufficient to improve the affinity of 33.
Benzimidazole-Based Inhibitors Are Not Aggregators
Various observations allow us to demonstrate that the DfrB1 inhibitors are not aggregation-based inhibitors.42 First, we have demonstrated selectivity, where DfrB1 inhibitors that are active between 1 and 10 μM do not significantly reduce the hDfr activity at concentrations that are 10- to 100-fold greater (Tables 1, 3 and 4). Second, inhibition curves displayed steepness consistent with soluble behavior (Figures 10A and 12A), rather than the unusually steep pitch associated with aggregation-based inhibition. Third, the competitive mode of inhibition determined by observation of the bound crystal structures with 1(20) or 3, and kinetics with 1(20) or 33, is not a characteristic of aggregators.42
DfrB1 Inhibitors Also Inhibit the Clinically Identified, TMP-Resistant Homologue DfrB4
We previously identified the DfrB4 coding sequence, flanked by further antibiotic resistance genes, in a TMP-resistant North American clinical sample.10 We further demonstrated that expression of DfrB4 in E. coli conferred complete resistance to the highest concentration of TMP that could be dissolved in the medium (600 μg/mL).10 DfrB4 shares 77% amino acid identity with DfrB1; the catalytic core is highly conserved, whereas the loops and termini differ.21,30,36,43 The key residues of DfrB1 responsible for binding the benzimidazole-type inhibitors, namely, the Lys32 network, YTT cluster, and active-site residues, are conserved in DfrB4, suggesting that DfrB4 may be inhibited by the same compounds. Here, we verified whether inhibitors of DfrB1 also inhibit DfrB4.
DfrB4 has not been biochemically characterized. We first confirmed that DfrB4 possesses the Dfr activity. Its kinetic parameters KMNADPH and kcat were similar to those of DfrB1, and KM of DfrB4 was threefold greater (Table 5). Both DfrB1 and DfrB4 exhibit impressively high resistance to TMP (Table 5).27 Both DfrBs have a catalytic efficiency of ≈100-fold lower than E. coli chromosomal Dfr; although inefficient, this is sufficient to ensure bacterial host survival in the presence of TMP.28
Table 5. Kinetic Parameters and Inhibition of DfrB1 and DfrB4 with Selected Inhibitorsa.
Five efficient DfrB1 inhibitors (1, 3, 33, 20b, and 31), representing different features, were assayed for inhibition of DfrB4 (Table 5). The Ki values were similar for both DfrBs (1.1- to 1.7-fold difference). Thus, both DfrBs are similarly inhibited by the simplest inhibitors, 1 and 3, and by V-shaped 33, tolerate bromination of the phenoxy moiety (20b), and accept the bulky tetrabenzimidazole-based 31.
The sequence differences between DfrB1 and DfrB4 have no significant impact on their catalytic activity, TMP resistance, or capacity to be inhibited by the benzimidazole-based compounds tested here. Importantly, this immediately suggests that the inhibitors reported here also inhibit all other known members of the DfrB family, as they share 77–99% of protein sequence identity.10
Conclusions
Our report has uncovered several features that contribute significantly to the inhibition of DfrB1. The inclusion of terminal carboxylates is a key feature, as their substitution abolished inhibition. The carboxylates establish frequent hydrogen bonds or electrostatic interactions with either the Lys32 network or the YTT cluster. The length of the inhibitor is a second key feature, to allow the above interactions to be established at both mouths of the active-site tunnel simultaneously. A third feature is the contribution of the benzimidazoles: although not crucial to binding, their impact is significant. A final feature is packing within the large, symmetrical active site: simultaneous binding of two linear bisbenzimidazole inhibitors could be substituted with one tetrabenzimidazole inhibitor (31), or, with little loss of affinity, by a single V-shaped bisbenzimidazole inhibitor (33) that was modeled in a twisted conformation. The weak contacts between the o-substituted central core of 33 and DfrB1 reveal its potential for future optimization. We have thus validated a series of features characterizing benzimidazole-based inhibitors that selectively inhibit the highly TMP-resistant DfrB1 and DfrB4 without affecting hDfr. We envision the future development of DfrB inhibitors as a dual inhibitor system, where anti-DfrB antibiotics would be coadministered with TMP, to inhibit both DfrBs and the bacterial chromosomal DfrAs in parallel.
Experimental Procedures
Materials
Dihydrofolate was synthesized as previously reported.44 Unless otherwise mentioned, DNA-modifying enzymes were purchased from Thermo Scientific (Waltham, MA, USA). Enterokinase was from Feldan (Québec, QC) or from ProSpec (USA). Isopropyl b-d-1-thiogalactopyranoside and other reagents for enzyme assays were from BioShop (Burlington, ON, Canada). DMSO was purchased from Fisher Scientific (Ottawa, ON, Canada), and β-NADPH was purchased from Alexis biochemicals (San Diego, CA).
