

Abstract
Fluoroquinolones are a class of widely prescribed antibiotics with a broad range of activity against Gram-positive, Gram-negative, and some atypical microbes. Unfortunately, these drugs are associated with significant adverse events including neuropathy, tendinopathy, cardiac rhythm abnormalities, and mental health side effects. The mechanism by which fluoroquinolones cause many of these toxicities is unknown. The antibacterial mechanism of action involves disruption of the catalytic mechanism of type-II topoisomerases in bacteria, namely topoisomerase IV and DNA gyrase. Fluoroquinolones inhibit the ability of the enzymes to ligate cleaved DNA and result in single- and double-stranded DNA breaks. Thus, there is an interest in investigating whether human topoisomerase II is involved in mediating the adverse events associated with quinolones. Previous studies demonstrate some response of human topoisomerase IIα and IIβ to high levels of ciprofloxacin. However, it is not clear whether the concentration of ciprofloxacin utilized in those studies corresponds to concentrations that would be routinely achievable in patients. Therefore, this study set out to examine three clinically relevant fluoroquinolones along with two older agents to determine whether these compounds display activity against topoisomerase IIα and IIβ at drug concentrations that more closely approximate typical patient plasma values. On the basis of our evidence, none of the quinolones studied were able to poison DNA cleavage by either human enzyme. Ciprofloxacin, desethylene-ciprofloxacin, and the recently removed from market gemifloxacin were able to inhibit topoisomerase II-mediated DNA relaxation at concentrations of 200–300 μM. On the basis of these data, we propose that human topoisomerase II is not likely to be the main cause of these adverse events and that additional targets need to be identified to clarify the mechanisms underlying quinolone toxicities.
Introduction
Fluoroquinolones are a widely used class of antimicrobial agents first introduced into clinical practice in the 1960s.1 Nalidixic acid, serendipitously isolated as an impurity in the synthesis of the antimalarial chloroquine, was the first quinolone compound and was found to exhibit bactericidal activity against Gram-negative bacteria.1,2 Although its mechanism of action was at the time ill-defined, its clinical application was suitable albeit limited to the treatment of urinary tract infections (UTIs).2,3
Subsequent development yielded additional related compounds that were fluorinated and which demonstrated a lower degree of resistance than did nalidixic acid. These early fluoroquinolones exhibited limited systemic bioavailability, consequent low systemic concentrations, and multiple daily dosing.1 The clinical utilization of these agents thus remained relatively confined to infections of the genitourinary tract.4,5
Structural modifications to the fluoroquinolone backbone continued to proliferate that resulted in compounds that displayed pharmacokinetic improvements (e.g., once-daily dosing) along with a significantly expanded spectra of antimicrobial activity that now, in addition to Gram-negative organisms, includes Gram-positive, atypical, and, for some quinolones, anaerobic organisms.1,2,4,5 From a class of agents that originated with a narrow clinical indication limited to UTI treatment, the fluoroquinolones are now routinely employed in the treatment of numerous infections including respiratory, genitourinary, gastrointestinal tract infections, skin and soft tissue, and bone infections.2 Fluoroquinolones represent one of the most highly prescribed antibiotic classes with approximately 30 million prescriptions issued in 2016.6 Ciprofloxacin, levofloxacin, and moxifloxacin are among the most commonly prescribed fluoroquinolones.
The development and expansion of the fluoroquinolone class—in both the number of agents and in their therapeutic utilization—has been accompanied by clinical enthusiasm. However, this justifiable optimism has been somewhat tempered by a history that includes a significant number of fluoroquinolone agents being withdrawn from the market because of significant and sometimes fatal toxicities.3,5 Over the past decade, the U.S. Food and Drug Administration (FDA) has released numerous safety warnings regarding serious adverse effects associated with currently available systemic fluoroquinolones with warnings issued to highlight the risk of tendonitis, tendon rupture, cardiac rhythm abnormalities, and central nervous system effects including seizures and peripheral neuropathy.7
The risk of peripheral neuropathy has been identified in the package insert of fluoroquinolones since 2004. However, in August 2013, because of a review by the FDA that identified the risk of this adverse effect to be rapid in the onset, potentially permanent, and often disabling, labeling changes were made to intensify and better characterize this warning.8 Symptoms of peripheral neuropathy described in the FDA report include “pain, burning, tingling, numbness, weakness, or a change in sensation to light touch, pain or temperature, or the sense of body position”.8 In a case-controlled study of men of 45–80 years old from 2001 to 2011, Etminan et al. validated that fluoroquinolone users were at increased risk of developing peripheral neuropathy.9 When stratified by the type of fluoroquinolone, the risk was similar. The exact mechanism(s) by which fluoroquinolones cause peripheral neuropathy and other adverse events remains yet to be elucidated.
