

ABSTRACT
Background
Total serum transforming growth factor‐beta 1 (tsTGF‐β1) is increased in patients with Marfan syndrome (MFS), but it has not been assessed in thoracic aortic aneurysm and dissection (TAAD), Loeys‐Dietz syndrome (LDS), and bicuspid aortic valve disease (BAVD).
Hypothesis
tsTGF‐β1 is increased in genetic aortic syndromes including TAAD, LDS, MFS, and BAVD.
Methods
We measured tsTGF‐β1 and performed sequencing of the genes FBN1, TGFBR1, and TGFBR2 in 317 consecutive patients with suspected or known genetic aortic syndrome (167 men, 150 women; mean age 43 ± 14 years). TAAD was diagnosed in 20, LDS in 20, MFS in 128, and BAVD in 30 patients, and genetic aortic syndrome was excluded in 119 patients.
Results
Elevated tsTGF‐β1 levels were associated with causative gene mutations (P = 0.008), genetic aortic syndrome (P = 0.009), and sporadic occurrence of genetic aortic syndrome (P = 0.048), whereas only genetic aortic syndrome qualified as an independent predictor of tsTGF‐β1 (P = 0.001). The tsTGF‐β1 levels were elevated in FBN1 and NOTCH1 mutations vs patients without mutations (both P = 0.004), and in NOTCH1 mutations vs ACTA2/MYH11 mutations (P = 0.015). Similarly, tsTGF‐β1 levels were elevated in MFS (P = 0.003) and in BAVD (P = 0.006) vs patients without genetic aortic syndrome. In contrast to specific clinical features of MFS, FBN1 in‐frame mutations (P = 0.019) were associated with increased tsTGF‐β1 levels.
Conclusions
tsTGF‐β1 is elevated in the entire spectrum of genetic aortic syndromes. However, gradual differences in the increases of tsTGF‐β1 levels may mirror different degrees of alteration of tsTGF‐β1 signaling in different genetic aortic syndromes.
Introduction
Marfan syndrome (MFS) is an autosomal‐dominant inherited disease of connective tissue caused by mutations of the gene fibrillin‐1 (FBN1), which encodes fibrillin‐1 monomers of extracellular microfibrils.1 The diagnosis of MFS is based on criteria of the revised Ghent nosology.2 Intact fibrillin‐1 controls the bioavailability of transforming growth factor‐beta 1 (TGF‐β1) through binding of the large latent complex in the extracellular matrix. Failure of defective fibrillin‐1 to bind latent TGF‐beta–binding protein (LTBP)3 results in increased TGF‐β1 activity, which was shown in both tissue and serum of mice with MFS.4 Patients with MFS also exhibited increased total TGF‐beta1 levels in serum (tsTGF‐β1),5, 6, 7, 8, 9 and their successful treatment of aortic aneurysm with beta‐adrenergic blockers (BAB), angiotensin receptor blockers (ARB), or angiotensin‐converting enzyme inhibitors (ACEi) was shown to result in commensurate decreases of tsTGF‐β1.5, 6, 7, 8, 9 These data nourish hope that tsTGF‐β1 may serve as a biomarker of MFS.10 Interestingly, TGF‐β1 signaling is also involved in other genetic aortic syndromes (GAS), such as Loeys‐Dietz syndrome (LDS) caused by mutations of TGFBR1, TGFBR2, or TGFB2,11, 12 isolated thoracic aortic aneurysm and dissection (TAAD) caused by mutations in ACTA2, SMAD3, or MYH11,13 and bicuspid aortic valve disease (BAVD), which in rare cases is caused by mutations in the NOTCH1 gene.14 Therefore, we hypothesized that tsTGF‐β1 was elevated in the entire spectrum of GAS comprising TAAD, LDS, MFS, and BAVD as compared to patients without GAS.
Methods
Patients
Between March 1, 2010 and February 28, 2013, we obtained blood samples to measure tsTGF‐β1 in 351 consecutive patients age >16 years who we evaluated for suspected or known GAS. We excluded 17 patients in whom we diagnosed genetic syndromes other than TAAD, LDS, MFS, and BAVD, and another 17 patients who did not complete clinical and molecular diagnostic workup. Thus, 317 patients constituted our study group. All patients had complete assessment of manifestations of MFS2, 15 and LDS,16, 17 including sequencing of FBN1, TGFBR1, and TGFBR2. We additionally sequenced ACTA2, COL3A1, MYH11, NOTCH1, SLC2A10, SMAD3, and TGFB2 genes in 22 patients with clinical features suggestive of mutations of the respective genes. The study was approved by the Hamburg ethical board.