General Procedure A
As previously described,45 the selected hydroxybenzaldehyde (2.0 equiv) and appropriate dibrominated compound (1.0 equiv) in a mixture of EtOH/H2O (9:1) containing NaOH (2.0 equiv) was heated under microwave irradiation in an Initiator Biotage oven for 20 min at 120 °C. After cooling, the precipitate was filtered, washed with EtOH/H2O, and dried in vacuo. The resulting bisbenzaldehydes were sufficiently pure to proceed to the next step.
General Procedure B
A mixture of the resulting bisbenzaldehyde (1.0 equiv), Na2S2O5 (1.0 equiv), 3,4-diaminobenzoic acid (2.0 equiv), and EtOH/H2O (3:1) was microwave irradiated (Biotage) for 15 min at 140 °C. After cooling, the precipitate was filtered, thoroughly washed with EtOH/H2O/ether, and dried in vacuo.
General Procedure C
As previously described,45 the selected phenol derivative (3.0 equiv) and appropriate dibromoalkane (1.0 equiv) were dissolved in a mixture of EtOH/H2O (9:1) containing NaOH (6.0 equiv). The mixture was heated under microwave irradiation in an Initiator Biotage oven for 20 min at 120 °C. After cooling, the precipitate was acidified with 6 mL of 37% HCl and thoroughly washed with water and ethanol.
General Procedure D
To a solution of the selected mono- or bis-acid (1.0 equiv), HATU (3.0 equiv), and DIPEA (10 equiv) stirred in DMF (0.1 M) at room temperature, the amine (2.2 equiv) was added, and the resulting reaction mixture was stirred at room temperature for 3 h. The resulting mixture was then diluted with ethyl acetate, washed with water and brine, dried over MgSO4, filtered, and concentrated. The crude residue obtained was purified by prep HPLC (Preparative HPLC-General Method A) to give the desired product.
General Procedure E
To a solution of the selected acid (1.0 equiv) and amine (1.1 equiv) in DMF (0.1 M) were added DIPEA (5 equiv) and HATU (1.5 equiv) at room temperature, and the resulting mixture was stirred for 3 h. The resulting mixture was then diluted with ethyl acetate, washed (water, brine), dried (MgSO4), filtered, and concentrated. The crude residue obtained was purified by prep HPLC (Preparative HPLC-General Method A) to give the title compound.
Analytical HPLC-General Method A
Analytical HPLC analyses were performed on an Agilent 1200 Series LC system with a Zorbax XDB-C18 column (4.6 × 30 mm, 3.5 μM) under the following conditions: flow rate 3 mL/min at room temperature; 10 μL of target compound solution in MeOH; solvent A 5% MeOH, 95% water + 0.05% TFA; solvent B 95% MeOH, 5% water + 0.05% TFA; 2 min gradient from 0 to 100% solvent B and 2 min 100% solvent B; and detection λ = 220/254 nm. The purity was determined by HPLC with UV detection at 220 nm.
Analytical HPLC-General Method B
Analytical HPLC analyses were also achieved on an Agilent 1200 Series LC system with a Kinetex C18 column (3.0 × 30 mm, 2.6 μM) under the following conditions: flow rate 1.5 mL/min at room temperature; 2 μL of target compound solution in MeOH; solvent A 5% MeOH, 95% water + 0.1% AcOH; solvent B 95% MeOH, 5% water + 0.1% AcOH; 0.5 min gradient from 0 to 100% solvent B and 1.5 min 100% solvent B; and detection λ = 220/254 nm. The purity was determined by HPLC with UV detection at 220 nm.
Preparative HPLC-General Method A
Preparative HPLC analyses were realized on an Agilent 1200 Series LC system with a Kinetex C18 column (21.2 × 100 mm, 5 μM) under the following conditions: 20 mL/min at room temperature; 900 μL of 25 mg/mL of compound in DMF; solvent A 5% MeOH, 95% water + 0.05% TFA; solvent B 95% MeOH, 5% water + 0.05% TFA; 2 min isocratic 30% solvent B followed by 8 min gradient to 100% solvent B and 3 min 100% solvent B; and detection λ = 254 nm.
