

ABSTRACT
Autophagy has a critical role in liver regeneration. However, no studies have demonstrated autophagic flux in the regenerating fatty liver. The aim of this study was to clarify the dynamics of autophagy in the regeneration of the fatty liver. Following 70% partial hepatectomy (PH) in db/db fatty mice, which is a non-alcoholic fatty liver disease (NAFLD) model, we investigated the survival rate and recovery of liver volume. Histological examination of the regenerating liver was examined using electron microscopy. The 7-day survival rate after PH in db/db mice was 20%, which was significantly lower than that in control mice (P< .01). Liver regeneration within 48 h after PH was significantly impaired in db/db mice (P< .05). The number of proliferating cell nuclear antigen (PCNA) positive cells and the expression levels of cell-cycle markers cyclins D, E, and A were lower in db/db mice compared with controls. In the regenerating liver, LC3-II level was higher in db/db mice, but p62 expression was increased and cathepsin D expression, a marker of autophagolysosome proteolysis, was decreased compared with controls. Additionally, electronic microscopy revealed that autophagosomes during liver regeneration in db/db mice were mainly located in lipid droplets. Our findings indicate that the different localization of autophagosomes in db/db mice compared with controls led to impairment of liver regeneration in the fatty liver.
KEYWORDS: autophagy, fatty liver, lipophagy, liver regeneration, proteolysis
INTRODUCTION
Obesity is a risk factor for non-alcoholic fatty liver disease (NAFLD).1 Obesity-related NAFLD can contribute to the development of chronic inflammation and oxidative stress, leading to an increased risk of liver dysfunction, progression to cirrhosis, and hepatocellular carcinoma.2 The liver has regenerative potential against damage, but this potential is impaired in the fatty liver.3–5 Postoperative regeneration and residual liver function influence the outcome of liver surgery6, and especially in fatty liver, the outcome is worse.7 Increased endoplasmic reticulum (ER) stress and oxidative stress impair fatty liver regeneration.4
Autophagy is a homeostatic mechanism that regulates the turnover of long-lived or damaged proteins and organelles, buffers intracellular constituents, and supplies amino acids taken from autophagolysosome degradation products.8 Autophagy can be induced by stress and has important roles in liver physiology and pathology.9,10
We previously reported that activation of autophagy is essential for the liver to regenerate.11 Mice with liver-specific knockout of autophagy-related gene 5 (Atg5) had worse survival and worse liver regeneration after partial hepatectomy (PH) compared with mice with normal livers. In NAFLD, impairment of autophagy plays an important role to exacerbate lipid metabolic disorders.12 Impairment of autophagy plays an important role in exacerbating lipid metabolic disorder, and thereby contributing to steatohepatitis in diabetes. In NAFLD mice, autophagic flux, such as a high level of LC3-II protein and increasing the number of autophagosomes, is accelerated under stress conditions13, but autophagic proteolysis is impaired because of catabolizing lipid droplets (lipophagy).14
Although the influence of autophagic flux on NAFLD was reported, its detailed dynamics during liver regeneration in NAFLD have not been reported. Therefore, the aim of this study was to clarify autophagic dynamics in liver regeneration in NAFLD.
RESULTS
Differences in characteristics and survival rates between m+/m+ and db/db mice
The db/db mice (body weight, 29.6–35.7 g; liver weight, 3.0–3.7 g) were used as NAFLD models in this experiment, and m+/m+ mice (body weight, 20.2–21.4 g; liver weight, 1.3–1.5 g) were used as the control. HE staining of liver slices before PH from a db/db mouse showed severe microvesicular steatosis (Fig. 1a). The survival rate of db/db mice after PH was significantly lower than that of m+/m+ mice (Fig. 1b). All 10 m+/m+ mice survived after PH, but 8 of 10 db/db mice died within 4 days after PH.
FIGURE 1.
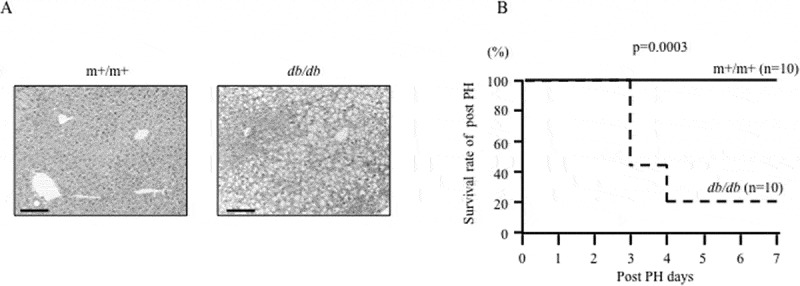
Pathological background of m+/m+ mice and db/db mice and the effect of partial hepatectomy (PH).
(a) HE staining of liver slices before PH from an m+/m+ mouse and a db/db fatty liver mouse. Scale bar, 50 μm. (b) Survival rate after PH. Ten mice underwent PH in each group.