Fluoroquinolones are known to disrupt the function of bacterial type-II topoisomerases, in particular, topoisomerase IV in Gram-positive and DNA gyrase in Gram-negative bacteria, although there are exceptions to this generalization.10,11 Topoisomerases are responsible for regulating the topology of DNA and are found across all three domains of life.12 Type-II topoisomerases alleviate positive supercoiling that occurs during transcription and replication and decatenate sister chromatids during replication termination by generating a transient double-stranded DNA break.13 These enzymes pass an intact DNA helix through the temporary double-stranded break (DSB) before ligating the DNA back together. Disruption of this mechanism has been used in both antimicrobial and anticancer therapeutics.10,14,15 Humans encode two isoforms of topoisomerase II: topoisomerase IIα and IIβ, which are known targets for anticancer agents.13,14,16 Both isoforms are found in the nucleus and the mitochondria.17−19
Oxidative stress and mitochondrial dysfunction have been detected in model systems in the presence of fluoroquinolones and have been offered as possible causes for several of the observed adverse events in patients.20−23 Authors of a recent study suggest that because bacterial type-II topoisomerases (similar to topoisomerase IV) are targeted by fluoroquinolones, the mitochondrial dysfunction implicated in some adverse events may be explained by interactions between fluoroquinolones and human topoisomerase IIα or IIβ in mitochondria.19 To that end, the authors provided evidence that at 80 μg/mL, ciprofloxacin may impair mtDNA replication.19 The authors also suggest that the effects on mitochondrial DNA replication are likely to be mediated by topoisomerase IIβ.19
Although the above study provides an interesting hypothesis, the only fluoroquinolone for which data were presented was ciprofloxacin. Additionally, very little data were provided to demonstrate whether this compound had a direct activity on human topoisomerase IIα or IIβ. Other studies in recent years have focused on human topoisomerase IIα and on either ciprofloxacin or moxifloxacin.24 Therefore, we set out to examine the activity of a series of fluoroquinolones at clinically relevant concentrations against both human topoisomerase IIα and IIβ.
Using a series of enzymological assays, we demonstrate that fluoroquinolones display a variable impact against human topoisomerase IIα and IIβ. For example, none of the compounds appeared to significantly increase the DNA cleavage with human topoisomerase IIα or IIβ, which suggests that these compounds do not mimic anticancer agents such as etoposide against the human enzymes. However, at higher drug concentrations, some fluoroquinolones, including gemifloxacin and ciprofloxacin, display inhibitory activity against human topoisomerase II-mediated DNA relaxation. A metabolite of ciprofloxacin also impacts topoisomerase II-mediated DNA relaxation, which indicates that some metabolites have activity against human enzymes. However, these in vitro effects are only seen at drug concentrations that are considerably more than an order of magnitude higher than the maximal patient plasma concentrations observed for the compounds. Thus, we propose that it is highly unlikely that patients experience high levels of drug required to see inhibition of human topoisomerase IIα and IIβ. Taken together, we hypothesize that while human topoisomerase II isoforms may have a role in fluoroquinolone toxicity, there are likely other targets also involved that need to be identified to find ways to protect patients from these serious toxicities.
Results and Discussion
Fluoroquinolones Do Not Increase DNA Cleavage by Human Topoisomerase IIα or IIβ
For the current study, we selected three clinically used fluoroquinolones (ciprofloxacin, levofloxacin, and moxifloxacin) along with one agent that has been removed from the market (gemifloxacin) and the original quinolone, nalidixic acid (Figure 1). To examine the impact of fluoroquinolones on human type-II topoisomerases, we performed a series of DNA cleavage assays. In these assays, purified topoisomerase IIα or IIβ is combined with a negatively supercoiled plasmid substrate (pBR322). The DNA cleavage is monitored by observing nicked and linearized plasmid molecules in an agarose gel. A previous study has shown that ciprofloxacin increases DNA cleavage by topoisomerase IV.25
Figure 1.

Structures of quinolones. The precursor quinolone, nalidixic acid, along with four prominent fluoroquinolones.