Purification of DfrB1 and DfrB4 for Kinetic Characterization
Histidine-tagged DfrB1 and DfrB4 (using previously reported constructs WT R67 DHFR pQE3228 and His6-DfrB4,10 respectively) were overexpressed and purified as described previously4,20 with the following modifications. The cell culture volume was 400 mL. During purification on His-Trap HP,4 the imidazole concentration for the plateau was 0.2 M and the elution concentration was 0.5 M. Fractions containing DfrB were identified according to activity assay and analysis on tricine sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE)46 and then concentrated using an Amicon concentrator (MWCO 3000, Millipore) for application on a Superose 12 column (1.6 cm × 55 cm) pre-equilibrated with 0.05 M potassium phosphate, pH 8.0. Fractions containing DfrB were identified as above. Sample purity was determined by tricine SDS-PAGE. The concentration was determined using the Bio-Rad protein assay (Bio-Rad, Hercules, CA).
Purification of the Human Dihydrofolate Reductase (hDfr)
Overexpression of the hexahistidine-tagged human Dfr (WT His6-hDHFR) and cell lysis was described previously.47 The lysate was applied on a 5 mL nickel-affinity column (GE Healthcare) with an ÅKTA fast-protein liquid chromatography system (Amersham Biosciences) at 0.5 mL/min. The column was washed with 5 column volumes (CV) of 0.1 M potassium phosphate buffer (pH 8.0) at 1 mL/min. A stepwise gradient of 0.1 M potassium phosphate buffer (pH 8.0) containing increasing concentrations of imidazole (0.02, 0.05, 0.06, 0.08, 0.1, 0.16 and 0.2 M; 2 CV per concentration) was applied. Fractions containing WT His6-hDHFR were identified according to the standard spectrophotometric DHFR activity assay and analysis on SDS-PAGE. Pooled fractions were concentrated using an Amicon concentrator (MWCO 10 000, Millipore) and further purified on a pre-equilibrated Superose 12 column (1.6 cm × 55 cm) with 0.1 M potassium phosphate buffer (pH 7.5). Fractions containing His6-hDHFR were identified as above. The His6-hDHFR concentration was determined using the Bio-Rad protein assay.
Construction of the DfrB1 Variant for Crystallization Purposes
To enable removal of the N-terminal 6-histidine tag from DfrB1 following its purification, we inserted an enterokinase cleavage site (DDDDK) into the construct WT R67 DHFR pQE32.28 A forward oligonucleotide primer (5′-CATGGATCCAAGACTACAAAGACGATGACGACAAGGTATTCCCATCGGACGCCAC-3′); Sigma-Aldrich (Oakville, ON) was designed to include the protease cleavage site (bold) and the DfrB1 sequence beginning at Val17, the probable chymotrypsin cleavage site, with the BamHI restriction site. A reverse primer removed the 13-residue C-terminal tail (ELGTPGRPAAKLN) that was previously introduced for other purposes28 and HindIII (underlined): 5′-GGGAAGCTTTTAGTTGATGCGTTCAAGCGCC-3′. The PCR product obtained with Phusion High-Fidelity DNA polymerase (Thermo Scientific (Waltham, MA, USA)) was digested with BamHI and HindIII, isolated on agarose gel, and extracted according to the “EZ-10 Spin Column PCR Products Purification Kit” protocol (Bio Basic, Burlington, ON, Canada). The insert was ligated to BamHI and HindIII-digested and dephosphorylated (FastAP Thermosensitive Alkaline Phosphatase, 30 min) pQE32 (Qiagen) with the DNA Ligation Kit Ver.2.1 (Takara, USA). After 30 min at 16 °C, the DNA ligation solutions were transformed into DH5α. DNA sequencing confirmed the expected construct DfrB1-His6V17Δ16/ENTK-pQE32. The DfrB1-His6V17Δ16/ENTK-pQE32 was transformed into E. coli BL21 pRep4 (Qiagen) for expression.
Purification and Crystallization of DfrB1 Soaked with 1
To crystallize DfrB1 in complex with 1, an identical protein expression and purification protocol was employed as described in ref (24). Following expression and purification, the INSF tandem dimer, as previously described,24 was concentrated to 20 mg/mL in 100 mM Tris pH 8.0. Immediately before crystallization, chymotrypsin was added to the sample in a ratio of 1:100 chymotrypsin/INSF, and the protein was diluted to 15 mg/mL using MPD, resulting in a final MPD concentration of 25%. Reservoirs were prepared using 750 μL of 100 mM sodium phosphate pH 7.6 and 60% MPD in a Greiner 24-well hanging-drop crystallization plate. On a siliconized glass cover slip (Hampton Research), 1.5 μL of protein was combined with 2.5 μL of reservoir solution and suspended over the well. The plate was incubated at 277 K, and crystals were obtained in a few days. Drops were soaked with 0.2 μL of a solution of 1 in 100% DMSO for a final concentration of 2 mM of 1.