Enzyme‐Linked Immunosorbent Assay for TGF‐β1
We collected venous blood samples in a serum separator tube and allowed samples to clot for 30 minutes at room temperature. For complete release of tsTGF‐β1, we incubated the probes overnight at 2 °C to 8 °C before centrifuging for 20 minutes at 2000 g. We removed the serum and stored it at −80 °C. We determined tsTGF‐β1 levels by using the Quantikine Human TGF‐beta 1 Immunoassay kit according to the manufacturer´s protocol (R&D Systems, Minneapolis, MN; catalog number DB100B). We ran all samples in duplicate. The detection limit of the assay was 4.61 pg/mL.
DNA Analysis
We extracted DNA from ethylenediaminetetraacetic acid blood samples, and we amplified and sequenced the entire coding and flanking intronic sequences of TGFBR1, TGFBR2, and FBN1 in all 317 patients, and the ACTA2, COL3A1, MYH11, NOTCH1, SLC2A10, SMAD3, and TGFB2 genes in 22 patients as detailed previously.11, 18, 19 To detect possible (partial) gene deletions or duplications, we used multiplex ligation‐dependent probe amplification analysis as described previously.11, 18, 19 All 123 FBN1 mutations fulfilled criteria for causality (Table 1).2
Table 1.
Correlation of Mutation Characteristics With tsTGF‐β1 Levels (ng/mL) in 123 Patients With Causative FBN1 Mutation
| FBN1 Characteristic | ||||
|---|---|---|---|---|
| Variable | Absent | Present | Spearman Correlation Coefficient (ρ) | P |
| PTC mutations | 42 | 37 | −0.314 | 0.005 |
| Splicing mutations | 109 | 14 | 0.026 | 0.773 |
| Any mutation affecting Cys | 70 | 39 | 0.064 | 0.511 |
| Missense mutation affecting Cys | 30 | 39 | −0.025 | 0.839 |
| Any mutation in cbEGF | 98 | 11 | 0.003 | 0.976 |
| Missense mutation in cbEGF | 58 | 11 | −0.038 | 0.758 |
| Any mutation in LTBP‐bd | 106 | 3 | 0.002 | 0.985 |
| Missense mutation in LTBP‐bd | 67 | 2 | 0.121 | 0.320 |
| Any mutation in exons 24–32 | 93 | 16 | −0.023 | 0.815 |
| Missense mutation in exons 24–32 | 59 | 10 | 0.054 | 0.661 |
Abbreviations: cbEGF, calcium‐binding epidermal growth factor; Cys, cysteine; LTBP‐bd, latent transforming growth factor‐β–binding protein; PTC, premature termination codon; tsTGF‐β1, total serum transforming growth factor‐beta 1.
Criteria of causality of the mutation were amino acid substitutions affecting a cysteine residue in 40, in‐frame and out‐of‐frame deletions/duplications/insertions in 22, other missense mutations in 18, nonsense mutations in 17, splice‐site alterations in 14, and mutations in a highly conserved residue in an epidermal growth factor‐like domain in 12 patients.
Final Diagnoses
We diagnosed MFS with a causative FBN1 mutation, aortic root dilatation ≥2 Z‐scores, ectopia lentis, a systematic score ≥7 points or a family history of MFS using Ghent criteria,2 LDS with a TGFBR1 or TGFBR2 mutation and ≥1 clinical manifestation of LDS, TAAD according to ACMG,20 and BAVD with a bicuspid aortic valve on echocardiography and exclusion of MFS and LDS. We designated the presence of any of these syndromes as GAS and absence of these syndromes as absence of GAS.
Clinical Variables
We employed standardized history, physical examination, transthoracic echocardiography and magnetic resonance imaging (MRI) to assess independent variables at the time of measuring tsTGF‐β1. We present Ghent systemic score points separately as skeletal scores and as nonskeletal scores, which comprised all nonskeletal features from the list of systemic Ghent features. We used stringent definitions for all clinical variables19, 21, 22, 23 and medications24 as described in the footnotes of Table 2 and Table 3, and as detailed previously.19 A list of detailed methods has been reviewed and will be made available upon request.
Table 2.
