

Abstract
The hydrogen storage properties of the Scandium (Sc) atom modified Boron (B) doped porous graphene (PG) system were studied based on the density functional theory (DFT). For a single Sc atom, the most stable adsorption position on B-PG is the boron-carbon hexagon center after doping with the B atom. The corresponding adsorption energy of Sc atoms was −4.004 eV. Meanwhile, five H2 molecules could be adsorbed around a Sc atom with the average adsorption energy of −0.515 eV/H2. Analyzing the density of states (DOS) and the charge population of the system, the adsorption of H2 molecules in Sc-B/PG system is mainly attributed to an orbital interaction between H and Sc atoms. For the H2 adsorption, the Coulomb attraction between H2 molecules (negatively charged) and Sc atoms (positively charged) also played a critical role. The largest hydrogen storage capacity structure was two Sc atoms located at two sides of the boron-carbon hexagon center in the Sc-B/PG system. Notably, the theoretical hydrogen storage capacity was 9.13 wt.% with an average adsorption energy of −0.225 eV/H2. B doped PG prevents the Sc atom aggregating and improves the hydrogen storage effectively because it can increase the adsorption energy of the Sc atom and H2 molecule.
Keywords: first principles, porous graphene, B doped Sc modified PG, hydrogen storage
1. Introduction
The development of human society faces the severe challenge of environmental pollution, which is urgently needed to explore more ideal energy materials to meet the growing energy demand [1,2,3]. Hydrogen (H2) is used as a promising energy carrier because of recycling, no pollution, high energy density and high calorific value [4,5,6]. Graphene is a two-dimensional material with a hexagonal honeycomb structure composed of sp2 hybrid orbits. Graphene-based materials possess large specific surface area, good adsorption kinetics, low density, high chemical stability and reversible hydrogen storage, which is a potential candidate in many areas, such as solid-state hydrogen storage, electronics and sensors [7,8,9,10,11,12,13,14]. However, due to a large surface inertness and weak binding ability to H2 molecules, clean graphene is difficult to become a promising hydrogen storage material [15,16,17,18,19].
Recently, transition metal atoms [20,21] modified graphene has attracted researchers’ attention since significantly increasing the adsorption energy of H2 molecules. Faye et al. [22] found that Pd double-sided modified graphene could adsorb up to eight H2 molecules using local density approximation (LDA) of Dmol3 software. The transition metal atoms Sc, Ti and V decorated on both sides of graphene could adsorb four H2 molecules with an average adsorption energy of 0.300–0.500 eV/H2 [23].
Yet the transition metal atom modified graphene is prone to aggregation, which reduces the adsorption site of H2 and weakens the hydrogen storage performance of graphene. Investigations have shown that the introduction of impurity atoms or vacancies can prevent the agglomeration of transition metal atoms and increase the hydrogen storage capacity of graphene [24]. There are two effective means available at present. The first method is a non-metal atom such as B, N etc. doped graphene [25,26,27,28]. Since the B atom has an empty p orbit, it can be used as an electron acceptor to obtain the electrons of other modified atoms, enhancing the adsorption energy of the modified atom [29]. Meanwhile, B-doped graphene has been successfully synthesized in experiments [30]. To introduce pore defects into graphene is another method [31,32]. Compared with non-defective graphene, there is no agglomeration between the metals because the metal atoms are closer to the substrate with a high metal binding energy. Moreover, the porous material adsorbs H2 through Van der Waals (VDW) interactions between the system and the H2 molecule, which is physical adsorption. Porous graphene (PG) [33] is a two-dimensional material synthesized in experiments with high specific surface area, which can store hydrogen efficiently [34]. Du [2] used local density approximation (LDA) of the VASP code to study the adsorption of H2 on the Li atom modified porous graphene. The hydrogen storage capacity of the system can reach 12.00 wt.%, and the average adsorption energy is about 0.243 eV. However, the LDA functional will overestimate the average binding energy of H2 molecules. Additionally, Reunchan [35] and Yuan [36] studied the hydrogen storage properties of Ca and Y atom-modified porous graphene by generalized gradient approximation (GGA) functional theory.
The research found that the combination of these two methods could avoid the aggregation of transition metal atoms on the PG surface, meet the requirement of the U.S. Department of Energy (DOE) for hydrogen storage, but also make the adsorption energy of H2 molecules in the range of reversible hydrogen storage [37,38]. Simultaneously, Sc modified porous graphene possessed a larger amount of hydrogen storage because of the lightest atomic mass in the transition metal atom. However, there are only a few reports on the adsorption mechanism and hydrogen storage capacity of Sc decorated B-doped PG.
Herein, we combined the Sc modification and the B doping methods to modify the porous graphene. In this paper, the most stable adsorption structure and adsorption energy of Sc decorated B-doped PG were manipulated via a first-principles calculation. In addition to this, the performance and mechanism of the H2 molecule adsorption in Sc-modified B-PG system were analyzed deeply. Based on the results, the largest hydrogen storage capacity structure was two Sc atoms located at two sides of the boron-carbon hexagon center in the Sc-B/PG system. Theoretically, the hydrogen storage capacity was 9.13 wt.% with an average adsorption energy of −0.225 eV/H2. B doped PG prevents the Sc atom aggregating and improves the hydrogen storage effectively because it can increase the adsorption energy of the Sc atom and H2 molecule.
2. Calculation Details
Based on the density functional theory, the GGA function of the CASTEP software package [39] was used to select the PBE exchange correlation gradient correction [40]. The Van der Waals correction (DFT-D method) was used in order to avoid the GGA function underestimation of energy in the calculation. All the atoms in the calculation were completely relaxed. The convergence criteria of the optimized structure were as follows: Each atom was subjected to a force below 0.01 eV/Å; the energy convergence tolerance was lower than 5.0 × eV/atom; the self-consistent field convergence threshold was selected as 1.0 × eV/atom between two steps. The vacuum layer was selected as 20 Å along the Z direction of the PG to eliminate the interlayer interaction. Considering the calculation accuracy and calculation cost, the cutoff energy was selected as 500 eV, and the K-point in the Brillouin zone was 7 × 7 × 1.