Purification of the DfrB1 Variant for Crystallization Purposes
The crystallization variant DfrB1 (DfrB1-His6V17Δ16/ENTK) was overexpressed and purified as previously described,4,20 with the modifications noted above for DfrB1 and DfrB4. DfrB1-His6V17Δ16/ENTK was concentrated to 2 mg/mL (24 mg in total) with an Amicon concentrator (MWCO 3000, Millipore), and it was incubated with enterokinase (13 U) for 20 h at 22 °C. The sample was concentrated and further purified on Superose 12 as described above, except that the column was linked at the bottom to His-Trap HP cartridge pre-equilibrated with 0.05 mM potassium phosphate, pH 8.0. This last step allows removal of uncleaved DfrB1-His6V17Δ16/ENTK. Fractions containing DfrB1 were identified and the purity was determined as above. The protein was concentrated with 0.05 mM potassium phosphate, pH 8.0–13.3 mg/mL for crystallization.
Crystallization of DfrB1-His6V17Δ16/ENTK was performed as previously described.29 Briefly, the protein concentration was adjusted from 13.3 to 10 mg/mL by addition of a final concentration of 25% MPD. Crystals were obtained using the hanging-drop vapor-diffusion method in a Greiner 24-well hanging-drop crystallization plate. Reservoirs were prepared in Eppendorf tubes with 100 mM Tris-Cl pH 8.0 55% MPD. On a siliconized glass cover slip (Hampton Research), 2.0 μL of protein solution was combined with 2.0 μL of the reservoir solution. The plate was incubated at 277 K, and crystals were obtained after 3–4 days. Drops were soaked with 0.4 μL of a solution of 3 in 100% DMSO for a final concentration of 12 mM of 3 five days prior to crystal harvest.
Data were collected under standard cryogenic conditions using a Rigaku MicroMax-007HF generator equipped with Varimax HF optics and a Saturn 944+ CCD detector. The data were processed using HKL-2000 (Table S1).48 The integrated and scaled data were imported into the CCP4 software suite.49 Iterative rounds of positional and B-factor refinement using Refmac50 and manual model building using Coot51 were then performed, using the coordinates from PDB ID 3SFM with all solvent and ligands removed24 as the starting model. As the 3SFM structure was crystallized in the same space group and with the same cell dimensions, care was taken to ensure that the same reflections were included in the Rfree test set. During the late stages of refinement, anisotropic B-factor refinement was introduced, and ligand and solvent molecules were incorporated into the model. Coordinates and structure factors were deposited to the PDB as PDB ID 6NXZ.
Kinetic Studies
For all kinetic assays, pH of solutions was 7. IC50 and Ki values were determined as previously described4,20 except that the reaction volume was 100 μL. The assays were conducted in Corning 96-well flat-bottomed polystyrene plates [Thermo Scientific (Waltham, MA, USA)]. Initial rates (first 15% of substrate consumption) were collected with a Beckman DTX 880 plate reader integrated with a Biomek NX automated liquid handling workstation (Beckman Coulter, Brea, CA). The length of compounds (4 to 9 and 12 to 18) was calculated with ChemDraw 3D (ChemOffice 2017, PerkinElmer Informatics). KMNADPH, KM, and kcat was determined as previously described.4
Building Small Molecule Input Database and Initial Pose Generation
All studied molecules were sketched in ChemDraw Professional 16. All atomistic 3D coordinates at pH7 were generated using the Database Wash application in MOE2018.52 We generated the DfrB1·(3)2 based on the 1,4-dioxobutan-2-ol central core of the crystal structure PDB ID 6NXZ as the starting point. The structure was prepared using the QuickPrep application with default parameters. The two molecules of 3 were built simultaneously in a stepwise manner using a standard structure-based drug design workflow. The generated DfrB1·(3)2 complex was used as the input structure to dock all remaining molecules.
Building Models of Protein·Inhibitor Complexes
Docking of inhibitors was performed using the Dock application in MOE2018.52 One molecule of 3 was replaced by a first molecule of the query inhibitor. Substitution of the second molecule of 3 by the second molecule of inhibitor followed. The final dimer complex was assembled from each docking run based on the best rmsd compared to 3. As per the Placement Method, the Triangle Matcher was used with 30 retained poses based on London dG scoring. Refinement was done in the rigid receptor mode, and the best 10 poses from the GBVI/WSA dG scoring function were written to the output database.