Total TGF‐β1 Serum Levels According to Patient Characteristics
| Total TGF‐β1 Serum Level, ng/mL | ||||
|---|---|---|---|---|
| Patient Characteristics | No. of Patientsa | Mean ± SD | Min – Max | P b |
| Causative gene mutation | 0.008 | |||
| No causative mutation | 159 | 34.7 ± 29.6 | 0.01–182 | |
| ACTA2, MYH11 c | 8 (4/4) | 39.5 ± 41.1 | 14.5–139.1 | |
| TGFBR1, TGFBR2, TGFB2 d | 20 (11/9) | 41.4 ± 26.7 | 6.4–102.8 | |
| FBN1 | 123 (54/69) | 45.2 ± 35.1 | 1.2–249.6 | |
| NOTCH1 | 7 (0/7) | 59 ± 9.8 | 39.9–69.6 | |
| Final diagnosis | 0.009 | |||
| No genetic aortic syndrome | 119 | 32 ± 26.8 | 1.5–135.1 | |
| Thoracic aortic aneurysm or dissection | 20 (14/6) | 39.1 ± 31.6 | 1.5–139.1 | |
| Loeys‐Dietz syndrome | 20 (11/9) | 41.4 ± 26.7 | 6.4–102.8 | |
| Marfan syndrome | 128 (55/73) | 44.4 ± 34.9 | 1.2–249.6 | |
| Bicuspid aortic valve disease | 30 (6/24) | 50.8 ± 37.4 | 0.02–182 | |
| Previous cardiovascular interventione | 0.748 | |||
| None | 160 (16.1%) | 39.3 ± 32.5 | 1.2–182 | |
| Aortic intervention | 142 (44.8%) | 39.7 ± 31.4 | 1.7–249.6 | |
| Isolated heart valve intervention | 15 (4.7%) | 46.3 ± 36.1 | 0.2–110.4 | |
| Aortic dissection | ||||
| None | 260 (82%) | 40.3 ± 30.8 | 0.2–182 | |
| History of aortic dissection | 57 (18%) | 38 ± 37.8 | 2.4–249.6 | 0.396f |
| Persistent dissecting aortic intimal flapg | 51 (16%) | 37.6 ± 39.1 | 2.4–249.6 | 0.281f |
| Total | 317 (90/108) | 39.8 ± 32.1 | 0.02–249.6 | |
Abbreviations: Max, maximum; Min, minimum; SD, standard deviation; TGF‐β1, transforming growth factor‐beta 1.
In parentheses are the number of mutations that occurred in families/number of sporadic mutations.
Kruskal‐Wallis test.
Seven ACTA2 mutations, and 1 MYH11 mutation.
Four TGFBR1 missense mutations, 15 TGFBR2 missense mutations, and 1 TGFB2 mutation.
Cardiovascular interventions comprised the presence of isolated aortic root replacement including conduit operations and David Procedure in 110 patients, isolated aortic stent‐graft implantation in 6, isolated aortic valve surgery in 8, isolated mitral valve surgery in 7, combined aortic root replacement and mitral valve surgery in 10, combined aortic root replacement and stent‐graft placement in 10, combined surgical replacement of thoracic descending aorta in 2, and combined replacement of the aortic root and of thoracic descending aorta in 4 patients.
The Mann–Whitney U test identified no differences of tsTGF‐β1 levels according to presence or absence of both history of aortic dissection and presence of a persistent dissecting aortic intimal flap.
Demonstration of a residual intimal flap in patients with a history of aortic dissection on magnetic resonance imaging at the time of tsTGF‐β1 measurement.
Table 3.
Correlation of Clinical Features With tsTGF‐β1 Levels (ng/mL) in 317 Patients
| Variable | Spearman Correlation Coefficient (ρ) | P |
|---|---|---|
| Age, y | −0.080 | 0.153 |
| Male | −0.001 | 0.992 |
| Body weight (kg) | 0.005 | 0.929 |
| Body height (m) | 0.029 | 0.608 |
| Body surface area (m2)a | 0.013 | 0.825 |
| Total cholesterol (mg/dL) | −0.045 | 0.444 |
| High‐density lipoprotein cholesterol (mg/dL) | 0.040 | 0.500 |
| Low‐density lipoprotein cholesterol (mg/dL) | −0.022 | 0.711 |
| Current smoking | 0.018 | 0.743 |
| β‐Adrenergic blocker | −0.034 | 0.547 |
| Angiotensin receptor blocker | 0.072 | 0.203 |
| ACEi | 0.026 | 0.650 |
| Any medication (BAB, ARB, ACEi)b | 0.067 | 0.235 |
| Systolic blood pressure (mm Hg) | 0.059 | 0.330 |
| Diastolic blood pressure (mm Hg) | 0.077 | 0.202 |
| Aortic root diameter (cm) | 0.030 | 0.688 |
| Aortic root Z‐scorec | 0.084 | 0.260 |
| LV ejection fraction (%)d | 0.048 | 0.564 |
| Indexed end‐systolic LV diameter (mm/m2)d | 0.016 | 0.861 |
| Indexed end‐diastolic LV diameter (mm/m2)d | 0.067 | 0.372 |
| Indexed left‐atrial diameter (mm/m2)d | 0.021 | 0.797 |
| Aorta ascenders diameter (cm)e | −0.102 | 0.086 |
| Aorta descenders diameter (cm)e | −0.089 | 0.145 |
| NT‐proBNP (pg/mL)f | −0.028 | 0.622 |
| Sporadic occurrence of syndromeg | 0.141 | 0.048 |
| Aortic root dilatationh | 0.059 | 0.296 |
| Ectopia lentis | −0.012 | 0.837 |
| Dural ectasia (Habermann) | 0.080 | 0.159 |
| Mitral valve prolapse | 0.054 | 0.335 |
| History of recurrent hernia | 0.110 | 0.051 |
| Skeletal score (points)i | 0.029 | 0.612 |
| Nonskeletal score (points)i | 0.063 | 0.264 |
| Systemic total score pointsi | 0.046 | 0.418 |
| Previous cardiovascular intervention | 0.040 | 0.473 |
| History of aortic dissection | −0.048 | 0.397 |
| Persistent dissecting aortic intimal flap | −0.061 | 0.282 |
| Causative gene mutation | 0.180 | 0.001 |
| Genetic aortic syndrome | 0.198 | <0.001 |
Abbreviations: ACEi, angiotensin‐converting enzyme inhibitor; ARB, angiotensin II receptor blocker; BAB, β‐adrenergic receptor blocker, LV, left ventricle; NT‐proBNP, N‐terminal pro‐brain natriuretic peptide serum levels; tsTGF‐β1, total serum transforming growth factor‐beta 1.