Formula (1) is used to define the average binding energy () of Sc atoms on B-PG:
| (1) |
among which , and represent the energy in the Sc-modified B-doped PG system, the B-doped PG system and a free Sc atom, respectively. Additionally, n represents the number of adsorbed Sc atoms.
The adsorption energy () and average adsorption energy () of the H2 molecule is defined as:
| (2) |
| (3) |
where , and respectively represent the total energy of the system with i, (i-1) H2 molecules adsorbed and the total energy of a free H2 molecule.
The formation energy [41] is defined as:
| (4) |
where is the energy of the doped porous graphene, and are the number of C, H and B atoms in the doped porous graphene, respectively. , , denote the chemical potential of C, H, B atoms, respectively (number of carbon atoms in graphene, 2, 12).
3. Results and Discussion
3.1. Single B Atom Doped Stable Position
Firstly, six structures of B doped PG were considered. The optimized structures and lattice parameters of 1~6 B-doped PG systems are shown in Table 1. The calculation results show that: As the number of doped B atoms increased, the formation of the doping system became larger and larger, and the energy barrier to be overcome by the doping increased, making doping difficult, which was consistent with Lu et al. [38]. Therefore, we only considered the case of doping one B atom. The optimized structure of a single B-doped PG system is shown in Table 1. The lattice constant was 7.57 Å, the B-H bond length was 1.19 Å, and the formation energy required for doping was 121.3 meV/atom. The lattice constant of 7.57 Å was significantly higher than that of clean porous graphene 7.49 Å [37], but it was consistent with the lattice constant of 7.57 Å obtained by Lu [38]. The increase in the lattice constant was due to the radius of the B atom being greater than the radius of the replaced C atom. The measured B-H bond length of 1.19 Å was completely consistent with that of Lu et al. [38]. The formation energy of the B-PG doping system was 121.30 meV/atom, which was also close to the result of 120.20 meV/atom [38], indicating that the calculation method and accuracy were feasible.
Table 1.
Optimized geometries, lattice constants, representative B-H bond lengths, and formation energies of the B-doped porous graphene (PG) system. Note: Gray, white and pink balls represent C, H and B atoms, respectively.
| 1B/PG | 2B/PG | 3B/PG | 4B/PG | 5B/PG | 6B/PG | |
|---|---|---|---|---|---|---|
| Top View |
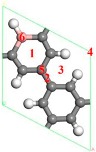
|
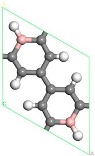
|
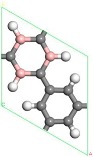
|
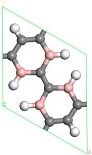
|
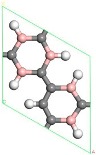
|
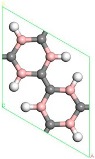
|
| a (Å) | 7.57 | 7.62 | 7.72 | 7.79 | 7.88 | 7.95 |
| (Å) | 1.19 | 1.19 | 1.20 | 1.19 | 1.20 | 1.21 |
| (meV/atom) | 121.30 | 168.03 | 247.61 | 291.03 | 377.73 | 440.99 |
In order to study the effect of B atom doping on the performance of porous graphene, the density of states of a single B atom doped porous graphene system was calculated. The results are shown in Figure 1. It can be seen that there was an energy band passing near the Fermi surface after doping B atoms compared with the clean porous graphene density of states; the change of density of states near the Fermi surface of the doping system mainly came from the contribution of the 2p orbit of the B atom. In addition, the 2p orbit of the B atom overlapped with the porous graphene in the vicinity of the Fermi surface. We obtained the B-PG system band gap of 0.349 eV, which was much smaller than the undoped PG’s band gap value of 2.399 eV. From Figure 1, in the interval of −3.0~−2.0 eV, the peak of PG shifted to the right due to the influence of the B atom 2p orbit. The doping system electrons were close to the Fermi level. This indicates that B atom doping increases the chemical activity of the PG system to some extent and can improve the hydrogen storage performance of the PG system.
Figure 1.

DOS of the porous graphene system with and without B-doped.
3.2. Single Sc Atom Modified B-PG System
3.2.1. Adsorption Structure of Single Sc Atom Modified B-PG
First, the adsorption of Sc atoms on porous graphene doped with a B atom was investigated. When only one Sc atom was modified, six different adsorption sites were considered. As shown in Table 1 on 1B/PG, adsorption sites were named 1, 2, 3, 4, 5, and 6, which represented the hole position of the C ring, C-C bridge, half C ring hole position, large hexagon hole position, C top position and B top position. Results indicated that position 1 was the most easily adsorbed position of a single Sc atom on B-PG, which was similar to the situation of the undoped-PG adsorbing Sc atom [42]. The optimized geometry is shown in Figure 2a. The Sc atom was located at the central hole of the Boron carbon six-membered ring and was slightly deviated. Simultaneously, the B-PG plane was slightly deformed, and the adsorption energy of the Sc atom on the B-PG was −4.004 eV. In our previous study, the adsorption energy of a single Sc atom on clean PG was −2.143 eV [42], which was much smaller than that of single Sc on B-PG of −4.004 eV. It can be seen that B doping can enhance the adsorption activity of the system; compared with pure porous graphene, the Sc atom was closer to the substrate and therefore had greater binding energy on the B-doped PG.
Figure 2.

Sc-B/PG system optimized geometry: (a) Single Sc atom modification; (b) two Sc atoms single-sided modification; (c) two Sc atoms double-sided modified in the same pore position. Gray, white, pink, and purple spheres represent C, H, B, and Sc atoms, respectively).
Figure 3 shows the partial densities of states (PDOS) of the Sc-B/PG system and the density of states of the clean PG system. It can be seen that the 2p orbit of the B atom and the 3d orbit of the Sc atom overlaps in the interval of −2.0~−1.0 eV, indicating that there was a strong interaction between the B atom and Sc atom. The 2p orbit of the C atom overlaps with the 3d orbit of the Sc atom in the range of −3.0~−1.0 eV. In the interval of −1.0 to 0.0 eV, the peaks of the orbits of C and Sc atoms also overlap, meaning that the C and Sc atoms have an existing strong interaction. In addition, comparing the total density of states of the Sc-B/PG system with the clean PG system, we can see that the 3d orbit of the Sc atom significantly changes the properties of the clean porous graphene near the Fermi surface; the peak of the Sc-B/PG system shifts to the left; and the electrons are close to the Fermi level, the system is more stable after doping the Sc and B atom.