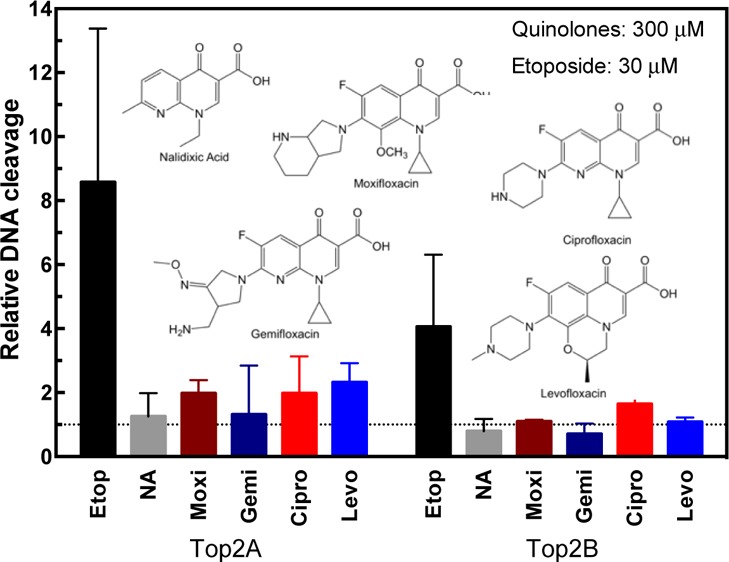
As seen in Figure 2, etoposide leads to an increase in both DNA nicks [single-strand break (SSB)] and DSBs. Drug concentrations for ciprofloxacin between 3 and 30 μM (∼1–10 μg/mL, see Table 1) are expected in patient serum samples; therefore, we tested a range up to 300 μM to determine whether there would be an effect on the enzyme. As seen from the quantified data, none of the fluoroquinolones caused a significant increase in DNA cleavage with either isoforms of topoisomerase II (Figure 2, lower panel). This finding is consistent with the previous data on ciprofloxacin and moxifloxacin from human enzymes.24 The nonfluorinated quinolone, nalidixic acid, also did not cause a significant increase in DNA cleavage. The lack of an increase in DNA cleavage supports the hypothesis that these agents do not act similar to an etoposide on human topoisomerase II, which prevents the enzyme from ligating cleaved DNA. This mechanism is referred to as “poisoning” and is found among several anticancer agents including etoposide.14 However, there are other agents that affect human topoisomerases without poisoning the DNA cleavage.16 Therefore, we explored the ability of fluoroquinolones to inhibit supercoiled DNA relaxation activity of the enzymes to determine whether they function as catalytic inhibitors of human topoisomerase IIα or IIβ.
Figure 2.

Plasmid DNA cleavage by topoisomerase IIα and IIβ in the presence of quinolones. A: Plasmid DNA was incubated in the absence (−T) or presence (+T) of topoisomerase IIα (Top2A, top gel) or β (Top2B, bottom gel). Linearized plasmid (Lin) is shown in the first lane. Reactions were incubated with 30 or 300 μM etoposide (Etop) as a positive control. Reactions were performed with 3, 30, or 300 μM of nalidixic acid (NA), moxifloxacin (Moxi), gemifloxacin (Gemi), ciprofloxacin (Cipro), or levofloxacin (Levo). Positions of SSB, DSB, and supercoiled (SC) DNA are denoted at left. Representative gels are shown, and experiments were performed at least three times. B: quantification of double-strand DNA cleavage relative to cleavage in the absence of quinolones is shown for both topoisomerase II isoforms. As a control, etoposide at 30 μM is shown compared with 300 μM of the quinolones. Dotted line represents the level of cleavage in the presence of dimethyl sulfoxide (DMSO). Error bars represent the standard deviation of three or more independent experiments.
Table 1. Plasmid DNA Relaxation Inhibition and Maximum Plasma Concentrations of Selected Fluoroquinolones.
| relaxation
inhibition in purified systema |
|||
|---|---|---|---|
| Top2A (μg/mL) | Top2B (μg/mL) | maximum plasma concentrationsb (μg/mL) | |
| gemifloxacin | 24 (50 μM) | 24 (50 μM) | 1.61 ± 0.51 |
| moxifloxacin | 6.1 ± 1.3 | ||
| levofloxacin | 9.3 ± 1.6 | ||
| ciprofloxacin | 110 (300 μM) | 110 (300 μM) | 5.4 |
| desethylene-ciprofloxacin | 110 (300 μM) | 74–110 (200–300 μM) | <0.81 |
Some Fluoroquinolones Inhibit Topoisomerase II-Mediated DNA Relaxation at High Concentrations
We also examined the ability of fluoroquinolones to impair the relaxation of negatively supercoiled pBR322 plasmid using the same concentration series as used in the DNA cleavage experiments. In the presence of ATP, human topoisomerase IIα and IIβ relax the plasmid, which is seen in Figure 3 by the conversion of supercoiled (SC) plasmid to relaxed (Rel) plasmid in the second lane (+T). Among the fluoroquinolones, the inhibition of relaxation is seen at concentrations around 300 μM only with gemifloxacin and ciprofloxacin. Interestingly, gemifloxacin was able to completely inhibit relaxation by 300 μM with both enzyme isoforms. Ciprofloxacin was only able to mildly inhibit relaxation at the same concentration, which is evidenced by the presence of lower DNA bands (topoisomers).
Figure 3.

Plasmid DNA relaxation mediated by topoisomerase IIα and IIβ in the presence of selected quinolones. Plasmid DNA was incubated with ATP in the absence (−T) or presence (+T) of topoisomerase IIα (Top2A, top gel) or β (Top2B, bottom gel). Reactions were performed with 3, 30, or 300 μM of nalidixic acid (NA), moxifloxacin (Moxi), gemifloxacin (Gemi), ciprofloxacin (Cipro), or levofloxacin (Levo). Positions of relaxed (Rel) and supercoiled (SC) plasmid are shown at left. Representative gels are shown, and experiments were performed at least three times.