Conformational Exploration of Ligand Dimers in the Tunnel
Conformational exploration of each inhibitor molecule bound to DfrB1 was performed using the LowModeMD method53 in MOE2018.52 All inhibitor atoms were kept free while receptor atoms within the active-site tunnel (all residues 4.5 Å from inhibitor molecules) were tethered with 10 kcal/mol starting from 0.25 Å deviation around the original coordinates. All residues 4.5 Å from the tethered atoms were fixed, and the rest of the system was set to inert. The Amber10:EHT forcefield was used for LowModeMD with R-Field solvation. The exploration was stopped after 10 000 conformations or after 100 consecutive conformations already present in the database. All conformations with an energy difference of 100 kcal/mol compared to the lowest energy conformation were retained.
rmsd Calculated for 1
To profile the similarity between the position of 1 in our simulation and in the crystal structure, rmsds were calculated for each of the 2992 conformations from the simulation of 1. From the crystal structure, the three additional subunits to create the complete tetramer were generated. Maintaining the inhibitor molecule in the position obtained from the simulation (i.e., without performing any superimposition), the rmsd over all inhibitor atoms present in the crystal structure were calculated against the inhibitor in each of the four possible symmetry-generated positions from the crystal structure, and the lowest of the four rmsd values was used. In addition, the rmsd was calculated after superimposing the simulated inhibitor molecules onto the crystal structure inhibitor molecule.
Identification of Ligand-Binding Mode
The protein ligand interaction fingerprint application in MOE201852 was used to study the average binding mode of each inhibitor bound to DfrB1. The calculation used the default parameters.
Acknowledgments
This work is dedicated to the memory of Elizabeth E. Howell. We thank Prof. Steven Laplante and Prof. Roberto Chica for the technical support, Nadia Nouasria for technical assistance, Éric Marsault for critical reading, the University of Montreal NMR laboratory, and Yves Gareau for assistance in analyzing NMR spectra. We also thank Simon Toulouse for assistance with drafting Figure S1. We thank Pawel Grochulski at the Canadian Light Source (beamline CMCF-ID) for collecting the data for PDB ID 6NY0. This work was supported by Natural Sciences and Engineering Research Council of Canada grants 227853 and 2018-04686 awarded to J.N.P., Canada Foundation for Innovation grant 11510 to J.N.P., CIHR operating grant MOP-13107 to A.M.B., and the Fondation Marcel et Rolande Gosselin to A.M.J.L.T. received scholarships from les Fonds Québécois pour la Recherche sur la Nature et les Technologies (FQRNT), Hydro-Québec and Faculté des études supérieures et post-doctorales de l’Université de Montréal (FESP), and PROTEO, the Québec Network for Research on Protein, Function, Engineering and Applications. B.J.Y. has held scholarships from the Natural Sciences and Engineering Research Council of Canada, McGill University, the CIHR Strategic Initiative in Chemical Biology, and the NSERC CREATE Program in Bionanomachines. D.B. received scholarship from PROTEO.
Glossary
Abbreviations
- AMT
aminopterin
- Dfr
dihydrofolate reductase
- DfrBs
type II Dfr
- DHF
dihydrofolate
- hDfr
human Dfr
- SUL
sulfonamide
- MOE
molecular operating environment
- MTX
methotrexate
- PLIF
protein ligand interaction fingerprint
- PMTX
pemetrexed
- TMP
trimethoprim
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsomega.9b00640.
Crystallographic data; analysis of molecular simulations of binding interactions; and additional kinetic data (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Giguère S.; Prescott J. F.; Dowling P. M.. Antimicrobial therapy in veterinary medicine; John Wiley & Sons Inc, 2013; Vol. 47, pp 256–257. [Google Scholar]
- World Health Organization, WHO . WHO list of critically important antimicrobials for human medicine.