Figure 3.

DOS of the Sc-B/PG system and PG system.
3.2.2. The Adsorption of H2 on a Single Sc Atom Modified B-PG System
A single Sc-modified B-doped porous graphene system can adsorb 5 H2 molecules, and the optimized structures are given in Figure 4. Moreover, Table 2 lists the adsorption and average adsorption energy of H2, the distance between Sc atom and H2 molecules, and the distance between the Sc and C atom on the B-PG. A single H2 molecule has multiple adsorption sites on Sc-modified B-PG, including the C-C bridge position, the C-H bridge position, the C atom top position, and the Sc atom top position. It has been discovered that the most stable adsorption position of the first H2 molecule is the top position of the C atom. This H2 molecule has an adsorption energy of −0.677 eV. Meanwhile, the H-H bond was stretched to 0.868 Å. To further explore the hydrogen storage behavior of the system, H2 molecules were continuously added to the system. Table 2 indicates that H2 molecules’ binding energies fluctuated with the number of adsorbed H2 molecules. The third H2 molecule adsorbed above the B-H bond and was slightly away from the hexagonal ring, so the adsorption energy decreased suddenly. However, the trend of was a complete contrast to the trend of the H2 molecule adsorption energy. That is, the smaller the distance between Sc and the B-PG substrate, the greater the adsorption energy of hydrogen, and vice versa because the addition of hydrogen affects the position of Sc, and the position of Sc in turn affects the adsorption of hydrogen. The system eventually reaches a steady state through this mutually adjusted process. As a result of the symmetry of the H2 molecular bonding configuration, all H2 molecules were symmetrically distributed on every side of the Sc atom when the fourth H2 was adsorbed. The distance from the H2 molecule to the Sc atom always increased, but when the fourth H2 molecule adsorbed, the suddenly decreased. At this time, the Sc atom was located at the center of the carbon ring doped by the B atom. Additionally, the first four H2 molecules were located on the same horizontal plane, which was parallel to the PG layer. Due to the limited space around the Sc atom and the repulsion between the adsorbed H2 molecules, the fifth H2 molecule moved to the upper layer after relaxation in Figure 4e. Therefore, the fifth H2 molecule had a H-H bond length of 0.762 Å, and it had a minimum adsorption energy of −0.187 eV, which was still bigger than −0.093 eV of the Sc-PG system without B doped [42]. The bond length of the adsorbed H2 molecule was in the range of 0.762~ 0.831 Å, and no dissociation of the H2 molecule was found. Therefore, the B doped PG decorated by Sc was more suitable for hydrogen storage at room temperature, and the practical application prospect was greater. The PG system modified by a single Sc atom can adsorb up to 5 H2 molecules due to the doping of B atom. In a single Sc-modified B-doped porous graphene system, the average adsorption energy (−0.515 eV/H2) and the hydrogen storage capacity (4.91 wt.%) are better than that of a Ti-PG system (−0.486 eV) [43].
Figure 4.

The optimized geometries of a single Sc-modified B-PG system with one H2 molecule (a), two H2 molecules (b), three H2 molecules (c), four H2 molecules (d) and five H2 molecules (e) adsorbed.
Table 2.
Calculated adsorption energy and average adsorption energy of H2, distance between Sc atom and H2 molecules, and minimum distance between Sc atom and C atom in Sc-modified B-PG system.
| Number of H2 | 1H2 | 2H2 | 3H2 | 4H2 | 5H2 |
|---|---|---|---|---|---|
| (eV) | −0.677 | −0.682 | −0.416 | −0.614 | −0.187 |
| (eV) | −0.677 | −0.680 | −0.592 | −0.597 | −0.515 |
| (Å) | 2.034 | 2.049 | 2.146 | 2.061 | 2.867 |
| (Å) | 2.274 | 2.229 | 2.380 | 2.333 | 2.385 |
Considering the interaction among the Sc atom with the adsorbed H2 molecule, we analyzed the partial densities of states (PDOS) of the H2 molecule and the Sc atom in Figure 5. It can be seen that the band broadening occurred in the H2 molecule from −11.0 to −8.0 eV. In the interval of −9.5~−7.5 eV, the 1s orbit of H2 overlapped with the 3d orbit of Sc, indicating that the 1s orbit of H2 and the 3d orbit of Sc had a existing interaction. When 1~4 H2 molecules were adsorbed on the Sc-decorated B-PG system, it could be seen that the peak of the H2 1s orbit overlapped with the peak of the Sc 3d orbit in the range of −3.0~0.0 eV, which was in accordance with the strong adsorption energy of the H2 molecules (except the fifth hydrogen).
Figure 5.

PDOS of Sc-decorated B-PG with one to five H2 molecules adsorbed.
To further investigate the combined effects of the B atom and Sc atom on H2 molecule adsorption, the PDOS of one H2 molecule adsorbed in a single Sc-modified B-PG system was analyzed (as shown in Figure 6). From 1.0 to 0.0 eV, there were overlapping peaks between B 2p orbit, Sc 3d orbit and 1s orbit of the H2 molecule, which means that B and Sc both played a role in H2 adsorption. In addition, it can be observed that the 2p orbit of B atoms overlapped with the 3d orbit of Sc atoms in most energy intervals, indicating that there was a strong interaction between B and Sc, which further explained why Sc had a higher binding energy in the B doping system.
Figure 6.

PDOS of Sc-decorated B-PG with one H2 molecules adsorbed.