Liver regeneration and hepatocyte proliferation after 70% partial hepatectomy
The liver regeneration rate after 70% PH was significantly inhibited in db/db mice compared with m+/m+ mice (Fig. 2a). In m+/m+ mice, the liver weight rapidly increased from 12 h after 70% PH (+18%), but the liver weight of db/db mice hardly changed in 24 h. Serum ALT concentrations at 12 h and 48 h after 70% PH were significantly higher in db/db mice compared with those in m+/m+ mice (Fig. 2b).
FIGURE 2.
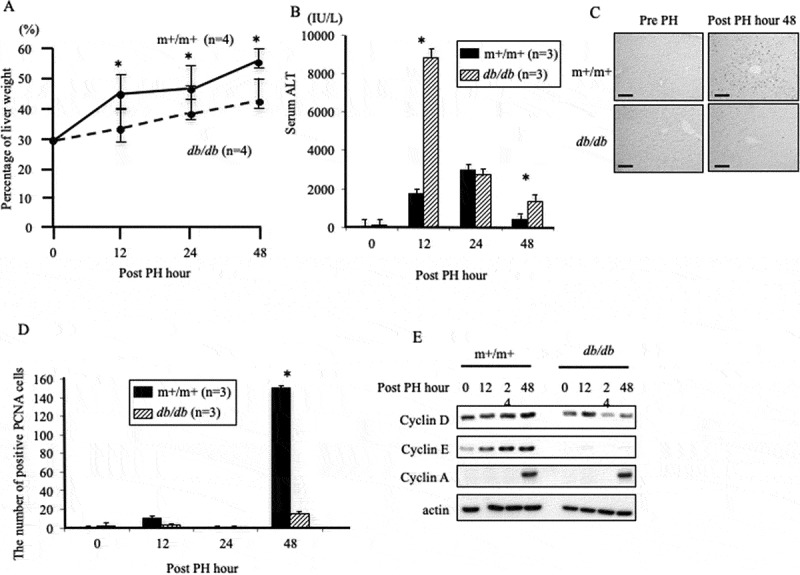
Liver regeneration and hepatocyte proliferation after 70% partial hepatectomy.
(a) The rate of liver regeneration after partial hepatectomy (PH). Data are representative of at least four independent experiments (*P< .05). (b) Serum alanine aminotransferase (ALT) levels were measured in each group of three mice (*P< .05). (c) Immunostaining of PCNA before and 48 h after PH (magnification, 200×). Scale bar, 50 μm. (d) The PCNA-positive cell number was measured in each group of three mice (*P< .05). (e) Protein levels of the cell-cycle markers, cyclins D, E, and A.
PCNA cells were observed in both m+/m+ and db/db mice after PH (Fig. 2c). PCNA-positive cell count in m+/m+ mice at 48 h after PH was significantly higher compared with that in db/db mice (Fig. 2d). Additionally, cyclin D and cyclin E protein levels after 70% PH in m+/m+ mice were higher compared with those in db/db mice (Fig. 2e). Cyclin D is initiated during G1 and drives the G1/S phase transition, and cyclin E is required for the transition from G1 to S phase. These results suggest that liver regeneration is suppressed in db/db mice compared with controls.
Autophagic activation in the regenerating liver
Next, we examined the involvement of pathways in steatotic liver regeneration. In m+/m+ mice, the peak LC3-II level was observed at 12 h after 70% PH, while the p62 protein, which regulates ubiquitinated protein, was increased gradually at least 48 h. However, in db/db mice, the LC3-II level was higher and p62 expression was notably increased in db/db mice compared with m+/m+ mice (Fig. 3), while Atg5-12 expression levels were nearly equal in both groups. These results suggest that autophagosome formation is not suppressed, but autophagic proteolysis is inhibited in db/db mice compared with controls. Thus, the cathepsin D protein level, a proteinase of lysosomes, was evaluated. As expected, cathepsin D expression was suppressed in db/db mice compared with controls (Fig. 3).
FIGURE 3.
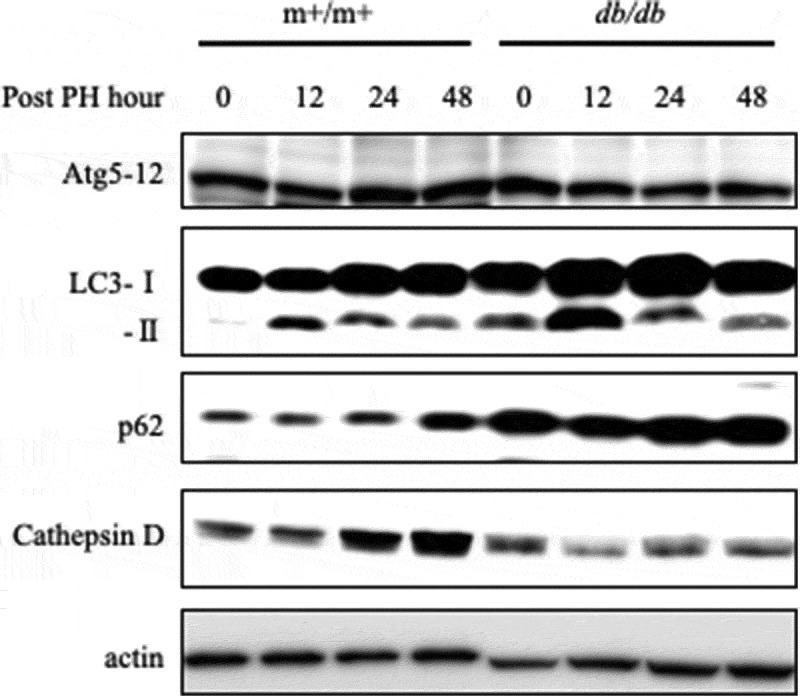
Suppression of proteinase activity in db/db mice.