To further test the impact on relaxation, an additional series was performed at a range of drug concentrations from 10 to 300 μM with gemifloxacin, ciprofloxacin, and a metabolite of ciprofloxacin, desethylene ciprofloxacin.26 Even though fluoroquinolones are active as administered, they do undergo metabolism, and we explored whether this metabolite displayed activity against human topoisomerase II. As seen in Figure 4A, ciprofloxacin and desethylene-ciprofloxacin display limited ability to inhibit relaxation below 200–300 μM. The impact of ciprofloxacin in the 200–300 μM range is consistent with the recently reported results at 80 μg/mL, which corresponds to ∼217 μM.19 In contrast, gemifloxacin displays a significant inhibition of relaxation at 50–100 μM with complete inhibition by 200 μM.
Figure 4.

Inhibition of relaxation of plasmid DNA in the presence of increasing concentrations of fluoroquinolones. A: Plasmid DNA was incubated with ATP in the absence (−T) or presence (+T) of topoisomerase IIα (Top2A, top gel) or IIβ (Top2B, bottom gel). Reactions were performed with 10–300 μM of gemifloxacin (Gemi), ciprofloxacin (Cipro), or desethylene-ciprofloxacin (D-Cipro). Positions of relaxed (Rel) and supercoiled (SC) plasmid are shown on the left. Representative gels are shown, and experiments were performed at least three times. B: Structure of desethylene-ciprofloxacin. C: Quantified plasmid double-stranded DNA cleavage with topoisomerase IIα (Top2A) or IIβ (Top2B) in the presence of 30 μM etoposide (Etop, black bars) or 300 μM desethylene-ciprofloxacin (D-Cipro, red bars) is shown. Dotted line represents the level of DNA cleavage in the presence of DMSO. Error bars represent the standard deviation of three or more independent experiments.
Although fluoroquinolones are shown to act as poisons (i.e., stabilize DNA cleavage) with bacterial type-II topoisomerases, they do not display the same ability against human type-II topoisomerases in the concentration ranges tested. As seen above, gemifloxacin does not poison either human isoform but is able to inhibit relaxation by both enzymes at >50 μM. This suggests that the mechanism of fluoroquinolone-induced inhibition for human enzymes is not the same as the mechanism seen with bacterial enzymes, which will be discussed further below. There are catalytic inhibitors of human type-II topoisomerases that block enzyme functions without stabilizing DNA cleavage.14,27,28 These often involve interactions with the ATPase domain, but they are not necessarily limited to that mechanism. Additional studies of gemifloxacin will be required to determine the mechanism behind the inhibition of human topoisomerase IIα and IIβ. Because gemifloxacin has been removed from the market, we did not pursue additional characterization of this compound against human enzymes.
Desethylene-Ciprofloxacin Does Not Poison Human Topoisomerase IIα or IIβ
To examine whether desethylene-ciprofloxacin (Figure 4B) could serve as a poison of human topoisomerase II, we performed DNA cleavage assays with this compound. As seen in Figure 4C, desethylene-ciprofloxacin does not increase double-stranded DNA cleavage by human topoisomerase IIα or IIβ even in the presence of 300 μM of the compound. The same results were observed at lower desethylene-ciprofloxacin concentrations (data not shown).
Although these results clarify that fluoroquinolones can have an impact on the overall catalytic activity of human topoisomerase IIα and IIβ, it is unclear whether the concentrations observed are clinically relevant. To that end, Table 1 summarizes both the results from plasmid DNA relaxation in a purified system discussed above and plasma concentrations reported in the package inserts for these drugs.29−32 As seen in Table 1, gemifloxacin at 24 μg/mL and above is expected to have a significant impact on relaxation, which implies there would be a lower impact at concentrations below this level. Conversely, ciprofloxacin and desethylene-ciprofloxacin would not be expected to have a significant impact on relaxation until levels above 74–110 μg/mL. As seen in Table 1, maximum plasma concentrations fall in the 1–10 μg/mL range. Plasma concentrations of these compounds would need to be 15–100-fold higher, depending on the fluoroquinolone, to observe an impact on topoisomerase II activity. These data indicate that the majority of patients treated with fluoroquinolones are highly unlikely to achieve plasma concentrations of these drugs that are high enough to cause significant disruption of topoisomerase II activity.