- Then R. L. History and future of antimicrobial diaminopyrimidines. J. Chemother. 1993, 5, 361–368. 10.1080/1120009X.1993.11741082. [DOI] [PubMed] [Google Scholar]
- Toulouse J. L.; Abraham S. M. J.; Kadnikova N.; Bastien D.; Gauchot V.; Schmitzer A. R.; Pelletier J. N. Investigation of classical organic and ionic liquid cosolvents for early-stage screening in fragment-based inhibitor design with unrelated bacterial and human dihydrofolate reductases. Assay Drug Dev. Technol. 2017, 15, 141–153. 10.1089/adt.2016.768. [DOI] [PubMed] [Google Scholar]
- Masters P. A.; O’Bryan T. A.; Zurlo J.; Miller D. Q.; Joshi N. Trimethoprim-sulfamethoxazole revisited. Int. Arch. Med. 2003, 163, 402. 10.1001/archinte.163.4.402. [DOI] [PubMed] [Google Scholar]
- Huovinen P.; Sundstrom L.; Swedberg G.; Skold O. Trimethoprim and sulfonamide resistance. Antimicrob. Agents Chemother. 1995, 39, 279–289. 10.1128/aac.39.2.279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broad D. F.; Smith J. T. Classification of trimethoprim-resistant dihydrofolate reductases mediated by R-plasmids using isoelectric focussing. Eur. J. Biochem. 1982, 125, 617–622. 10.1111/j.1432-1033.1982.tb06727.x. [DOI] [PubMed] [Google Scholar]
- Howell E. E. Searching sequence space: Two different approaches to dihydrofolate reductase catalysis. ChemBioChem 2005, 6, 590–600. 10.1002/cbic.200400237. [DOI] [PubMed] [Google Scholar]
- Huovinen P. Trimethoprim resistance. Antimicrob. Agents Chemother. 1987, 31, 1451–1456. 10.1128/aac.31.10.1451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toulouse J. L.; Edens T. J.; Alejaldre L.; Manges A. R.; Pelletier J. N. Integron-associated DfrB4, a previously uncharacterized member of the trimethoprim-resistant dihydrofolate reductase B family, is a clinically identified emergent source of antibiotic resistance. Antimicrob. Agents Chemother. 2017, 61, 1–5. 10.1128/aac.02665-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alonso H.; Gready J. E. Integron-sequestered dihydrofolate reductase: a recently redeployed enzyme. Trends Microbiol. 2006, 14, 236–242. 10.1016/j.tim.2006.03.003. [DOI] [PubMed] [Google Scholar]
- Ho J. M. W.; Juurlink D. N. Considerations when prescribing trimethoprim-sulfamethoxazole. Can. Med. Assoc. J. 2011, 183, 1851–1858. 10.1503/cmaj.111152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadlec K.; Kehrenberg C.; Schwarz S. Molecular basis of resistance to trimethoprim, chloramphenicol and sulphonamides in Bordetella bronchiseptica. J. Antimicrob. Chemother. 2005, 56, 485–490. 10.1093/jac/dki262. [DOI] [PubMed] [Google Scholar]
- L’Abée-Lund T. M.; Sørum H. Class 1 integrons mediate antibiotic resistance in the fish pathogen Aeromonas salmonicida worldwide. Microb. Drug Resist. 2001, 7, 263–272. 10.1089/10766290152652819. [DOI] [PubMed] [Google Scholar]
- Sunde M. Prevalence and characterization of class 1 and class 2 integrons in Escherichia coli isolated from meat and meat products of Norwegian origin. J. Antimicrob. Chemother. 2005, 56, 1019–1024. 10.1093/jac/dki377. [DOI] [PubMed] [Google Scholar]
- Cabello F. C. Heavy use of prophylactic antibiotics in aquaculture: a growing problem for human and animal health and for the environment. Environ. Microbiol. 2006, 8, 1137–1144. 10.1111/j.1462-2920.2006.01054.x. [DOI] [PubMed] [Google Scholar]
- Yassin A. K.; Gong J.; Kelly P.; Lu G.; Guardabassi L.; Wei L.; Han X.; Qiu H.; Price S.; Cheng D.; Wang C. Antimicrobial resistance in clinical Escherichia coli isolates from poultry and livestock, China. PLoS One 2017, 12, e0185326 10.1371/journal.pone.0185326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sköld O. Resistance to trimethoprim and sulfonamides. Vet. Res. 2001, 32, 261–273. 10.1051/vetres:2001123. [DOI] [PubMed] [Google Scholar]
- Stapleton P. J.; Lundon D. J.; McWade R.; Scanlon N.; Hannan M. M.; O’Kelly F.; Lynch M. Antibiotic resistance patterns of Escherichia coli urinary isolates and comparison with antibiotic consumption data over 10 years, 2005-2014. Ir. J. Med. Sci. 2017, 186, 733–741. 10.1007/s11845-016-1538-z. [DOI] [PubMed] [Google Scholar]