Du Bois and Du Bois.21
A total of 154 patients were on any medication at the time of TGF‐β1 measurement including isolated or combined intake of BAB in 105, ARB in 58, and ACEi in 40 patients as described previously.24
Assessment according to Devereux.22
Two‐dimensional echocardiography according to Simpson's rule and current guidelines.23
Measurements were obtained on magnetic resonance imaging at the time of tsTGF‐β1 measurements.
Electrochemiluminescence sandwich immunoassay (Roche Diagnostics GmbH, Mannheim, Germany) on an Elecsys System 2010.
This variable was considered only in 198 patients with thoracic aortic aneurysm and dissection, Loeys‐Dietz syndrome, Marfan syndrome, and bicuspid aortic valve disease, where we defined sporadic occurrence in all patients with exclusion of diagnostic criteria of his or her genetic aortic syndrome in his or her parent, child, or sibling.
Aortic root Z‐score ≥2 or previous aortic root replacement.
According to Ghent criteria as detailed previously.2
Genetic Variables
To assess FBN1 mutation characteristics we compared both premature termination codon (PTC) mutations, and splicing mutations vs all other mutations. We classified mutations as follows: elimination or creation of a cysteine, localization in a calcium‐binding epidermal growth factor‐like domain, in an LTBP‐like binding domain, or in the so‐called neonatal region spanning exons 24 to 32, where we counted only coding exons. We compared any exon mutation with the respective characteristic vs all other exon mutations, and then we compared only missense mutations with the respective characteristic vs all other missense mutations (Table 1).1
Data Analysis
We expressed quantitative data as means and qualitative data as numbers. The Kolmogorov‐Smirnov test rejected normal distribution of tsTGF‐β1 values in our 317 patients (P < 0.001), and therefore we employed the Kruskal‐Wallis as a global test to identify differences between more than 2 groups, using and the Mann–Whitney U test for differences between pairs of groups (Table 2 and Table 4, Figure 1). We employed the Spearman correlation coefficient (ρ) to identify an association of independent variables with tsTGF‐β1 levels (Table 3 and Table 1, Figure 1). We performed multivariate testing of variables with univariate P values < 0.05 using multiple linear regression analysis with backward elimination, where variables with P > 0.1 were removed from the model. We considered P values <0.05 as significant, and we used SPSS software (SPSS for Windows, version 17.0; SPSS Inc., Chicago, IL) for statistical analyses.
Table 4.
Total TGFβ1 Serum Levels According to Aortic Features in 119 Patients Without Genetic Aortic Syndrome
| Total TGF‐β1, ng/mL | ||||
|---|---|---|---|---|
| Aortic Feature | Absence/Presence | Absence | Presence | P a |
| Aortic root dilatation | 74/45 | 30.6 ± 28.6 | 34.4 ± 23.9 | 0.144 |
| Previous aortic surgery or intervention | 83/36 | 31.1 ± 28.9 | 34.2 ± 21.3 | 0.120 |
| History of aortic dissection | 101/18 | 32.4 ± 27.8 | 30.1 ± 21.2 | 0.959 |
| Persistent dissecting aortic intimal flap | 102/17 | 32.2 ± 27.7 | 30.9 ± 21.6 | 0.832 |
| Any aortic featureb | 70/49 | 31.4 ± 29.1 | 32.9 ± 23.5 | 0.301 |
Abbreviations: TGF‐β1, transforming growth factor‐beta 1.
Mann–Whitney U test.
Any aortic feature comprised aortic dissection, aortic surgery or aortic intervention, or presence of aortic root dilatation, or combinations.
Figure 1.