Another observation is in order regarding these data. Structural and biochemical studies of the binding site of fluoroquinolones on topoisomerase IV indicate that a critical enzyme/drug interaction occurs that involves a metal-ion bridge.33,34 According to the data, this interaction involves two key residues in the bacterial enzymes.33,34 An invariant Ser (sometimes Thr) and Asp/Glu are found in GyrA and ParC/GrlA, which are subunits of the heterotetrameric (A2:B2) structure of DNA gyrase and topoisomerase IV, respectively. Absence of these residues can contribute to the resistance to the poisoning of bacterial topoisomerases by fluoroquinolones. This enzyme/drug interaction mechanism is also supported by biochemical evidence with DNA gyrase, which suggests that this is a generalized mechanism for bacterial type-II topoisomerases.35,36
As seen in Figure 5, these residues are shared among representative bacterial species that are known to be susceptible to fluoroquinolones. As has been pointed out previously, human type-II topoisomerases lack these residues.24 To this end, Aldred et al. demonstrated that mutations at these positions in human topoisomerase IIα render this isoform susceptible to poisoning by ciprofloxacin.24 Thus, based on sequence analysis, we would not expect wild-type human topoisomerase IIα and IIβ to be poisoned by fluoroquinolones if the mechanism of poisoning uses the same interactions and metal-ion bridges found in bacterial enzymes. Our data demonstrate among a group of clinically approved agents that human topoisomerase IIα and IIβ are not poisoned by these compounds, as predicted by the analysis of the amino acid sequence data.
Figure 5.

Alignment of selected enzyme sequence at the region known to bind to quinolones. Highlighted in red are the positions of the Ser and Asp/Glu residues. Shown are sequences for DNA Gyrase GyrA and topoisomerase IV ParC/GrlA subunits from Escherichia coli (Ec), Staphylococcus aureus (Sa), Streptococcus pneumonia (Sp), Pseudomonas aeruginosa (Pa), and Haemophilus influenza (Hi). Also shown are sequences for Homo sapiens topoisomerase II (hTIIα and hTIIβ), which lack the residues involved in the quinolone salt bridge.
Conclusions
Fluoroquinolones represent an important class of widely prescribed pharmaceuticals with a broad range of activity in both Gram-positive and Gram-negative infections. Unfortunately, a small percentage of patients experience severe adverse reactions to these agents including but not limited to peripheral neuropathies, tendon rupture, heart rhythm abnormalities, and various mental health side effects, as discussed above. Evidence suggests that mitochondrial dysfunction may play a role in some of these events such as the neuropathies and issues related to muscles.20−23 To that end, it has been proposed that fluoroquinolone-mediated disruption of human topoisomerase IIα and IIβ, which are present in the mitochondria, could be contributing to these toxicities.19
On the basis of the evidence we have provided using three clinically relevant fluoroquinolones, we were unable to observe an impact on human topoisomerase IIα or IIβ at clinically relevant drug concentrations. Our results are consistent with a recent study that indicated that human topoisomerase IIα and β can be inhibited by 80 μg/mL ciprofloxacin, which as they demonstrate could lead to disruption of mitochondrial DNA replication.19 Given the high concentration used in this previous study, it is unlikely that patients will achieve sufficient drug levels given the clinical data to have this type of impact on topoisomerase II in the mitochondria.
The adverse events associated with fluoroquinolones are significant and warrant further investigation. Although it is possible that human topoisomerase IIα and/or IIβ could play a role in fluoroquinolone adverse events, the evidence presented here suggests that inhibition of human topoisomerase II is not likely to explain the types of adverse events observed clinically. Thus, we propose that additional potential drug targets should be identified and explored. For example, previous studies demonstrating increased reactive oxygen species and decreased mitochondrial protein expression in response to fluoroquinolones provide valuable leads for identifying targets in humans cells.22,37 Therefore, efforts to study these adverse events should focus on identifying other enzymes and pathways impacted by these compounds.
Methods
Enzymes and Materials
Wild-type human topoisomerase II (TOP2A) and IIβ (TOP2B) were expressed in Saccharomyces cerevisiae JEL1top1 cells and purified as described previously.28 Both enzymes contain a C-terminal His-tag for purification purposes. The enzymes were stored at −80 °C as a 1 mg/mL (4 μM for wild type) stock in 50 mM Tris-HCl, pH 7.7, 0.1 mM EDTA, 750 mM KCl, 5% glycerol, and <40 μM dithiothreitol (DTT) (carried from the enzyme preparation).
Negatively supercoiled pBR322 DNA was prepared from E. coli using a Plasmid Mega Kit (Qiagen) with some modification of the manufacturer’s protocol. Etoposide (Sigma) and quinolones (Cayman Chemical and LKT) were stored at 4 °C as 20 or 30 mM stock solutions in 100% DMSO, respectively.