- Bastien D.; Ebert M. C. C. J. C.; Forge D.; Toulouse J.; Kadnikova N.; Perron F.; Mayence A.; Huang T. L.; Vanden Eynde J. J.; Pelletier J. N. Fragment-based design of symmetrical bis-benzimidazoles as selective inhibitors of the trimethoprim-resistant, type II R67 dihydrofolate reductase. J. Med. Chem. 2012, 55, 3182–3192. 10.1021/jm201645r. [DOI] [PubMed] [Google Scholar]
- Duff M. R.; Chopra S.; Strader M. B.; Agarwal P. K.; Howell E. E. Tales of dihydrofolate binding to R67 dihydrofolate reductase. Biochemistry 2016, 55, 133–145. 10.1021/acs.biochem.5b00981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fling M. E.; Elwell L. P. Protein expression in Escherichia coli minicells containing recombinant plasmids specifying trimethoprim-resistant dihydrofolate reductases. J. Bacteriol. 1980, 141, 779–785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narayana N. High-resolution structure of a plasmid-encoded dihydrofolate reductase: Pentagonal network of water molecules in the D2-symmetric active site. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2006, 62, 695–706. 10.1107/s0907444906014764. [DOI] [PubMed] [Google Scholar]
- Yachnin B. J.; Colin D. Y.; Volpato J. P.; Ebert M.; Pelletier J. N.; Berghuis A. M. Novel crystallization conditions for tandem variant R67 DHFR yield a wild-type crystal structure. Acta Crystallogr., Sect. F: Struct. Biol. Cryst. Commun. 2011, 67, 1316–1322. 10.1107/s1744309111030417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthews D. A.; Smith S. L.; Baccanari D. P.; Burchall J. J.; Oatley S. J.; Kraut J. Crystal structure of a novel trimethoprim-resistant dihydrofolate reductase specified in Escherichia coli by R-plasmid R67. Biochemistry 1986, 25, 4194–4204. 10.1021/bi00363a005. [DOI] [PubMed] [Google Scholar]
- Carrillo N.; Ceccarelli E. A.; Roveri O. A. Usefulness of kinetic enzyme parameters in biotechnological practice. Biotechnol. Genet. Eng. Rev. 2010, 27, 367–382. 10.1080/02648725.2010.10648157. [DOI] [PubMed] [Google Scholar]
- Jackson M.; Chopra S.; Smiley R. D.; Maynord P. O. N.; Rosowsky A.; London R. E.; Levy L.; Kalman T. I.; Howell E. E. Calorimetric studies of ligand binding in R67 dihydrofolate reductase. Biochemistry 2005, 44, 12420–12433. 10.1021/bi050881s. [DOI] [PubMed] [Google Scholar]
- Schmitzer A. R.; Lépine F.; Pelletier J. N. Combinatorial exploration of the catalytic site of a drug-resistant dihydrofolate reductase: Creating alternative functional configurations. Protein Eng., Des. Sel. 2004, 17, 809–819. 10.1093/protein/gzh090. [DOI] [PubMed] [Google Scholar]
- Krahn J. M.; Jackson M. R.; DeRose E. F.; Howell E. E.; London R. E. Crystal structure of a type II dihydrofolate reductase catalytic ternary complex. Biochemistry 2007, 46, 14878–14888. 10.1021/bi701532r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamath G.; Howell E. E.; Agarwal P. K. The tail wagging the dog: Insights into catalysis in R67 dihydrofolate reductase. Biochemistry 2010, 49, 9078–9088. 10.1021/bi1007222. [DOI] [PubMed] [Google Scholar]
- Assaraf Y. G. Molecular basis of antifolate resistance. Cancer Metastasis Rev. 2007, 26, 153–181. 10.1007/s10555-007-9049-z. [DOI] [PubMed] [Google Scholar]
- Lele A. C.; Mishra D. A.; Kamil T. K.; Bhakta S.; Degani M. S. Repositioning of DHFR Inhibitors. Curr. Top. Med. Chem. 2016, 16, 2125–2143. 10.2174/1568026616666160216152540. [DOI] [PubMed] [Google Scholar]
- Piper J. R.; McCaleb G. S.; Montgomery J. A.; Kisliuk R. L.; Gaumont Y.; Sirotnak F. M. Syntheses and antifolate activity of 5-methyl-5-deaza analogues of aminopterin, methotrexate, folic acid, and N10-methylfolic acid. J. Med. Chem. 1986, 29, 1080–1087. 10.1021/jm00156a029. [DOI] [PubMed] [Google Scholar]
- Kisliuk R. Deaza analogs of folic acid as antitumor agents. Curr. Pharm. Des. 2003, 9, 2615–2625. 10.2174/1381612033453695. [DOI] [PubMed] [Google Scholar]
- Chopra S.; Dooling R. M.; Horner C. G.; Howell E. E. A balancing act between net uptake of water during dihydrofolate binding and net release of water upon NADPH binding in R67 dihydrofolate reductase. J. Biol. Chem. 2008, 283, 4690–4698. 10.1074/jbc.m709443200. [DOI] [PubMed] [Google Scholar]