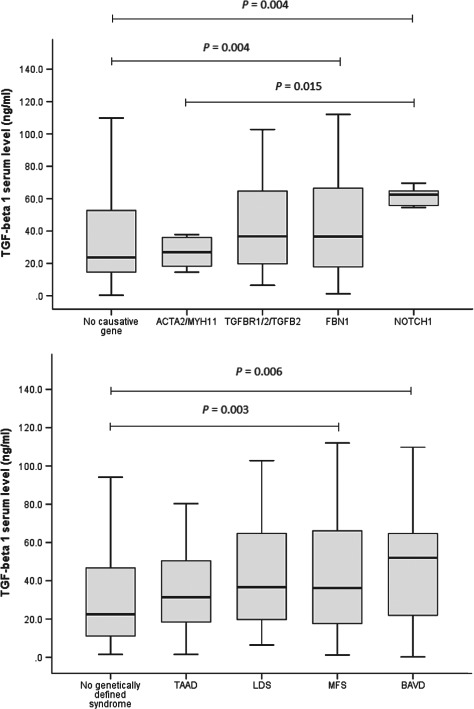
The box and whiskers plots of total serum transforming growth factor‐β1 (tsTGF‐β1) identify higher levels in FBN1 mutations (P = 0.004) and in NOTCH1 mutations (P = 0.004) than in patients with no mutations, and higher levels in NOTCH1 mutations than in ACTA2/MYH11 mutations (P = 0.015; upper panel). Similarly, tsTGF‐β1 levels are higher in Marfan syndrome (MFS) (P = 0.003) and in bicuspid aortic valve disease (BAVD) than in patients with no genetic aortic syndrome (P = 0.006; lower panel). Abbreviations: LDS, Loeys‐Dietz syndrome; TAAD, thoracic aortic aneurysm and dissection.
Results
Baseline Characteristics
We measured tsTGF‐β1 levels in 167 men and 150 women at a mean age of 43 ± 14 years (range, 17–75 years). Of these, 158 patients had a causative mutation (49.8%), 198 had GAS (62.5%), 157 had previously undergone cardiovascular surgery (49.5%) including aortic surgery or intervention in 142 (44.8%), and 57 patients had a history of aortic dissection (18%), with an intimal flap still present on current MRI in 51 patients (16.1%; Table 2).
The tsTGF‐β1 Levels According to Patient Characteristics
The tsTGF‐β1 levels were increased with both presence of a causative mutation (P = 0.008) and with presence of GAS (P = 0.009; Table 2). These tsTGF‐β1 levels were elevated in FBN1 mutations and in NOTCH1 mutations vs patients without mutations (both P = 0.004) and in NOTCH1 mutations versus ACTA2/MYH11 mutations (P = 0.015; Figure 1, upper panel). Similarly, tsTGF‐β1 levels were elevated in MFS (P = 0.003) and in BAVD (P = 0.006) vs patients without GAS (Figure 1, lower panel). Conversely, previous cardiovascular intervention (P = 0.748), a history of aortic dissection (P = 0.396), and a persistent dissecting aortic intimal flap (P = 0.281) were not correlated with increased tsTGF‐β1 levels (Table 2).
The tsTGF‐β1 Levels in Patients Without GAS
In the 119 patients without GAS, aortic root dilatation was present in 45 (37.8%), previous aortic surgery or intervention in 36 (30.3%), a history with aortic dissection in 18 (15.1%), and a persistent dissecting aortic intimal flap in 17 patients (14.3%). Interestingly, however, none of these features related to increased tsTGF‐β1 serum levels (Table 4).
The tsTGF‐β1 Levels: Univariate Associations
Besides causative gene mutations (P = 0.001) and GAS (P < 0.001), only sporadic occurrence of GAS versus familial occurrence of GAS (P = 0.048) presented as an additional clinical feature with relationship to increased tsTGF‐β1. All other features did not relate to tsTGF‐β1 levels (Table 3).
The tsTGF‐β1 Levels: Multivariate Testing
Because sporadic occurrence of GAS was exclusively assessed in 198 patients with GAS, we analyzed those 3 variables with univariate association with tsTGF‐β1 (P < 0.05) in 2 separate models. In the first model we included the 2 variables of GAS and causative gene mutation and identified GAS (regression coefficient b = 12.497 with 95% confidence interval [CI]: 5.288 to 19.706; P = 0.001) as the exclusive predictor of tsTGF‐β1. In the second model we included the 2 variables causative gene mutation and sporadic occurrence of GAS, where a causative gene mutation was excluded (P = 0.654) with identification of a weak association with sporadic occurrence of GAS (b = 8.495; 95% CI: −1.102 to 18.092; P = 0.082).
The tsTGF‐β1 Levels' Association With FBN1 Mutation Characteristics
In‐frame mutations related to higher tsTGF‐β1 levels than PTC mutations in the subgroup 123 patients with causative FBN1 mutation (P = 0.005), whereas all other FBN1 mutation characteristics were unrelated to tsTGF‐β1 levels (Table 1).