Topoisomerase IIα-Mediated Relaxation of Plasmid DNA
Reaction mixtures contained 22 nM wild-type topoisomerase IIα, 5 nM negatively supercoiled pBR322 DNA, and 1 mM ATP in 20 μL of 10 mM Tris-HCl, pH 7.9, 175 mM KCl, 0.1 mM Na2EDTA, 5 mM MgCl2, and 2.5% glycerol. Assays were started by the addition of enzyme, and DNA relaxation mixtures were incubated for 15 min at 37 °C. DNA relaxation reactions were carried out in the presence of 1% DMSO (control), etoposide, or increasing concentrations of fluoroquinolones. DNA relaxation was stopped by the addition of 3 μL of stop solution (77.5 mM Na2EDTA, 0.77% SDS). Samples were mixed with 2 μL of agarose gel-loading buffer, heated for 2 min at 45 °C, and subjected to gel electrophoresis in 1% TBE agarose gels. The agarose gel was then stained in ethidium bromide for 15–30 min. DNA bands were visualized by UV light using a Bio-Rad ChemiDoc MP Imaging System and Image Lab Software (Hercules, CA). DNA relaxation was monitored by the conversion of supercoiled plasmid DNA to relaxed topoisomers.
Topoisomerase IIα-Mediated Cleavage of Plasmid DNA
Plasmid DNA cleavage reactions were performed using the procedure of Fortune and Osheroff.38 Reaction mixtures contained 150 nM of wild-type TOP2A or TOP2B and 5 nM negatively supercoiled pBR322 DNA in 20 μL of 10 mM Tris-HCl, pH 7.9, 100 mM KCl, 1 mM EDTA, 5 mM MgCl2, and 2.5% glycerol. Final reaction mixtures contained <1 μM DTT, which represents the residual DTT carried along from the enzyme preparation. Unless stated otherwise, assays were started by the addition of enzyme, and DNA cleavage mixtures were incubated for 6 min at 37 °C. DNA cleavage reactions were carried out in the absence of compound (1% DMSO solution as a control) or in the presence of etoposide, or increasing concentrations of fluoroquinolones DNA cleavage complexes were trapped by the addition of 2 μL of 5% SDS followed by 2 μL of 250 mM Na2EDTA, pH 8.0. Proteinase K was added (2 μL of a 0.8 mg/mL solution), and reaction mixtures were incubated for 30 min at 37 °C to digest topoisomerase IIα. Samples were mixed with 2 μL of agarose gel loading buffer (60% sucrose in 10 mM Tris-HCl, pH 7.9), heated for 2 min at 45 °C, and subjected to electrophoresis in 1% agarose gels in 40 mM Trisacetate, pH 8.3, and 2 mM EDTA containing 0.5 μg/mL ethidium bromide. Double-stranded DNA cleavage was monitored by the conversion of negatively supercoiled plasmid DNA to linear molecules. DNA bands were visualized by UV light and quantified using a Bio-Rad ChemiDoc MP Imaging System and Image Lab Software (Hercules, CA). Results were plotted using GraphPad Prism 6 (La Jolla, CA). The relative DNA cleavage was calculated by setting the DNA cleavage levels in the presence of DMSO to 1.
Acknowledgments
We thank Drs. Neil Osheroff and Elizabeth Gibson for their critical review of the manuscript and many helpful discussions. We thank Jo Ann Byl for providing the moxifloxacin. We also thank Dr. James Dewar for helpful discussions. We thank Dr. Anni Andersen for providing the expression vectors for TOP2A and TOP2B. This work was supported by funds from the Lipscomb University College of Pharmacy and Health Sciences and the Center for Science and Culture. C.A.F. and K.G.H. were participants in the Pharmaceutical Sciences Summer Research Program of the Lipscomb University College of Pharmacy.
Glossary
Abbreviations
- DSB
double-stranded DNA break
- EDTA
ethylenediaminetetraacetic acid
- FDA
U.S. Food and Drug Administration
- SSB
single-stranded DNA break
- TOP2
topoisomerase II
The authors declare no competing financial interest.
References
- Emmerson A. M.; Jones A. M. The quinolones: decades of development and use. J. Antimicrob. Chemother. 2003, 51, 13–20. 10.1093/jac/dkg208. [DOI] [PubMed] [Google Scholar]
- Mitscher L. A. Bacterial topoisomerase inhibitors: quinolone and pyridone antibacterial agents. Chem. Rev. 2005, 105, 559–592. 10.1021/cr030101q. [DOI] [PubMed] [Google Scholar]
- Bisacchi G. S. Origins of the Quinolone Class of Antibacterials: An Expanded “Discovery Story”. J. Med. Chem. 2015, 58, 4874–4882. 10.1021/jm501881c. [DOI] [PubMed] [Google Scholar]
- Andersson M. I.; MacGowan A. P. Development of the quinolones. J. Antimicrob. Chemother. 2003, 51, 1–11. 10.1093/jac/dkg212. [DOI] [PubMed] [Google Scholar]
- Appelbaum P. C.; Hunter P. A. The fluoroquinolone antibacterials: past, present and future perspectives. Int. J. Antimicrob. Agents 2000, 16, 5–15. 10.1016/s0924-8579(00)00192-8. [DOI] [PubMed] [Google Scholar]
- CDC Outpatient Antibiotic Prescriptions—United States, 2016. https://www.cdc.gov/antibiotic-use/community/programs-measurement/state-local-activities/outpatient-antibiotic-prescriptions-US-2016.html (accessed Dec 3, 2018).