- Park H.; Zhuang P.; Nichols R.; Howell E. E. Mechanistic Studies of R67 Dihydrofolate Reductase. J. Biol. Chem. 1997, 272, 2252–2258. 10.1074/jbc.272.4.2252. [DOI] [PubMed] [Google Scholar]
- Bradrick T. D.; Beechem J. M.; Howell E. E. Unusual binding stoichiometries and cooperativity are observed during binary and ternary complex formation in the single active pore of R67 dihydrofolate reductase, a D2 symmetric protein. Biochemistry 1996, 35, 11414–11424. 10.1021/bi960205d. [DOI] [PubMed] [Google Scholar]
- Cappoen D.; Forge D.; Vercammen F.; Mathys V.; Kiass M.; Roupie V.; Anthonissen R.; Verschaeve L.; Vanden Eynde J. J.; Huygen K. Biological evaluation of bisbenzaldehydes against four Mycobacterium species. Eur. J. Med. Chem. 2013, 63, 731–738. 10.1016/j.ejmech.2013.03.023. [DOI] [PubMed] [Google Scholar]
- Lu Y.; Liu Y.; Xu Z.; Li H.; Liu H.; Zhu W. Halogen bonding for rational drug design and new drug discovery. Expert Opin. Drug Discovery 2012, 7, 375–383. 10.1517/17460441.2012.678829. [DOI] [PubMed] [Google Scholar]
- Parisini E.; Metrangolo P.; Pilati T.; Resnati G.; Terraneo G. Halogen bonding in halocarbon-protein complexes: a structural survey. Chem. Soc. Rev. 2011, 40, 2267–2278. 10.1039/c0cs00177e. [DOI] [PubMed] [Google Scholar]
- Segel I. H.Enzyme Kinetics: Behavior and Analysis of Rapid Equilibrium and Steady State Enzyme Systems; Wiley: New York, 1993; p 957. [Google Scholar]
- Shoichet B. K. Screening in a spirit haunted world. Drug Discovery Today 2006, 11, 607–615. 10.1016/j.drudis.2006.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hicks S. N.; Smiley R. D.; Stinnett L. G.; Minor K. H.; Howell E. E. Role of Lys-32 residues in R67 dihydrofolate reductase probed by asymmetric mutations. J. Biol. Chem. 2004, 279, 46995–47002. 10.1074/jbc.m404484200. [DOI] [PubMed] [Google Scholar]
- Blakley R. L. Crystalline dihydropteroylglutamic acid. Nature 1960, 188, 231–232. 10.1038/188231a0. [DOI] [Google Scholar]
- Mayence A.; Pietka A.; Collins M. S.; Cushion M. T.; Tekwani B. L.; Huang T. L.; Vanden Eynde J. J. Novel bisbenzimidazoles with antileishmanial effectiveness. Bioorg. Med. Chem. Lett. 2008, 18, 2658–2661. 10.1016/j.bmcl.2008.03.020. [DOI] [PubMed] [Google Scholar]
- Schägger H.; von Jagow G. Tricine-sodium dodecyl sulfate-polyacrylamide gel electrophoresis for the separation of proteins in the range from 1 to 100 kDa. Anal. Biochem. 1987, 166, 368–379. 10.1016/0003-2697(87)90587-2. [DOI] [PubMed] [Google Scholar]
- Volpato J. P.; Fossati E.; Pelletier J. N. Increasing methotrexate resistance by combination of active-site mutations in human dihydrofolate reductase. J. Mol. Biol. 2007, 373, 599–611. 10.1016/j.jmb.2007.07.076. [DOI] [PubMed] [Google Scholar]
- Otwinowski Z.; Minor W. [20] Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997, 276, 307–326. 10.1016/s0076-6879(97)76066-x. [DOI] [PubMed] [Google Scholar]
- Winn M. D.; Ballard C. C.; Cowtan K. D.; Dodson E. J.; Emsley P.; Evans P. R.; Keegan R. M.; Krissinel E. B.; Leslie A. G.; McCoy A.; McNicholas S. J.; Murshudov G. N.; Pannu N. S.; Potterton E. A.; Powell H. R.; Read R. J.; Vagin A.; Wilson K. S. Overview of the CCP4 suite and current developments. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2011, 67, 235–242. 10.1107/s0907444910045749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murshudov G. N.; Vagin A. A.; Dodson E. J. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr., Sect. D: Biol. Crystallogr. 1997, 53, 240–255. 10.1107/s0907444996012255. [DOI] [PubMed] [Google Scholar]
- Emsley P.; Lohkamp B.; Scott W. G.; Cowtan K. Features and development of Coot. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2010, 66, 486–501. 10.1107/s0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ULC, Chemical Computing Group . Molecular Operating Environment (MOE), 2013; Vol. 08.
- Labute P. LowModeMD-Implicit low mode velocity filtering applied to conformational search of macrocycles and protein loops. J. Chem. Inf. Model. 2010, 50, 792–800. 10.1021/ci900508k. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.