Expression of the autophagy-related genes Atg5, LC3-I, LC3-II, p62, and Cathepsin D was determined by western blotting in each group at 0–48 h after PH.
Localization of autophagosomes in regenerating liver
Using electron microscopy, we determined the number of autophagosomes and their localization in the regenerating liver of m+/m+ and db/db mice (Fig. 4a). The formation of autophagic vacuoles (Avi) and degradative autophagic vacuoles (AVd) were regarded as indicative of autophagosome activation. The number of autophagosomes before PH in db/db mice was higher compared with m+/m+ mice (Fig. 4b). During liver regeneration, the number of autophagosomes was increased in both groups, but their localization differed. In m+/m+ mice, most autophagosomes were in the cytoplasm, while in db/db mice, autophagosomes were incorporated into lipid droplets (Fig. 4(c,d)). Immunofluorescence staining of LC3 revealed that LC3-positive foci in lipid droplets were increased in the regenerating liver of db/db mice at 12 h after PH (Fig. 5).
FIGURE 4.
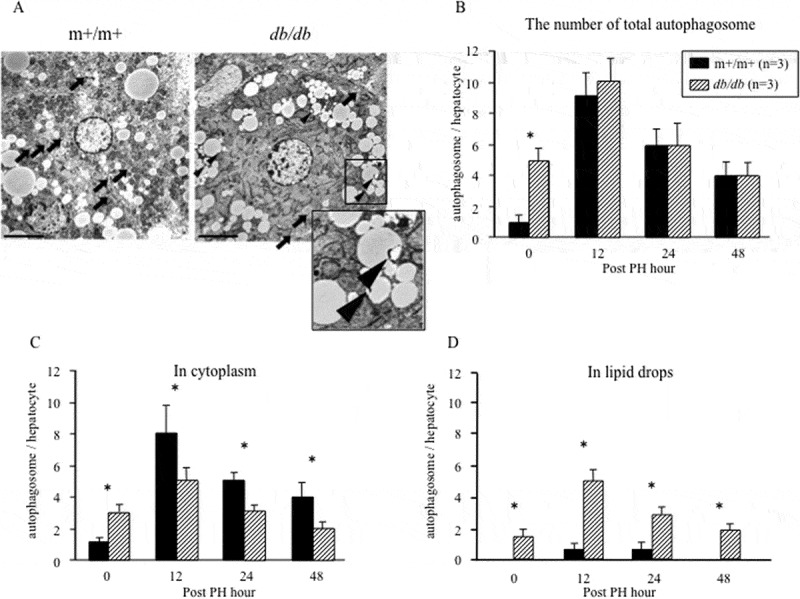
Localization of autophagosomes of electron microscopy.
(a) Autophagosomes in the regenerating liver (12 h after PH) detected by electron microscopy (magnification, 3000×). Arrows, autophagosomes in the cytoplasm; arrowheads, autophagosomes in lipid droplets. Scale bar, 5.0 μm. The number of (b) total autophagosomes, (c) autophagosomes in the cytoplasm, and (d) autophagosomes in lipid droplets were measured three times independently in m+/m+ mice and db/db mice (*P< .05).
FIGURE 5.
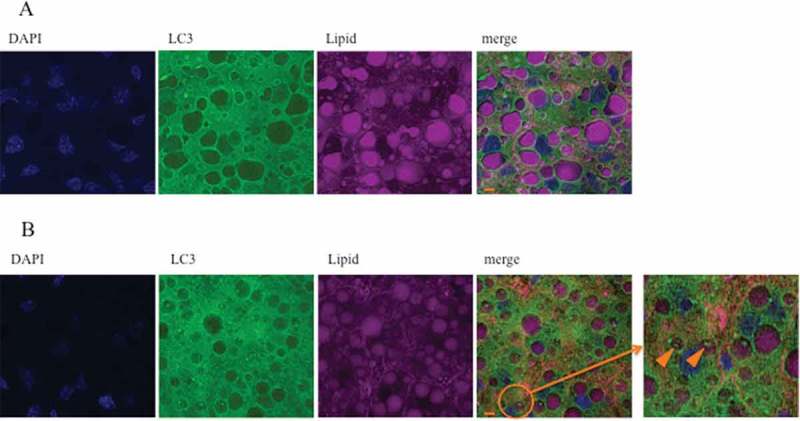
Localization of autophagosomes of immunofluorescent staining.