- FDA Drug Safety Communication: FDA updates warnings for oral and injectable fluoroquinolone antibiotics due to disabling side effects. https://www.fda.gov/Drugs/DrugSafety/ucm511530.htm (accessed Nov 30, 2018).
- “FDA Drug Safety Communication: FDA advises restricting fluoroquinolone antibiotic use for certain uncomplicated infections; warns about disabling side effects that can occur together.” http://www.fda.gov/Drugs/DrugSafety/ucm500143.htm (accessed Dec 3, 2018).
- Etminan M.; Brophy J. M.; Samii A. Oral fluoroquinolone use and risk of peripheral neuropathy: a pharmacoepidemiologic study. Neurology 2014, 83, 1261–1263. 10.1212/wnl.0000000000000846. [DOI] [PubMed] [Google Scholar]
- Aldred K. J.; Kerns R. J.; Osheroff N. Mechanism of quinolone action and resistance. Biochemistry 2014, 53, 1565–1574. 10.1021/bi5000564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson E. G.; Ashley R. E.; Kerns R. J.; Osheroff N.. Bacterial Type II Topoisomerases and Target-Mediated Drug Resistance. In Antimicrobial Resistance in the 21st Century. Emerging Infectious Diseases of the 21st Century, Fong I., Shlaes D., Drlica K., Eds.; Springer: Cham, 2018. [Google Scholar]
- Champoux J. J. DNA topoisomerases: structure, function, and mechanism. Annu. Rev. Biochem. 2001, 70, 369–413. 10.1146/annurev.biochem.70.1.369. [DOI] [PubMed] [Google Scholar]
- Nitiss J. L. DNA topoisomerase II and its growing repertoire of biological functions. Nat. Rev. Cancer 2009, 9, 327–337. 10.1038/nrc2608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy M. B.; Mercer S. L.; Deweese J. E.. Inhibitors and Poisons of Mammalian Type II Topoisomerases. In Advances in Molecular Toxicology, Fishbein J. C.; Heilman J., Eds.; Academic Press: Cambridge, MA, 2017; Vol. 11, pp 203–240. [Google Scholar]
- Deweese J. E.; Osheroff N. The DNA cleavage reaction of topoisomerase II: wolf in sheep’s clothing. Nucleic Acids Res. 2008, 37, 738–748. 10.1093/nar/gkn937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nitiss J. L. Targeting DNA topoisomerase II in cancer chemotherapy. Nat. Rev. Cancer 2009, 9, 338–350. 10.1038/nrc2607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H.; Zhang Y.-W.; Yasukawa T.; Dalla Rosa I.; Khiati S.; Pommier Y. Increased negative supercoiling of mtDNA in TOP1mt knockout mice and presence of topoisomerases II and II in vertebrate mitochondria. Nucleic Acids Res. 2014, 42, 7259–7267. 10.1093/nar/gku384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pommier Y.; Sun Y.; Huang S.-y. N.; Nitiss J. L. Roles of eukaryotic topoisomerases in transcription, replication and genomic stability. Nat. Rev. Mol. Cell Biol. 2016, 17, 703–721. 10.1038/nrm.2016.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hangas A.; Aasumets K.; Kekäläinen N. J.; Paloheinä M.; Pohjoismäki J. L.; Gerhold J. M.; Goffart S. Ciprofloxacin impairs mitochondrial DNA replication initiation through inhibition of Topoisomerase 2. Nucleic Acids Res 2018, 46, 9625–9636. 10.1093/nar/gky793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernard-Beaubois K.; Hecquet C.; Hayem G.; Rat P.; Adolphe M. In vitro study of cytotoxicity of quinolones on rabbit tenocytes. Cell Biol. Toxicol. 1998, 14, 283–292. 10.1023/a:1007435025616. [DOI] [PubMed] [Google Scholar]
- Lowes D. A.; Wallace C.; Murphy M. P.; Webster N. R.; Galley H. F. The mitochondria targeted antioxidant MitoQ protects against fluoroquinolone-induced oxidative stress and mitochondrial membrane damage in human Achilles tendon cells. Free Radic. Res. 2009, 43, 323–328. 10.1080/10715760902736275. [DOI] [PubMed] [Google Scholar]