Discussion
The major result of this study was corroboration of the hypothesis that GAS was associated with elevated tsTGF‐β1 levels. These tsTGF‐β1 levels were elevated in FBN1 mutations and in NOTCH1 mutations vs patients without mutations, and in NOTCH1 mutations vs ACTA2/MYH11 mutations. Similarly, tsTGF‐β1 levels were elevated in MFS and in BAVD vs patients without GAS. Besides GAS, only sporadic occurrence of GAS related marginally to increased tsTGF‐β1 levels. Other clinical features including aortic diameters, Z‐scores, clinical features of MFS, or aortic dissection were unrelated to tsTGF‐β1 levels.
GAS exhibited increased tsTGF‐β1 levels as compared to patients without GAS. Thus, increased tsTGF‐β1 may generally indicate presence of GAS rather than a single disease entity such as MFS. Gillis et al reviewed the complex metabolism of GAS where they argued that TGF‐β1 dysregulation was the common pathogenetic mechanism of all types of GAS.14 Therefore, our findings support the view of GAS as a TGF‐β1‐related disease.
However, our results exhibited gradual differences of tsTGF‐β1 elevations across different GAS, where the highest levels were present in BAVD and the lowest levels in TAAD. These differences were significant between tsTGF‐β1 in NOTCH1 mutations vs ACTA2/MYH11 mutations. This finding fits into the current understanding of different mechanisms of aneurysm formation in different gens.14 NOTCH1 mutations cause BAVD, and relate to both alteration of Notch signaling in genetic aortic aneurysms25 and to increased TGF‐β1 signaling.26 Conversely, ACTA2/MYH11 mutations affect the contractile apparatus of the vascular smooth muscle cells rather than TGF‐β1 signaling. Therefore, lower TGF‐β1 levels may be expected from the underlying major pathomechanism despite some evidence for upregulation of TGF‐β1 signaling in ACTA2 and MYH11 mutations with TAAD.27 Finally, our finding of increased tsTGF‐β1 in LDS is novel and supports LDS as a disease with upregulation of TGF‐β1 signaling.14
The tsTGF‐β1 levels were increased in sporadic GAS, which may reflect a generally more severe disease in de novo mutations. Interestingly, we did not identify any other clinical features that related to increased tsTGF‐β1. This argues for GAS as a systemic disease with multifocal rather than organ‐specific dysregulation of TGF‐β1.
Franken et al reported increased tsTGF‐β1 in 28 MFS patients with previous aortic root replacement vs 71 MFS patients without such surgery and a correlation of tsTGF‐β1 with aortic root diameters.9 However, in a larger study on tsTGF‐β1 in MFS patients, the GenTAC Consortium did not find correlations between tsTGF‐β1 levels and aortic root diameters or aortic root Z‐scores.6 Our study extends these GenTAC Consortium findings in MFS to the entire spectrum of GAS, where tsTGF‐β1 levels were unrelated to aortic root replacement, aortic dissection, or aortic root dilatation. Medications such as BAB, ACEi, or ARB were found to impact tsTGF‐β1 levels in treatment studies of MFS.6, 9, 28 However, our study was not designed to assess medication effects; patients were on various drugs, at different dosages and combinations and types of a BAB, ARB, and ACEi over different time periods, and included treatment of various diseases rather than MFS alone. Therefore, our finding that medications did not relate significantly to tsTGF‐β1 levels does not exclude that a BAB, ARB, or ACEi modulates tsTGF‐β1 signaling in GAS.
Although the mechanisms by which FBN1 mutations cause increased TGF‐β1 signaling are currently unclear, it has been surmised that domain‐specific functions of FBN1 are needed for its modulation of TGF‐β1 signaling.14 Our findings support this view, because the relationship of FBN1 in‐frame mutations with higher tsTGF‐β1 levels than in frameshift and PTC mutations suggests that haploinsufficiency of FBN1 has less impact on TGF‐β1 signaling than expression of mutant FBN1 proteins. In line with this, specific missense mutations in exons 41 and 42 that encode the TGF‐β–binding protein‐like domain 5 associate with higher TGF‐β levels in acromicric and geleophysic dysplasias compared with controls.29
Study Limits
Some potential limits of the study require elucidation. The total of 351 persons who we evaluated for GAS were likely to represent the prevalence of GAS in the general Hamburg metropolitan population.30 Because we only excluded a small fraction of patients (34 patients, 10.8%) from this study, our patients were likely to represent GAS in the general Hamburg population without major bias.