LC3-II foci localized in lipid droplets (enclosed by the orange circle) were more reduced in m+/m+ mice (a) compared with db/db mice (b) at 12 h after PH, as detected by immunofluorescent staining (magnification 40×). Scale bar, 100 μm.
DISCUSSION
There has been no detailed report on the influence of autophagy on liver regeneration in the fatty liver. This is the first report that investigated the dynamics of autophagy in fatty liver regeneration. We found that liver regeneration after 70% PH was decreased, and protein degradation was suppressed by autophagy during liver regeneration in the fatty liver compared with normal livers. Additionally, we acknowledged that autophagosome localization is different from that in the normal liver, and it occurs within lipid droplets in fatty liver regeneration.
In the fatty liver, liver regeneration after liver resection is decreased compared with controls.3–5 This causes severe complications such as liver failure and is often a serious problem in clinical practice for patients who undergo major hepatectomy or living donor liver transplantation.15,16 In this study, the survival rate after 70% PH was significantly decreased in db/db mice compared with livers from normal mice because of decreased liver regeneration, and these results are consistent with those of previous reports.3 After PH in db/db mice, serum ALT was elevated because of prolonged hepatocellular injury and nuclear reduction, such as decreased PCNA expression, and decreased cell-cycle regulatory protein levels (cyclin D, E, A) was also observed. This indicated that the early phase of liver regeneration was impaired, and we focused on autophagy as a cause of this impaired liver regeneration. Autophagy was reported to be closely related to the accumulation of lipids in the liver, and suppression of autophagy leads to the progression of fatty liver.17,18 Once fatty liver has developed, hepatocytes undergo cytotoxicity because of excessive overload of fatty acids.19–21 To maintain homeostasis in hepatocytes, a certain level of autophagy is induced in the fatty liver. Singh et al. showed that autophagy induced in fatty liver has autophagosome structures within lipid droplets at electron microscopic level.17 This was called lipophagy and that its localization was reported to be different from that of the previously described autophagy. There are several reports on lipophagy, but the significance of autophagy in liver regeneration in NAFLD is not clear. There has been no detailed study on the localization of lipid droplets and autophagosomes during liver regeneration. Thus, we investigated the relationship between lipids and autophagy during hepatic regeneration in a mouse hepatectomy model using electron microscopy and fluorescent staining, especially focusing on localization.
When autophagy is induced by stress such as starvation in hepatocytes, intracellular proteolysis occurs and energy is produced.22,23 We showed that postoperative expression of LC3-II gradually increased after 18 h and decreased after 48 h in normal liver, and, in Atg5 KO mice, liver regeneration was significantly impaired, damaged proteins accumulated, and hepatocytes were in the senescent state. Because Atg5KO mice that underwent up to 90% PH had liver failure and died after 24 h, we suggest that autophagy is an essential phenomenon for the early phase of liver regeneration.11 In the fatty liver, LC3-II expression is increased during liver regeneration compared with the normal liver, but p62 expression that is selectively reduced by autophagy was increased. mRNA expressions of p62 were nearly equal during liver regeneration (12, 24, 48 h after hepatectomy) between +/+ and db/db mice (data are not shown). Tanaka S, et al. showed, in NAFLD model, the expression level of LC3-II and p62 increased, but there was no difference in the level of p62 mRNA expression24, this result was the same as ours. The most important is that proteolysis of p62 was inhibited, not expression level of mRNA. Also, Lin CW, et al. reported that induced autophagic flux by amiodarone in hepatectomy-treated mice performed, the protein expression level of LC3-II was higher and p62 expression decreased. Then, in addition, treatment with Chloroquine, an autophagy inhibitor leading to a disruption of lysosome-autophagosome fusion and lysosomal protein degradation, the LC3-II level was higher and p62 expression was notably increased.25 It is the same as our result of LC3 and p62 expression. This indicated that the later stage of autophagy flux is inhibited in db/db mice, which was a remarkable difference from normal liver regeneration.
In the fatty liver, proteolysis that occurs in the autolysosome, which is the final process of autophagy, is suppressed.13 As well, Quan W, et al. reported autophagic proteolysis is impaired because of catabolizing lipid droplets in fatty liver.14 We examined the expression of cathepsin D, a proteolytic enzyme of lysosome, to confirm that degradation of autolysosomal proteolysis occurs in fatty liver regeneration. Cathepsin D expression was then significantly decreased in the fatty liver, and it was found that proteolysis by the autolysosome was also suppressed in the fatty liver during liver regeneration.