The adsorption mechanism could be better understood by analyzing the charge density difference. Figure 7 shows the electronic charge density difference of the system after H2 molecules adsorbed. The blue and yellow isosurfaces represented the electron accumulation and electron loss regions, respectively, and the isosurface unit was 0.007 e/. It can be observed from Figure 7a,b that the yellow electron-depleting region was concentrated between the Sc atom and the B-PG, meaning that there was a large amount of charge transfer from the Sc atom to the B-PG. We also analyzed the Mulliken charge population before and after the adsorption of the Sc atom in the B-PG system. We found that the B atom 2p orbit and the C atom had an accumulation of electrons, while the 4s orbit of Sc lost electrons by 1.5 e. Combined with the PDOS of Sc, B and C atom in Figure 3, it was found that the 4s orbit of Sc transferred some electrons to the 2p orbit of B and C, respectively. The charge population indicated that the 3d orbit of Sc obtained 0.45 e electrons, which meant that the B-PG transferred electrons to the Sc atom. On the whole, the Sc carries a positive charge, and the B-PG layer carries a negative charge. Therefore, an electric field is formed between the Sc atom and the B-PG. The adsorption energy of Sc atoms on B-PG was large due to the strong electrostatic Coulomb interaction between Sc and B-PG, and the orbital interaction between Sc, B and C atoms. Since the Sc atom has a positive charge, this will attract the negative charge in H2 and accumulate near the metal. It can also be seen from Figure 7 that the blue electron region was concentrated between H2 and Sc. Near the side of the Sc atom, polarization occurred in the perpendicular direction of the H2 molecule. It has been indicated that the H2 molecule undergoes charge redistribution due to the electrostatic field between the Sc atom and the B-PG. Combined with the Mulliken charge population of the 1Sc-B-PG system it adsorbed two H2 molecules, the H atoms in the polarized H2 molecule were negatively charged at −0.17 e and −0.16 e. Conversely, the Sc atom was positively charged at 1.94 e. Therefore, there was a Coulomb attraction between the H2 molecule and the Sc atom. The C atom also had a partial charge transfer on B-PG, indicating that these C atoms also play a role on the H2 molecule adsorption. Consequently, the adsorption of H2 molecules in the Sc-modified B-PG system is mainly attributed to the orbital interaction between H and Sc atoms. Furthermore, the Coulomb attraction between H2 molecules (negatively charged) and Sc atoms (positively charged) strengthen the adsorption of H2 molecules.
Figure 7.

Electronic charge density difference for the Sc-B/PG system in the presence of (a) one H2 molecule; (b) two H2 molecules; (c) five H2 molecules.
To better understand the bonding environment of all B-H bonds in this structure after adsorption, we calculated a Bond Population of B-H bond and Mulliken charge Population of B, H, and Sc atoms in B-PG, Sc-B/PG and H2 adsorbed Sc-B/PG system, where H was the H atom in the first to fifth hydrogen molecules adsorbed. As shown in Table 3, the B-H population gradually increased as the hydrogen molecules continued to adsorb. After Sc atom modification, the positive charge of B atom decreased rapidly, indicating that Sc was stable on B-PG. Thereafter, the positive charge of the B atom fluctuated with the adsorption of hydrogen. The positive charge of Sc in the hydrogen storage process had been increasing. After the first H2 molecule was adsorbed by the Sc-B/PG structure, the Sc atom was charged to 1.72 e, which is greater than that of 1.56 e in the undoped B atom system [42]. It was shown that after doping the B atom, the catalytic effect of Sc atoms in hydrogen storage was more obvious. As the hydrogen molecules continuously adsorbed, the H atom in H2 molecules started to be negatively charged and the charge amount gradually decreased. The H atom of the first H2 molecule after doping B atoms was charged −0.17 e, and its absolute value was greater than that of −0.13 e in the undoped system [42]. This indicated that doping B atom enhanced the interaction between H2, Sc atoms and PG, which was more conducive to hydrogen storage.
Table 3.
Calculated Bond Population of the B-H bond and Mulliken Charge Population of B, H, Sc atoms in B-PG, Sc-B/PG and H2 adsorbed Sc-B/PG system.
| System | B-PG | Sc-B/PG | 1H2 | 2H2 | 3H2 | 4H2 | 5H2 |
|---|---|---|---|---|---|---|---|
| B-H | 1.06 | 1.04 | 1.03 | 1.04 | 1.05 | 1.05 | 1.06 |
| B (e) | 0.33 | 0.04 | 0.04 | 0.03 | 0.06 | 0.06 | 0.05 |
| Sc (e) | None | 1.15 | 1.72 | 1.94 | 2.18 | 2.46 | 2.64 |
| H (e) | None | None | −0.17 | −0.16 | −0.13 | −0.16 | −0.10 |
3.3. Two Sc Atoms Modified B-PG System
3.3.1. Adsorption Structure of two Sc Atoms Modified B-PG
Transition metal atoms are prone to aggregation on the surface of graphene, resulting in a decrease in the free surface area of the graphene. Metal aggregation is mainly attributed to the high cohesive energy of the atoms [44,45,46]. The cohesive energy of the Sc atom is 3.900 eV [47], each Sc atom is equivalent to an H2 adsorption site. To further increase the amount of hydrogen storage, a second Sc atom is added to the same side of the system. Figure 2b shows one of the most stable geometries for adsorbing two Sc atoms on B-PG. It can be seen that the second Sc atom tended to adsorb at the center of the other carbon ring instead of agglomeration with the first Sc atom, which was attributed to the Coulomb repulsion between the two Sc atoms (the two Sc atoms have a positive charge of 0.90 e, 0.97 e, respectively), and there was a strong interaction between the Sc atom and the B-PG. The average binding energy of the two Sc atoms on B-PG was −4.069 eV, and its absolute value was greater than the cohesive energy of the Sc atom, which was 3.900 eV. This means that the adsorption structure of the two Sc atoms on PG is more stable than that of the single Sc atom on B-PG.
The structure of two Sc atoms on both sides of B-PG was also studied to open up enough space to adsorb H2 molecules. Each transition metal atom is an active adsorption site. The double-sided modification of B-PG by the transition metal atom can raise the hydrogen storage area. On account of Figure 2a, the second Sc atom had four adsorption sites on the opposite side of the B-PG, including two carbon ring centers, C-C bridge site, and half C ring pore sites on the opposite side. The most stable adsorption structure was obtained as shown in Figure 2c. The average binding energy of two Sc atoms was −4.085 eV, larger than that of two Sc atoms in the clean PG system. Therefore, the presence of B atoms in PG prevented the Sc atoms from aggregating. In addition, it can be seen from Figure 2a,b that the B-PG system underwent slight deformation after Sc atoms were adsorbed. However, Figure 2c did not undergo any deformation, which was related to the symmetric distribution of Sc atoms.