We identified significantly higher tsTGF‐β1 levels in patients with GAS as in patients without GAS. Patients without GAS were not healthy, but 49 patients exhibited aortic features including a history of aortic dissection, aortic surgery or intervention, or aortic root dilatation (41.2%; Table 4). However, patients without GAS exhibited mean tsTGF‐β1 levels of 32 ng/mL, which was lower than the normal mean tsTGF‐β1 levels of 39.6 ng/mL provided by the manufacturer of our TGF‐β1 essay.31 Thus, the presence of aortic disease alone without a genetic cause does not seem to increase tsTGF‐β1 levels as compared to a normal healthy population.
Similarly, we excluded mutations in TGFBR1, TGFBR2, ACTA2, NOTCH1, and TGFB2 only in those 22 of 119 patients without GAS who exhibited specific clinical features of syndromes associated with the respective genes. Therefore, we may have missed GAS in some patients despite extensive clinical and molecular diagnostic workup. This may explain higher maximum tsTGF‐β1 levels of 135.1 ng/mL in our patients without GAS as compared to maximum levels of 63.4 ng/mL reported in healthy volunteers.31
Moreover, tsTGF‐β1 measurements can be distorted by platelet degranulation. However, we followed the manufacturer's recommendations for elimination of platelets. Comparison with tsTGF‐β1 values from other studies of MFS was not possible, because the methods differed across studies (see Supporting Information, Table, in the online version of this article).
Conclusion
TsTGF‐β1 is elevated in the entire spectrum of GAS. Therefore, dysregulation of TGF‐β1 signaling may be a common pathogenetic mechanism of GAS. However, gradual differences of tsTGF‐β1 with higher levels in NOTCH1 mutations and BAVD, and lower levels in ACTA2/MYH11 mutations and TAAD, may mirror different degrees of tsTGF‐β1 signaling alterations in GAS. Because no single clinical feature related to increased tsTGF‐β1 levels, GAS appears as a systemic disease with multifocal rather than organ‐specific dysregulation of TGF‐β1. Finally, tsTGF‐β1 may emerge as a diagnostic marker of GAS. However, larger studies with completely healthy controls are needed to establish diagnostic thresholds of tsTGF‐β1 levels.
Supporting information
Review of published results on measurements of circulating tsTGF‐beta1 in Marfan patients
Mathias Hillebrand, MD, and Nathalie Millot, MD, contributed equally to this work.
The authors have no funding, financial relationships, or conflicts of interest to disclose.
References
- 1. Faivre L, Collod‐Beroud G, Loeys BL, et al. Effect of mutation type and location on clinical outcome in 1,013 probands with Marfan syndrome or related phenotypes and FBN1 mutations: an international study. Am J Hum Genet. 2007;81:454–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Loeys BL, Dietz HC, Braverman AC, et al. The revised Ghent nosology for the Marfan syndrome. J Med Genet. 2010;47:476–485. [DOI] [PubMed] [Google Scholar]
- 3. Doyle JJ, Gerber EE, Dietz HC. Matrix‐dependent perturbation of TGFbeta signaling and disease. FEBS Lett. 2012;586:2003–2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Habashi JP, Judge DP, Holm TM, et al. Losartan, an AT1 antagonist, prevents aortic aneurysm in a mouse model of Marfan syndrome. Science. 2006;312:117–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ahimastos AA, Aggarwal A, D'Orsa KM, et al. Effect of perindopril on large artery stiffness and aortic root diameter in patients with Marfan syndrome: a randomized controlled trial. JAMA. 2007;298:1539–1547. [DOI] [PubMed] [Google Scholar]
- 6. Matt P, Schoenhoff F, Habashi J, et al. Circulating transforming growth factor‐beta in Marfan syndrome. Circulation. 2009;120:526–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kim KL, Yang JH, Song SH, et al. Positive correlation between the dysregulation of transforming growth factor‐beta1 and aneurysmal pathological changes in patients with Marfan syndrome. Circ J . 2013;77:952–958. [DOI] [PubMed] [Google Scholar]
- 8. Ogawa N, Imai Y, Nishimura H, et al. Circulating transforming growth factor beta‐1 level in Japanese patients with Marfan syndrome. Int Heart J . 2013;54:23–26. [DOI] [PubMed] [Google Scholar]
- 9. Franken R, den Hartog AW, de Waard V, et al. Circulating transforming growth factor‐beta as a prognostic biomarker in Marfan syndrome. Int J Cardiol. 2013;168:2441–2446. [DOI] [PubMed] [Google Scholar]
- 10. van Bogerijen GHW, Tolenaar JL, Grassi V, et al. Biomarkers in TAA—the holy grail. Prog Cardiovasc Dis. 2013;56:109–115. [DOI] [PubMed] [Google Scholar]