We focused on autophagosome localization. Under the condition of excessive fat, phagocytosis of lipid droplets by the autophagosome can be seen. Electron microscopic findings confirmed the double membrane structure of the autophagosome in the cytoplasm during liver regeneration of a normal liver, whereas the same structure was recognized in lipid droplets in fatty liver regeneration. This was similar to the electron microscopic findings of lipophagy reported by Singh et al.17 Because the LC3 dot could be confirmed in lipid droplets using fluorescent immunostaining, lipophagy, in which localization of the autophagosome increased within lipid droplets during liver regeneration in the fatty liver, was thought to be occurring. In the fatty liver, we speculated that autophagy during liver regeneration did not undergo proteolysis contrary to normal liver, but that it was likely to undergo lipolysis. In the situation where liver regeneration, division, and proliferation of hepatocytes are highly controlled, remodeling of intracellular proteins, including renewal of proteins and decomposition of unnecessary proteins into bulk, might be essential. Induction of autophagy leads to the promotion of liver regeneration.24 However, it is possible that the difference in autophagosome localization between the normal liver and fatty liver is responsible for liver regeneration in the fatty liver.
We also observed SIRT1, p-AMPK/AMPK, regulating autophagy at up-stream of LC3 (Figure S1). SIRT1 regulates autophagy by interacting with autophagy-related genes Atg5, Atg7, and Atg8 and deacetylating them.26 And, under stress condition, SIRT1which regulates autophagy exerts a protective role.27 In db/db mice, the expression level of SIRT1 was higher compared with m+/m+ mice. We considered SIRT1 promoted autophagy to protect from liver dysfunction by hepatectomy. AMPK plays a role as a key activator of the signaling network by autophagy. The expression level of p-AMPK was higher in the normal liver during liver regeneration. Tsuchihashi N, et al. reported RNAi-mediated knockdown of AMPKα reduced the expression of LC3-II to less than si-control. However, knockdown of Atg7 leads to the early induction of pAMPK. This result indicates AMPK activity could be negatively regulated by Atg.728 Our study revealed the expression level of LC3-II was higher in db/db mice, though, p-AMPK was inverse. This negative feedback might influence these expressions.
In liver regeneration, lipid accumulation is required in normal livers.29,30 Even in this study, normal livers had an accumulation of lipids in the cells during liver regeneration. However, unlike the fatty liver, lipophagy was not induced in situations where autophagy was induced, despite the lipid overload. The cause of different localization in normal and fatty livers requires further study.
In conclusion, the different localization of autophagosomes was revealed. The difference might impair liver regeneration in the fatty liver.
MATERIALS AND METHODS
Animal studies
Six-week-old male C57BL/KsJ db/db mice, which have abnormal leptin receptors, were used as a model of liver steatosis and diabetes, and were purchased from CLEA Japan, Inc. (Tokyo, Japan). C57BL/KsJ m+/m+ mice were used as controls. All animals were acclimatized to the environment for 1 week before the experiment. Mice were anesthetized with ether and 70% PH was performed by removing the left lateral and median lobes after mid-ventral laparotomy.31,32 Anesthesia and analgesics were used for all surgical experiments to minimize any suffering of the mice. Mice were anesthetized by intraperitoneal injection 0.1 mL/10 g (body weight) of medetomidine (1 mg/mL; 0.75 mL), midazolam (5 mg/mL; 2 mL), butorphanol (5 mg/mL; 2.5 mL), and distilled water (19.75 mL). Pentazocine (10 mg/kg) was used as a pain reliever. At the indicated times after hepatectomy, the mice were euthanized for blood and tissue collection by deep anesthesia using isoflurane. After excision, the liver weight-to-body weight ratio was measured, and liver specimens were stored in liquid nitrogen or fixed in 4% paraformaldehyde. The samples, including livers and blood, were collected from the mice at the indicated time points and pooled for the subsequent experiments. Serum concentrations of alanine aminotransferase (ALT) were measured.
We monitored the health of mice four times a day after surgery. The mice were euthanized when abnormal symptoms such as general fatigue, decreased activity, frequent vomiting, or respiratory distress were observed. All efforts were made to minimize suffering. Mice were fed a laboratory diet with water and maintained in a room with alternating 12-h light/dark cycles. All animal experiments were approved by the Kyushu University Animal Experimental Committee, and the care of the animals was in accordance with institutional guidelines. The study protocol conformed to the ethical guidelines of the Declaration of Helsinki.
Calculation of regeneration
Liver regeneration was evaluated using the remnant liver weight. The total liver regeneration rate was calculated using the following equation: 100 × (C – (A – B))/A; where B is the excised liver weight at the time of PH, A is the estimated total liver weight (= B× 100/70), and C is the weight of the regenerated liver at the time of sacrifice.
Antibodies
Rabbit monoclonal anti-LC3 (Cell Signaling Technology, Cat# 4599), anti-Atg5 (Cell Signaling Technology, Cat# 12994), anti-p62 (Cell Signaling Technology, Cat# 5114), and β-actin antibodies (Cell Signaling Technology, Cat# 5125) were purchased. Rabbit polyclonal anti-cyclin A (Santa Cruz Biotechnology, Cat# sc-751), anti-cyclin D (Santa Cruz Biotechnology, Cat# sc-25765), anti-cyclin E (Santa Cruz Biotechnology, Cat# sc-481), and anti-cathepsin D (Santa Cruz Biotechnology, Cat# sc-10725) antibodies were purchased.