3.3.2. Hydrogen Storage of two Sc Atoms Modified B-PG
The B-PG system with two Sc atoms modified on one side can adsorb 10 H2 molecules. The optimized geometry structure is shown in Figure 8. The average adsorption energy of the B-PG system was about −0.339 eV/H2 with the hydrogen storage capacity (7.73 wt.%). Because the adsorption energy of two Sc atoms on B-PG was larger than their cohesion energy, the distance between them was close without bonding.
Figure 8.
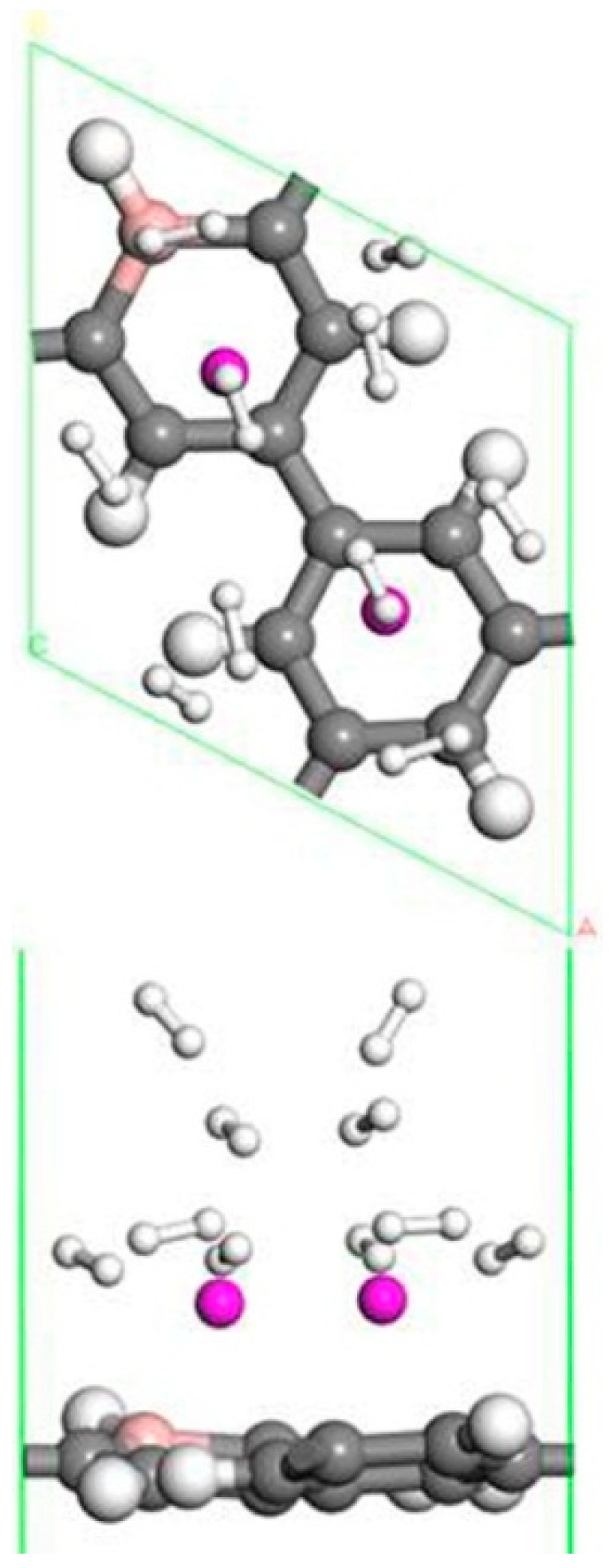
The optimized geometries for ten H2 molecules adsorbed on two Sc atoms single-sided modified B-PG.
Figure 2c is a stable structure of two Sc atoms modified B-PG on both sides, and the structure after hydrogen storage is shown in Figure 9. The distance between the two Sc atoms was large enough to prevent the aggregation where double-sided modified B-PG was concerned. There was no deformation that occurred in the B doped porous graphene surface, which was different from the distortion in the single Sc modified B-PG after hydrogen storage. The first eight H2 molecules were symmetrically distributed, but the ninth to twelfth H2 molecules gradually moved away from the Sc atoms and tended to adsorb to the top of the C-H bond of adjacent carbon rings. Therefore, the average adsorption energy of H2 decreased. Two Sc double-sided modified B-PG systems can adsorb up to 12 H2 molecules with an average adsorption energy of −0.225 eV/H2. According to the standards of the U.S. Department of Energy and the International Energy Agency, the hydrogen storage capacity of ideal hydrogen storage materials should be greater than 5.50 wt.%, and the adsorption energy between H2 molecules and materials should be from 0.200 to 0.700 eV [48,49]. Therefore, the average adsorption energy of the H2 molecule was also in the range of reversible hydrogen storage. The corresponding theoretical hydrogen storage capacity is 9.13 wt.%, which is higher than the storage hydrogen content of the Sc-PG system 9.09 wt.% [42] and is greater than the amount of hydrogen storage of the Y-PG system 7.87 wt.% [36]. The interaction between the H2 molecule, the Sc atom and the B-PG system in the two Sc atoms modified the B-PG system and was similar to the single Sc atom modification B-PG system, and is not described here. By comparing Figure 4e, Figure 8 and Figure 9f, it can be seen that the single Sc atom modified B-PG and the two Sc atoms single-sided modified B-PG adsorbed H2 molecules were layered. However, there was no hydrogen stratification with two Sc double-sided decorated B-PG. Moreover, the latter two H2 molecules adsorbed above the C-H bond of another carbon six-membered ring. Therefore, the hydrogen storage spaces were larger in the Sc double-sided modification B-PG system than the Sc single-sided modification. In addition, the H2 molecules around a single Sc were parallel to the B-PG plane. When the two Sc atoms were modified on the same side, the H2 molecule changed from parallel to vertical, but they were not completely perpendicular to the B-PG surface due to the interaction. The perpendicular adsorption of H2 molecules is more likely to occur when two Sc atoms are modified at double sides, which is due to the interaction of H2 molecules on both sides of the B-PG. In summary, the two Sc located in the same boron-carbon ring position on the opposite side of B-PG is the most suitable structure for hydrogen storage, and Sc modified B-PG is a promising hydrogen storage material.