- 11. Leutermann R, Sheikhzadeh S, Brockstadt L, et al. A 1‐bp duplication in TGFB2 in three family members with a syndromic form of thoracic aortic aneurysm. Eur J Hum Genet. 2014;22:944–948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Maccarrick G, Black JH III, Bowdin S, et al. Loeys‐Dietz syndrome: a primer for diagnosis and management [published online ahead of print February 27, 2014]. Genet Med. doi: 10.1038/gim.2014.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Pyeritz RE. Heritable thoracic aortic disorders. Curr Opin Cardiol. 2014;29:97–102. [DOI] [PubMed] [Google Scholar]
- 14. Gillis E, Van Laer L, Loeys BL. Genetics of thoracic aortic aneurysm: at the crossroad of transforming growth factor‐β signaling and vascular smooth muscle cell contractility. Circ Res. 2013;113:327–340. [DOI] [PubMed] [Google Scholar]
- 15. De Paepe A, Devereux RB, Dietz HC, et al. Revised diagnostic criteria for the Marfan syndrome. Am J Med Genet. 1996;62:417–426. [DOI] [PubMed] [Google Scholar]
- 16. Loeys BL, Chen J, Neptune ER, et al. A syndrome of altered cardiovascular, craniofacial, neurocognitive and skeletal development caused by mutations in TGFBR1 or TGFBR2. Nat Genet. 2005;37:275–281. [DOI] [PubMed] [Google Scholar]
- 17. Loeys BL, Schwarze U, Holm T, et al. Aneurysm syndromes caused by mutations in the TGF‐beta receptor. N Engl J Med. 2006;355:788–798. [DOI] [PubMed] [Google Scholar]
- 18. Sheikhzadeh S, Brockstaedt L, Habermann CR, et al. Dural ectasia in Loeys‐Dietz syndrome: comprehensive study of 30 patients with a TGFBR1 or TGFBR2 mutation [published online ahead of print October 28, 2013]. Clin Genet. doi: 10.1111/cge.12308. [DOI] [PubMed] [Google Scholar]
- 19. Sheikhzadeh S, Sondermann C, Rybczynski M, et al. Comprehensive analysis of dural ectasia in 150 patients with a causative FBN1 mutation [published online ahead of print August 29, 2013]. Clin Genet. doi: 10.1111/cge.12264. [DOI] [PubMed] [Google Scholar]
- 20. Pyeritz RE. Evaluation of the adolescent or adult with some features of Marfan syndrome. Genet Med. 2012;14:171–177. [DOI] [PubMed] [Google Scholar]
- 21. Du Bois D, Du Bois EF. A formula to estimate the approximate surface area if height and weight be known. 1916. Nutrition. 1989;5:303–311; discussion 312–313. [PubMed] [Google Scholar]
- 22. Devereux RB, de Simone G, Arnett DK, et al. Normal limits in relation to age, body size and gender of two‐dimensional echocardiographic aortic root dimensions in persons ≥15 years of age. Am J Cardiol. 2012;110:1189–1194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Lang RM, Bierig M, Devereux RB, et al. Recommendations for chamber quantification. Eur J Echocardiogr. 2006;7:79–108. [DOI] [PubMed] [Google Scholar]
- 24. Mortensen K, Aydin MA, Rybczynski M, et al. Augmentation index relates to progression of aortic disease in adults with Marfan syndrome. Am J Hypertens. 2009;22:971–979. [DOI] [PubMed] [Google Scholar]
- 25. Zou S, Ren P, Nguyen M, et al. Notch signaling in descending thoracic aortic aneurysm and dissection. PLoS One. 2012;7:e52833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Gomez D, Al Haj Zen A, Borges LF, et al. Syndromic and non‐syndromic aneurysms of the human ascending aorta share activation of the Smad2 pathway. J Pathol . 2009;218:131–142. [DOI] [PubMed] [Google Scholar]
- 27. Renard M, Callewaert B, Baetens M, et al. Novel MYH11 and ACTA2 mutations reveal a role for enhanced TGFbeta signaling in FTAAD. Int J Cardiol. 2013;165:314–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ahimastos AA, Dart AM, Kingwell BA. Angiotensin II blockade in Marfan's syndrome. N Engl J Med. 2008;359:1732; author reply 1733–1734. [DOI] [PubMed] [Google Scholar]
- 29. Le Goff C, Mahaut C, Wang LW, et al. Mutations in the TGFbeta binding‐protein‐like domain 5 of FBN1 are responsible for acromicric and geleophysic dysplasias. Am J Hum Genet. 2011;89:7–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Rybczynski M, Bernhardt AM, Rehder U, et al. The spectrum of syndromes and manifestations in individuals screened for suspected Marfan syndrome. Am J Med Genet A. 2008;146A:3157–3166. [DOI] [PubMed] [Google Scholar]
- 31. Quantikine ELISA Human TGF‐beta 1 Immunoassay [package insert]. Minneapolis, MN: R&D Systems; 2013. http://www.rndsystems.com/pdf/DB100B.pdf. Accessed February 19, 2013. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Review of published results on measurements of circulating tsTGF‐beta1 in Marfan patients