Western blotting
For western blotting, liver tissue was frozen and homogenized in NETN buffer on ice. After 10 min of centrifugation at 15,000 g at 4°C, the supernatant was collected and boiled for 30 min with 2× sample buffer at 70°C. Then, 20 µg of protein extract was electrophoresed on an SDS-polyacrylamide electrophoresis gel and transferred to a PVDF membrane. After 30 min of incubation with Blocking One buffer (NACALAI TESQUE, Cat# 05999) to block non-specific binding, the membranes were exposed to specific primary antibody at 4°C overnight. The next day, membranes were washed and exposed to a secondary antibody for 1 h. Antigen–antibody binding was visualized using an enhanced chemiluminescence detection system (ECL Plus Western Blotting Detection Reagents; GE Healthcare, Buckinghamshire, UK).
Electron microscopy
Livers were immediately fixed in 2.5% glutaraldehyde, 0.1 mol/L phosphate-buffered saline (PBS; pH 7.4), for 2 h at 4°C, post-fixed in 2% osmium tetroxide for 2 h, dehydrated in an ethanol/propylene oxide series, and embedded in epoxy resin. Sections (70 nm thick) were cut using an ultramicrotome and double-stained with uranyl acetate and lead solution.33,34 Electron micrographs were taken using a Hitachi H-7500 transmission electron microscope. To quantify autophagosomes, high-powered micrographs (at 8000–10,000× magnification) of 10 single cells from multiple distinct low-powered fields were obtained for each specimen. Maturation from the phagophore to the autophagolysosome is a dynamic and continuous process, and autophagosomes or early autophagic vacuoles (AVi) and degradative autophagic vacuoles (AVd) can be observed by electron microscopy. AVi can be identified based on their morphologically intact cytoplasm and the presence of ribosomes and rough ER. AVd can be identified based on the presence of a limiting membrane that is partially visible as two bilayers separated by a narrow electrolucent cleft as a double membrane. These features are classified as markers of autophagy.
Histology and immunohistochemical analysis
Tissues were post-fixed overnight in 4% paraformaldehyde in phosphate buffer (pH 7.4). After embedding in paraffin, 3-µm-thick sections were prepared. Sections were stained with hematoxylin-eosin or with specific antibodies, as previously described.35 For immunohistochemical staining, the sections were deparaffinized and autoclaved at 121°C for 15 min in 0.01 mol/L citrate-buffered saline (pH 7.0) for antigen retrieval. Proliferating cell nuclear antigen (PCNA) staining was performed using the rabbit polyclonal anti-PCNA antibody (Santa Cruz Biotechnology, Cat# sc-7907). Ten visual fields per sample were surveyed in each of three samples to determine the mean percentage of PCNA-positive cells.
Immunofluorescence studies
munofluorescence analyses were performed as follows: Liver tissues were frozen in O.C.T. compound. Sections (10 μm) were fixed in 4% paraformaldehyde for 10 min and then washed with PBS-glycine (10 mM) and immersed in 0.1% Saponin in PBS. Sections were then washed again in PBS-glycine (10 mM) and immersed in 3% BSA in PBS (blocking of nonspecific reaction). The sections were incubated in specific antibody at 4°C overnight. The next day, the sections were washed in PBS-glycine (10 mM) and exposed to a secondary antibody for 1 h. Finally, LipidTOX Deep Red (Thermo Fisher Scientific, Cat# 34477) was used according to the manufacturer’s protocol. All fluorescence images were visualized using a BZ-8100 fluorescence microscope (Keyence, Tokyo, Japan).
Statistical analysis
All statistical analyses were performed using JMP statistical software version 9 (SAS Institute Inc., Cary, NC, USA). Parametric paired t-tests were used to compare paired samples, and continuous variables were expressed as the mean ± standard deviation and compared using the nonparametric Wilcoxon test for independent samples. Survival was calculated using the Kaplan–Meier product-limited method; differences in survival between the groups were compared using the log-rank test. P values < .05 were considered significant.
Acknowledgments
We thank Jodi Smith, Ph.D., from Edanz Group (www.edanzediting.com/ac) for editing a draft of this manuscript.
Authors’ Contributions
Y.M., T.Y., and M.M. designed this study. Y.M., T.Y., T.T., K.T., and T.F. performed the research. S.I., T.I., and Y.S. analyzed the data. M.M. wrote the paper. All authors critically reviewed the manuscript.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Supplementary material
Supplemental data for this article can be accessed on the publisher’s website.