Figure 9.

Optimized geometries of two Sc double-sided modified B-PG systems with two H2 molecules (a), four H2 molecules (b), six H2 molecules (c), eight H2 molecules (d), ten H2 molecules (e), and twelve H2 molecules (f) adsorbed.
4. Conclusions
We unambiguously investigated the hydrogen storage of the B atom doped Sc decorated PG. With an average adsorption energy (−0.515 eV/H2), the most stable adsorption position could adsorb five H2 molecules on the center of the boron-carbon ring for a single Sc atom system. By adding a second Sc atom to another side of the PG system, the hydrogen storage capacity effectively could be improved. When the two Sc atoms were located at both sides of the central boron-carbon hexagon, the theoretical hydrogen storage capacity could reach the maximum (~9.13 wt.%), which could adsorb twelve H2 molecules. At the same time, the two Sc atoms structure was the most suitable hydrogen storage for the Sc-B/PG system, possessing the average adsorption energy (−0.225 eV/H2). Furthermore, the adsorption of these H2 molecules in the Sc-modified B/PG system was also responsible for two aspects via the analysis of DOS and charge population: (i) The orbital interaction between H and Sc atoms; (ii) the Coulomb attraction between H2 molecules (negatively charge) and Sc atoms (positively charge). The H2 molecules were negatively charged due to polarization of the electrostatic field between Sc and B-PG. Therefore, Sc-modified B-doped porous graphene is expected to be used in the field of hydrogen storage.
Acknowledgments
The authors gratefully acknowledge the HongLiu first-class disciplines Development Program of the Lanzhou University of Technology.
Author Contributions
We would like to give great thanks to Y.C. because he designed the project. J.W. performed the calculations and prepared the manuscript, C.Z. revised the paper, L.Y. and M.Z. analyzed the data, and all authors discussed the results and commented on the manuscript.
Funding
This work was supported by the National Natural Science Foundation of China (No. 51562022) and the State Key Laboratory of Advanced Processing and Recycling of Non-ferrous Metals, Lanzhou University of Technology (No. SKLAB02014004).
Conflicts of Interest
The authors declare no conflicts of interest.
Footnotes
Sample Availability: Samples of the compounds are not available from the authors.
References
- 1.Lei Y., Shevlin S.A., Zhu W., Guo Z.X. Hydrogen-induced magnetization and tunable hydrogen storage in graphitic structures. Phys. Rev. B. 2008;77 doi: 10.1103/PhysRevB.77.134114. [DOI] [Google Scholar]
- 2.Du A., Zhu Z., Smith S.C. Multifunctional Porous Graphene for Nanoelectronics and Hydrogen Storage: New Properties Revealed by First Principle Calculations. J. Am. Chem. Soc. 2010;132:2876–2877. doi: 10.1021/ja100156d. [DOI] [PubMed] [Google Scholar]
- 3.Wang Y., Meng Z., Liu Y., You D., Wu K., Lv J., Wang X., Deng K., Rao D., Lu R. Lithium decoration of three dimensional boron-doped graphene frameworks for high-capacity hydrogen storage. Appl. Phys. Lett. 2015;106:63901. doi: 10.1063/1.4907975. [DOI] [Google Scholar]
- 4.Hussain T., Pathak B., Ramzan M., Maark T.A., Ahuja R. Calcium doped graphane as a hydrogen storage material. Appl. Phys. Lett. 2012;100:183902. doi: 10.1063/1.4710526. [DOI] [Google Scholar]
- 5.Song N., Wang Y., Zheng Y., Zhang J., Xu B., Sun Q., Jia Y. New template for Li and Ca decoration and hydrogen adsorption on graphene-like SiC: A first-principles study. Comput. Mater. Sci. 2015;99:150–155. doi: 10.1016/j.commatsci.2014.12.016. [DOI] [Google Scholar]
- 6.Gao Y., Zhao N., Li J., Liu E., He C., Shi C. Hydrogen spillover storage on Ca-decorated graphene. Int. J. Hydrog. Energy. 2012;37:11835–11841. doi: 10.1016/j.ijhydene.2012.05.029. [DOI] [Google Scholar]
- 7.Seenithurai S., Kodi Pandyan R., Vinodh Kumar S., Mahendran M. H2 adsorption in Ni and passivated Ni doped 4 Å single walled carbon nanotube. Int. J. Hydrog. Energy. 2013;38:7376–7381. doi: 10.1016/j.ijhydene.2013.04.085. [DOI] [Google Scholar]
- 8.Bhattacharya A., Bhattacharya S., Majumder C., Das G.P. Transition-Metal Decoration Enhanced Room-Temperature Hydrogen Storage in a Defect-Modulated Graphene Sheet. J. Phys. Chem. C. 2010;114:10297–10301. doi: 10.1021/jp100230c. [DOI] [Google Scholar]
- 9.Ramos-Castillo C.M., Reveles J.U., Cifuentes-Quintal M.E., Zope R.R., de Coss R. Ti4- and Ni4-Doped Defective Graphene Nanoplatelets as Efficient Materials for Hydrogen Storage. J. Phys. Chem. C. 2016;120:5001–5009. doi: 10.1021/acs.jpcc.5b12711. [DOI] [Google Scholar]
- 10.Ramos-Castillo C.M., Reveles J.U., Zope R.R., de Coss R. Palladium Clusters Supported on Graphene Monovacancies for Hydrogen Storage. J. Phys. Chem. C. 2015;119:8402–8409. doi: 10.1021/acs.jpcc.5b02358. [DOI] [Google Scholar]