- Kalghatgi S.; Spina C. S.; Costello J. C.; Liesa M.; Morones-Ramirez J. R.; Slomovic S.; Molina A.; Shirihai O. S.; Collins J. J. Bactericidal antibiotics induce mitochondrial dysfunction and oxidative damage in Mammalian cells. Sci. Transl. Med. 2013, 5, 192ra85. 10.1126/scitranslmed.3006055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ilgin S.; Can O. D.; Atli O.; Ucel U. I.; Sener E.; Guven I. Ciprofloxacin-induced neurotoxicity: evaluation of possible underlying mechanisms. Toxicol. Mech. Methods 2015, 25, 374–381. 10.3109/15376516.2015.1026008. [DOI] [PubMed] [Google Scholar]
- Aldred K. J.; Schwanz H. A.; Li G.; McPherson S. A.; Turnbough C. L. Jr.; Kerns R. J.; Osheroff N. Overcoming target-mediated quinolone resistance in topoisomerase IV by introducing metal-ion-independent drug-enzyme interactions. ACS Chem. Biol. 2013, 8, 2660–2668. 10.1021/cb400592n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson V. E.; Gootz T. D.; Osheroff N. Topoisomerase IV catalysis and the mechanism of quinolone action. J. Biol. Chem. 1998, 273, 17879–17885. 10.1074/jbc.273.28.17879. [DOI] [PubMed] [Google Scholar]
- Bergan T.; Thorsteinsson S. B.; Rohwedder R.; Scholl H. Elimination of ciprofloxacin and three major metabolites and consequences of reduced renal function. Chemotherapy 2009, 35, 393–405. 10.1159/000238702. [DOI] [PubMed] [Google Scholar]
- Roca J.; Ishida R.; Berger J. M.; Andoh T.; Wang J. C. Antitumor bisdioxopiperazines inhibit yeast DNA topoisomerase II by trapping the enzyme in the form of a closed protein clamp. Proc. Natl. Acad. Sci. U.S.A. 1994, 91, 1781–1785. 10.1073/pnas.91.5.1781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Regal K. M.; Mercer S. L.; Deweese J. E. HU-331 Is a Catalytic Inhibitor of Topoisomerase IIα. Chem. Res. Toxicol. 2014, 27, 2044–2051. 10.1021/tx500245m. [DOI] [PubMed] [Google Scholar]
- Cipro (ciprofloxacin hydrochloride) [package insert]; Bayer HealthCare: West Haven, CT, 2004. [Google Scholar]
- Levaquin (levofloxacin) [package insert]; Janssen Pharmaceuticals, Inc.: Titusville, NJ, 2007. [Google Scholar]
- Factive (gemifloxacin mesylate) [package insert]; Oscient Pharmaceuticals: Waltham, MA, 2008. [Google Scholar]
- Avelox (moxifloxacin hydrochloride) [package insert]; Bayer HealthCare: Whippany: New Jersey, 2016. [Google Scholar]
- Wohlkonig A.; Chan P. F.; Fosberry A. P.; Homes P.; Huang J.; Kranz M.; Leydon V. R.; Miles T. J.; Pearson N. D.; Perera R. L.; Shillings A. J.; Gwynn M. N.; Bax B. D. Structural basis of quinolone inhibition of type IIA topoisomerases and target-mediated resistance. Nat. Struct. Mol. Biol. 2010, 17, 1152–1153. 10.1038/nsmb.1892. [DOI] [PubMed] [Google Scholar]
- Aldred K. J.; McPherson S. A.; Turnbough C. L. Jr.; Kerns R. J.; Osheroff N. Topoisomerase IV-quinolone interactions are mediated through a water-metal ion bridge: mechanistic basis of quinolone resistance. Nucleic Acids Res. 2013, 41, 4628–4639. 10.1093/nar/gkt124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aldred K. J.; Blower T. R.; Kerns R. J.; Berger J. M.; Osheroff N. Fluoroquinolone interactions withMycobacterium tuberculosisgyrase: Enhancing drug activity against wild-type and resistant gyrase. Proc. Natl. Acad. Sci. U.S.A. 2016, 113, E839–E846. 10.1073/pnas.1525055113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashley R. E.; Lindsey R. H. Jr.; McPherson S. A.; Turnbough C. L. Jr.; Kerns R. J.; Osheroff N. Interactions between Quinolones and Bacillus anthracis Gyrase and the Basis of Drug Resistance. Biochemistry 2017, 56, 4191–4200. 10.1021/acs.biochem.7b00203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nadanaciva S.; Dillman K.; Gebhard D. F.; Shrikhande A.; Will Y. High-content screening for compounds that affect mtDNA-encoded protein levels in eukaryotic cells. J. Biomol. Screening 2010, 15, 937–948. 10.1177/1087057110373547. [DOI] [PubMed] [Google Scholar]
- Fortune J. M.; Osheroff N. Merbarone Inhibits the Catalytic Activity of Human Topoisomerase IIα by Blocking DNA Cleavage. J. Biol. Chem. 1998, 273, 17643–17650. 10.1074/jbc.273.28.17643. [DOI] [PubMed] [Google Scholar]