Abbreviations
- ALT
alanine aminotransferase
- Atg
autophagy-related gene
- AVi
autophagic vacuole
- AVd
degradative autophagic vacuole
- ER
endoplasmic reticulum
- LC3
microtubule-associated protein 1 light chain 3
- NAFLD
non-alcoholic fatty liver disease
- PCNA
proliferating cell nuclear antigen
- PH
partial hepatectomy
References
- 1.Heijden RA, Sheedfar F, Morrison MC, Hommelberg PP, Kor D, Kloosterhuis NJ, Gruben N, Youssef SA, Bruin A, Hofker MH, et al. High-fat diet induced obesity primes inflammation in adipose tissue prior to liver in C57BL/6j mice. Aging. 2015;7:256–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Starley BQ, Calcagno CJ, Harrison SA.. Nonalcoholic fatty liver disease and hepatocellular carcinoma: a weighty connection. Hepatology. 2010;51:1820–32. [DOI] [PubMed] [Google Scholar]
- 3.Murata H, Yagi T, Iwagaki H, Ogino T, Sadamori H, Matsukawa H, Umeda Y, Haga S, Tanaka N, Ozaki M.. Mechanism of impaired regeneration of fatty liver in mouse partial hepatectomy model. J Gastroenterol Hepatol. 2007;22:2173–80. [DOI] [PubMed] [Google Scholar]
- 4.Hamano M, Ezaki H, Kiso S, Furuta K, Egawa M, Kizu T, Chatani N, Kamada Y, Yoshida Y, Takehara T. Lipid overloading during liver regeneration causes delayed hepatocyte DNA replication by increasing ER stress in mice with simple hepatic steatosis. J Gastroenterol. 2014;49:305–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hoppe S, von Loeffelholz C, Lock JF, Doecke S, Sinn BV, Rieger A, Malinowski M, Pfeiffer AF, Neuhaus P, Stockmann M. Nonalcoholic steatohepatits and liver steatosis modify partial hepatectomy recovery. J Invest Surg. 2015;28:24–31. [DOI] [PubMed] [Google Scholar]
- 6.Stockmann M, Lock JF, Riecke B, Heyne K, Martus P, Fricke M, Lehmann S, Niehues SM, Schwabe M, Lemke AJ, et al. Prediction of postoperative outcome after hepatectomy with a new bedside test for maximal liver function capacity. Ann Surg. 2009;250:119–25. [DOI] [PubMed] [Google Scholar]
- 7.Truant S, Bouras AF, Petrovai G, Buob D, Ernst O, Boleslawski E, Hebbar M, Pruvot FR. Volumetric gain of the liver after major hepatectomy in obese patients: a case-matched study in 84 patients. Ann Surg. 2013;258:696–702. [DOI] [PubMed] [Google Scholar]
- 8.Mizushima N, Komatsu M. Autophagy: renovation of cells and tissues. Cell. 2011;147:728–41. [DOI] [PubMed] [Google Scholar]
- 9.Singh R, Cuervo AM. Autophagy in the cellular energetic balance. Cell Metab. 2011;13:495–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ding WX, Ni HM, Li M, Liao Y, Chen X, Stolz DB, Dorn GW, Yin XM. Nix is critical to two distinct phases of mitophagy, reactive oxygen species-mediated autophagy induction and Parkin-ubiquitin-p62-mediated mitochondrial priming. J Biol Chem. 2010;285:27879–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Toshima T, Shirabe K, Fukuhara T, Ikegami T, Yoshizumi T, Soejima Y, Ikeda T, Okano S, Maehara Y. Suppression of autophagy during liver regeneration impairs energy charge and hepatocyte senescence in mice. Hepatology. 2014;60:290–300. [DOI] [PubMed] [Google Scholar]
- 12.Zhang Q, Li Y, Liang T, Lu X, Zhang C, Liu X, Jiang X, Martin RC, Cheng M, Cai L. ER stress and autophagy dysfunction contribute to fatty liver in diabetic mice. Int J Biol Sci. 2015;11:559–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Inami Y, Yamashina S, Izumi K, Ueno T, Tanida I, Ikejima K, Watanabe S. Hepatic steatosis inhibits autophagic proteolysis via impairment of autophagosomal acidification and cathepsin expression. Biochem Biophys Res Commun. 2011;412:618–25. [DOI] [PubMed] [Google Scholar]
- 14.Quan W, Hur KY, Lim Y, Oh SH, Lee JC, Kim KH, Kim GH, Kim SW, Kim HL, Lee MK, et al. Autophagy deficiency in beta cells leads to compromised unfolded protein response and progression from obesity to diabetes in mice. Diabetologia. 2012;55:392–403. [DOI] [PubMed] [Google Scholar]
- 15.Clavien PA, Oberkofler CE, Raptis DA, Lehmann K, Rickenbacher A, El-Badry AM. What is critical for liver surgery and partial liver transplantation: size or quality? Hepatology. 2010;52:715–29. [DOI] [PubMed] [Google Scholar]
- 16.Kauffmann R, Fong Y. Post-hepatectomy liver failure. Hepatobiliary Surg Nutr. 2014;3:238–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Singh R, Kaushik S, Wang Y, Xiang Y, Novak I, Komatsu M, Tanaka K, Cuervo AM, Czaja MJ. Autophagy regulates lipid metabolism. Nature. 2009;458:1131–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Christian P, Sacco J, Adeli K. Autophagy: emerging roles in lipid homeostasis and metabolic control. Biochim Biophys Acta. 2013;1831:819–24. [DOI] [PubMed] [Google Scholar]