- 11.Lebon A., Carrete J., Gallego L.J., Vega A. Ti-decorated zigzag graphene nanoribbons for hydrogen storage. A van der Waals-corrected density-functional study. Int. J. Hydrog. Energy. 2015;40:4960–4968. doi: 10.1016/j.ijhydene.2014.12.134. [DOI] [Google Scholar]
- 12.Chu S., Hu L., Hu X., Yang M., Deng J. Titanium-embedded graphene as high-capacity hydrogen-storage media. Int. J. Hydrog. Energy. 2011;36:12324–12328. doi: 10.1016/j.ijhydene.2011.07.015. [DOI] [Google Scholar]
- 13.Liu Y., Ren L., He Y., Cheng H.-P. Titanium-decorated graphene for high-capacity hydrogen storage studied by density functional simulations. J. Phys. Condens. Matter. 2010;22:445301. doi: 10.1088/0953-8984/22/44/445301. [DOI] [PubMed] [Google Scholar]
- 14.Chi S.-S., Liu Y., Zhao N., Guo X., Nan C.-W., Fan L.-Z. Solid polymer electrolyte soft interface layer with 3D lithium anode for all-solid-state lithium batteries. Energy Storage Mater. 2019;17:309–316. doi: 10.1016/j.ensm.2018.07.004. [DOI] [Google Scholar]
- 15.Luo Z., Fan X., Pan R., An Y. A first-principles study of Sc-decorated graphene with pyridinic-N defects for hydrogen storage. Int. J. Hydrog. Energy. 2017;42:3106–3113. doi: 10.1016/j.ijhydene.2016.11.039. [DOI] [Google Scholar]
- 16.Zhang H., Xia G., Zhang J., Sun D., Guo Z., Yu X. Graphene-Tailored Thermodynamics and Kinetics to Fabricate Metal Borohydride Nanoparticles with High Purity and Enhanced Reversibility. Adv. Energy Mater. 2018;8:1702975. doi: 10.1002/aenm.201702975. [DOI] [Google Scholar]
- 17.Chong L., Zeng X., Ding W., Liu D.-J., Zou J. NaBH4 in “Graphene Wrapper:” Significantly Enhanced Hydrogen Storage Capacity and Regenerability through Nanoencapsulation. Adv. Mater. 2015;27:5070–5074. doi: 10.1002/adma.201500831. [DOI] [PubMed] [Google Scholar]
- 18.Xia G., Tan Y., Chen X., Sun D., Guo Z., Liu H., Ouyang L., Zhu M., Yu X. Monodisperse Magnesium Hydride Nanoparticles Uniformly Self-Assembled on Graphene. Adv. Mater. 2015;27:5981–5988. doi: 10.1002/adma.201502005. [DOI] [PubMed] [Google Scholar]
- 19.Yang H., Lombardo L., Luo W., Kim W., Züttel A. Hydrogen storage properties of various carbon supported NaBH4 prepared via metathesis. Int. J. Hydrog. Energy. 2018;43:7108–7116. doi: 10.1016/j.ijhydene.2018.02.142. [DOI] [Google Scholar]
- 20.Venkataramanan N.S., Suvitha A., Mizuseki H., Kawazoe Y. A theoretical study of the effects of transition metal dopants on the adsorption and dissociation of hydrogen on nickel clusters. Int. J. Quantum Chem. 2013;113:1940–1948. doi: 10.1002/qua.24418. [DOI] [Google Scholar]
- 21.Rojas M.I., Leiva E.P.M. Density functional theory study of a graphene sheet modified with titanium in contact with different adsorbates. Phys. Rev. B. 2007;76 doi: 10.1103/PhysRevB.76.155415. [DOI] [Google Scholar]
- 22.Faye O., Szpunar J.A., Szpunar B., Beye A.C. Hydrogen adsorption and storage on Palladium—Functionalized graphene with NH-dopant: A first principles calculation. Appl. Surf. Sci. 2017;392:362–374. doi: 10.1016/j.apsusc.2016.09.032. [DOI] [Google Scholar]
- 23.Durgun E., Ciraci S., Yildirim T. Functionalization of carbon-based nanostructures with light transition-metal atoms for hydrogen storage. Phys. Rev. B. 2008;77 doi: 10.1103/PhysRevB.77.085405. [DOI] [Google Scholar]
- 24.Kim G., Jhi S.-H., Park N., Louie S.G., Cohen M.L. Optimization of metal dispersion in doped graphitic materials for hydrogen storage. Phys. Rev. B. 2008;78 doi: 10.1103/PhysRevB.78.085408. [DOI] [Google Scholar]
- 25.Huang H.-W., Hsieh H.-J., Lin I.-H., Tong Y.-J., Chen H.-T. Hydrogen Adsorption and Storage in Heteroatoms (B, N) Modified Carbon-Based Materials Decorated with Alkali Metals: A Computational Study. J. Phys. Chem. C. 2015;119:7662–7669. doi: 10.1021/acs.jpcc.5b01416. [DOI] [Google Scholar]
- 26.Nachimuthu S., Lai P.-J., Jiang J.-C. Efficient hydrogen storage in boron doped graphene decorated by transition metals—A first-principles study. Carbon. 2014;73:132–140. doi: 10.1016/j.carbon.2014.02.048. [DOI] [Google Scholar]
- 27.Liu W., Liu Y., Wang R. Prediction of hydrogen storage on Y-decorated graphene: A density functional theory study. Appl. Surf. Sci. 2014;296:204–208. doi: 10.1016/j.apsusc.2014.01.087. [DOI] [Google Scholar]
- 28.Tokarev A., Avdeenkov A.V., Langmi H., Bessarabov D.G. Modeling hydrogen storage in boron-substituted graphene decorated with potassium metal atoms: Hydrogen storage in boron-substituted graphene with potassium atoms. Int. J. Energy Res. 2015;39:524–528. doi: 10.1002/er.3268. [DOI] [Google Scholar]
- 29.Beheshti E., Nojeh A., Servati P. A first-principles study of calcium-decorated, boron-doped graphene for high capacity hydrogen storage. Carbon. 2011;49:1561–1567. doi: 10.1016/j.carbon.2010.12.023. [DOI] [Google Scholar]