- 19.Hatting M, Zhao G, Schumacher F, Sellge G, Al Masaoudi M, Gaβler N, Boekschoten M, Muller M, Liedtke C, Cubero FJ, et al. Hepatocyte caspase-8 is an essential modulator of steatohepatitis in rodents. Hepatology. 2013;57:2189–201. [DOI] [PubMed] [Google Scholar]
- 20.Csak T, Ganz M, Pespisa J, Kodys K, Dolganiuc A, Szabo G. Fatty acid and endotoxin activate inflammasomes in mouse hepatocytes that release danger signals to stimulate immune cells. Hepatology. 2011;54:133–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Leroux A, Ferrere G, Godie V, Cailleux F, Renoud ML, Gaudin F, Naveau S, Prevot S, Makhzami S, Perlmuter G, et al. Toxic lipids stored by Kupffer cells correlates with their pro-inflammatory phenotype at an early stage of steatohepatitis. J Hepatol. 2012;57:141–49. [DOI] [PubMed] [Google Scholar]
- 22.Levine B, Mizushima N, Virgin HW. Autophagy in immunity and inflammation. Nature. 2011;469:323–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Komatsu M, Waguri S, Koike M, Sou YS, Ueno T, Hara T, Mizushima N, Iwata J, Ezaki J, Murata S, et al. Homeostatic levels of p62 control cytoplasmic inclusion body formation in autophagy-deficient mice. Cell. 2007;131:1149–63. [DOI] [PubMed] [Google Scholar]
- 24.Tanaka S, Hikita H, Tatsumi T, Sakamori R, Nozaki Y, Sakane S, Shiode Y, Nakabori T, Saito Y, Hiramatsu N, et al. Rubicon inhibits autophagy and accelerates hepatocyte apoptosis and lipid accumulation in nonalcoholic fatty liver disease in mice. Hepatology. 2016;64:1994–2014. [DOI] [PubMed] [Google Scholar]
- 25.Lin CW, Chen YS, Lin CC, Chen YJ, Lo GH, Lee PH, Kuo PL, Dai CY, Huang JF, Chung WL, et al. Amiodarone as an autophagy promoter reduces liver injury and enhances liver regeneration and survival in mice after partial hepatectomy. Sci Rep. 2015;30:15807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lee IH, Cao L, Mostoslavsky R, Lombard DB, Liu J, Bruns NE, Tsokos M, Alt FW, Finkei T. A role for the NAD-dependent deacetylase Sirt1 in the regulation of autophagy. Proc Natl Acad Sci U S A. 2008;105:3374–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Luo G, Jian Z, Zhu Y, Zhu Y, Chen B, Ma R, Tang F, Xiao Y. Sirt1 promotes autophagy and inhibits apoptosis to protect cardiomyocytes from hypoxic stress. Int J Mol Med. 2019;43:2033–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tsuchihashi NA, Hayashi K, Dan K, Goto F, Nomura Y, Fujioka M, Kanzaki S, Komune S, Ogawa K. Autophagy through 4EBP1 and AMPK regulates oxidative stress-induced premature senescence in auditory cells. Oncotarget. 2015;28:3644–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Newberry EP, Kennedy SM, Xie Y, Luo J, Stanley SE, Semenkovich CF, Crooke RM, Graham MJ, Davidson NO. Altered hepatic triglyceride content after partial hepatectomy without impaired liver regeneration in multiple murine genetic models. Hepatology. 2008;48:1097–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shteyer E, Liao Y, Muglia LJ, Hruz PW, Rudnick DA. Disruption of hepatic adipogenesis is associated with impaired liver regeneration in mice. Hepatology. 2004;40:1322–32. [DOI] [PubMed] [Google Scholar]
- 31.Lin T, Ibrahim W, Peng CY, Finegold MJ, Tsai RY. A novel role of nucleostemin in maintaining the genome integrity of dividing hepatocytes during mouse liver development and regeneration. Hepatology. 2013;58:2176–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Borude P, Edwards G, Walesky C, Li F, Ma X, Kong B, Guo GL, Apte U. Hepatocyte-specific deletion of farnesoid X receptor delays but does not inhibit liver regeneration after partial hepatectomy in mice. Hepatology. 2012;56:2344–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Catanese MT, Uryu K, Kopp M, Edwards TJ, Andrus L, Rice WJ, Silvestry M, Kuhn RJ, Rice CM. Ultrastructural analysis of hepatitis C virus particles. Proc Natl Acad Sci U S A. 2013;110:9505–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hamasaki M, Furuta N, Matsuda A, Nezu A, Yamamoto A, Fujita N, Oomori H, Noda T, Haraguchi T, Hiraoka Y, et al. Autophagosomes form at ER-mitochondria contact sites. Nature. 2013;495:389–93. [DOI] [PubMed] [Google Scholar]
- 35.Mano Y, Aishima S, Fukuhara T, Tanaka Y, Kubo Y, Motomura T, Toshima T, Iguchi T, Shirabe K, Maehara Y, et al. Decreased roundabout 1 expression promotes development of intrahepatic cholangiocarcinoma. Hum Pathol. 2013;44:2419–26. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.