- 30.Wang H., Zhou Y., Wu D., Liao L., Zhao S., Peng H., Liu Z. Synthesis of Boron-Doped Graphene Monolayers Using the Sole Solid Feedstock by Chemical Vapor Deposition. Small. 2013;9:1316–1320. doi: 10.1002/smll.201203021. [DOI] [PubMed] [Google Scholar]
- 31.Rangel E., Ramírez-Arellano J.M., Carrillo I., Magana L.F. Hydrogen adsorption around lithium atoms anchored on graphene vacancies. Int. J. Hydrog. Energy. 2011;36:13657–13662. doi: 10.1016/j.ijhydene.2011.08.023. [DOI] [Google Scholar]
- 32.Choudhary A., Malakkal L., Siripurapu R.K., Szpunar B., Szpunar J. First principles calculations of hydrogen storage on Cu and Pd-decorated graphene. Int. J. Hydrog. Energy. 2016;41:17652–17656. doi: 10.1016/j.ijhydene.2016.07.147. [DOI] [Google Scholar]
- 33.Bieri M., Treier M., Cai J., Aït-Mansour K., Ruffieux P., Gröning O., Gröning P., Kastler M., Rieger R., Feng X., et al. Porous graphenes: two-dimensional polymer synthesis with atomic precision. Chem. Commun. 2009:6919–6921. doi: 10.1039/b915190g. [DOI] [PubMed] [Google Scholar]
- 34.Huang C., Wu H., Deng K., Tang W., Kan E. Improved permeability and selectivity in porous graphene for hydrogen purification. Phys Chem Chem Phys. 2014;16:25755–25759. doi: 10.1039/C4CP04385E. [DOI] [PubMed] [Google Scholar]
- 35.Reunchan P., Jhi S.-H. Metal-dispersed porous graphene for hydrogen storage. Appl. Phys. Lett. 2011;98:93103. doi: 10.1063/1.3560468. [DOI] [Google Scholar]
- 36.Yuan L., Chen Y., Kang L., Zhang C., Wang D., Wang C., Zhang M., Wu X. First-principles investigation of hydrogen storage capacity of Y-decorated porous graphene. Appl. Surf. Sci. 2017;399:463–468. doi: 10.1016/j.apsusc.2016.12.054. [DOI] [Google Scholar]
- 37.Lu R., Rao D., Meng Z., Zhang X., Xu G., Liu Y., Kan E., Xiao C., Deng K. Boron-substituted graphyne as a versatile material with high storage capacities of Li and H2: a multiscale theoretical study. Phys. Chem. Chem. Phys. 2013;15:16120. doi: 10.1039/c3cp52364k. [DOI] [PubMed] [Google Scholar]
- 38.Lu R., Rao D., Lu Z., Qian J., Li F., Wu H., Wang Y., Xiao C., Deng K., Kan E., et al. Prominently Improved Hydrogen Purification and Dispersive Metal Binding for Hydrogen Storage by Substitutional Doping in Porous Graphene. J. Phys. Chem. C. 2012;116:21291–21296. doi: 10.1021/jp308195m. [DOI] [Google Scholar]
- 39.Sun J., Wang H.-T., He J., Tian Y. Ab initio investigations of optical properties of the high-pressure phases of ZnO. Phys. Rev. B. 2005;71 doi: 10.1103/PhysRevB.71.125132. [DOI] [Google Scholar]
- 40.Rao D., Lu R., Meng Z., Wang Y., Lu Z., Liu Y., Chen X., Kan E., Xiao C., Deng K., et al. Electronic properties and hydrogen storage application of designed porous nanotubes from a polyphenylene network. Int. J. Hydrog. Energy. 2014;39:18966–18975. doi: 10.1016/j.ijhydene.2014.09.112. [DOI] [Google Scholar]
- 41.Rao D. Ph.D. Thesis. Nanjing University of Science Technology; Nanjing, China: 2013. Improve hydrogen storage capacities of several porous materials; p. 68. [Google Scholar]
- 42.Chen Y., Wang J., Yuan L., Zhang M., Zhang C. Sc-Decorated Porous Graphene for High-Capacity Hydrogen Storage: First-Principles Calculations. Materials. 2017;10:894. doi: 10.3390/ma10080894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yuan L., Kang L., Chen Y., Wang D., Gong J., Wang C., Zhang M., Wu X. Hydrogen storage capacity on Ti-decorated porous graphene: First-principles investigation. Appl. Surf. Sci. 2018;434:843–849. doi: 10.1016/j.apsusc.2017.10.231. [DOI] [Google Scholar]
- 44.Knick C.R. Master’s Thesis. Wright State University; Dayton, OH, USA: 2012. Modeling the Exfoliation Rate of Graphene Nanoplatelet Production and Application for Hydrogen Storage; pp. 52–60. [Google Scholar]
- 45.Ao Z., Dou S., Xu Z., Jiang Q., Wang G. Hydrogen storage in porous graphene with Al decoration. Int. J. Hydrog. Energy. 2014;39:16244–16251. doi: 10.1016/j.ijhydene.2014.01.044. [DOI] [Google Scholar]
- 46.Nam H., Jang S., Lee W. Strategies for the Modification of Adhesion at Graphene/Al Interfaces: A First Principles Study. J. Nanosci. Nanotechnol. 2014;14:7674–7678. doi: 10.1166/jnn.2014.9473. [DOI] [PubMed] [Google Scholar]
- 47.Chen M., Yang X.-B., Cui J., Tang J.-J., Gan L.-Y., Zhu M., Zhao Y.-J. Stability of transition metals on Mg (0001) surfaces and their effects on hydrogen adsorption. Int. J. Hydrog. Energy. 2012;37:309–317. doi: 10.1016/j.ijhydene.2011.09.065. [DOI] [Google Scholar]
- 48.Zhang Z.W., Li J.C., Jiang Q. Hydrogen Adsorption on Eu/SWCNT Systems: A DFT Study. J. Phys. Chem. C. 2010;114:7733–7737. doi: 10.1021/jp100017y. [DOI] [Google Scholar]
- 49.Sahaym U., Norton M.G. Advances in the application of nanotechnology in enabling a “hydrogen economy”. J. Mater. Sci. 2008;43:5395–5429. doi: 10.1007/s10853-008-2749-0. [DOI] [Google Scholar]