

Abstract
Background:
Anomalous origin of the left coronary artery from the pulmonary artery (ALCAPA) is a rare congenital coronary abnormality associated with early infant mortality and adult sudden death. As it predominantly presents in the first year of life, diagnosis in living adults is extremely rare. Current management is based on limited case series or extrapolated from pediatric cases. Modern advances in noninvasive cardiac imaging have substantially increased the number of diagnoses, uncovering a large adult population that has not been reviewed.
Hypothesis:
The availability of newer diagnostic modalities correlates with an increasing incidence in an older cohort, and true association between sudden death and ALCAPA may be lower, especially among older patients.
Methods:
A comprehensive literature search was performed for all case reports of ALCAPA on MEDLINE and PubMed using the keywords ALCAPA, Bland‐White‐Garland, and coronary anomaly; and augmented by references from published case reports from 1908 to 2008. All adult cases, defined by age 18 years and older, were reviewed for this article.
Results:
One hundred fifty‐one adult cases of ALCAPA are described, in addition to the case of an asymptomatic 53‐year‐old woman. The average reported age was 41 years old with the oldest being 83. Sixty‐six percent of the patients presented with symptoms of angina, dyspnea, palpitations, or fatigue; 17% presented with ventricular arrhythmia, syncope, or sudden death; and 14% were asymptomatic. Twelve percent were diagnosed at autopsy. The majority had some form of surgical correction during their clinical course.
Conclusions:
ALCAPA is a rare and life‐threatening condition in adults. The availability of newer, less invasive diagnostic modalities has resulted in more frequent identification of this condition in an older cohort. © 2011 Wiley Periodicals, Inc.
The authors have no funding, financial relationships, or conflicts of interest to disclose.
Introduction
Anomalous origin of the left coronary artery from the pulmonary artery (ALCAPA) is a rare congenital coronary abnormality associated with early infant mortality and adult sudden death. The incidence of ALCAPA is estimated at 1/300 000 live births comprising between 0.24% and 0.46% of congenital cardiac disease.1, 2, 3, 4 The first reported cases were described by Konstantinowitsch in 1906 of a 2‐day‐old infant5 and Abrikossoff in 1911 of a 5‐month‐old infant.6 Without treatment, approximately 90% of infants die within the first year of life.7 As it predominantly presents in the first year of life, diagnosis in living adults is extremely rare and most information is thus derived from individual case reports or extrapolated from pediatric cases.
Modern advances in noninvasive cardiac imaging have substantially increased the number of diagnosed cases of adults with benign clinical outcomes. Early autopsy studies reported the average age of sudden death in untreated adult ALCAPA patients was 35 years old,8, 9, 10 prompting a recommendation that all diagnosed adults undergo surgical treatment. Nevertheless, with the increased diagnosis of adult ALCAPA, the true association between sudden death and ALCAPA may be lower, especially among older patients.
We report the case of an asymptomatic 53‐year‐old woman diagnosed by multislice computed tomography (MSCT) and present a comprehensive review of 151 adult cases of ALCAPA as reported in the world literature, including those diagnosed with newer modalities of cardiac computed tomography (CT) and magnetic resonance imaging (MRI).
Methods
A comprehensive literature search was performed for all case reports of ALCAPA in MEDLINE and PubMed, using the keywords ALCAPA, Bland‐White‐Garland, and coronary anomaly. This list was augmented by references from published case reports dating from 1908 to 2008. All adult cases, defined by age 18 years and older, were reviewed for inclusion in this article. The use of secondary review articles to obtain clinical details was limited to 19 foreign language cases. All other data was extracted from primary case reports of angiographically or autopsy‐confirmed ALCAPA. Translation of non‐English language reports was obtained using WorldLingo.com software (WorldLingo Translations LLC, Las Vegas, NV; http://www.worldlingo.com).
Results
Case Report
A 53‐year‐old mother of 3 with no known cardiac history presented for evaluation prior to elective gynecologic surgery. She has no significant cardiovascular risk factors and denied any cardiovascular symptoms. Her physical examination was unremarkable, including the absence of a murmur. A baseline electrocardiogram (ECG) revealed anteroseptal Q‐waves prompting an exercise stress echocardiogram. This study revealed inferolateral ST depressions with the echocardiography demonstrating inferior, inferolateral, and inferoseptal wall hypokinesis. Subsequent coronary angiography (Figure 1A and 1B) demonstrated an inability to visualize the left coronary artery (LCA). The right coronary artery (RCA) was a large vessel with the suggestion of right to left coronary filling of the left anterior descending (LAD) artery and main pulmonary artery (PA). Because of the poor angiographic visualization and the suspicion of ALCAPA, cardiac MSCT imaging was obtained (Figure 2A–2E). This confirmed a dilated RCA originating from the right coronary cusp with extensive apical collateralization to the LAD. The LCA can be traced to its origination from the posterior aspect of the main PA.
Figure 1.
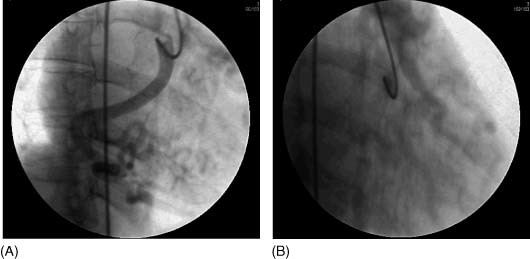
Coronary angiography at 30 degrees left anterior oblique and 20 degrees right anterior oblique. (A) Poor opacification of a dilated and tortuous right coronary artery (RCA) shows a patent vessel with extensive posterior and apical collateralization. (B) Retrograde filling of the left anterior descending artery and main pulmonary artery are suggested on delayed imaging after contrast injection of the RCA.
Figure 2.
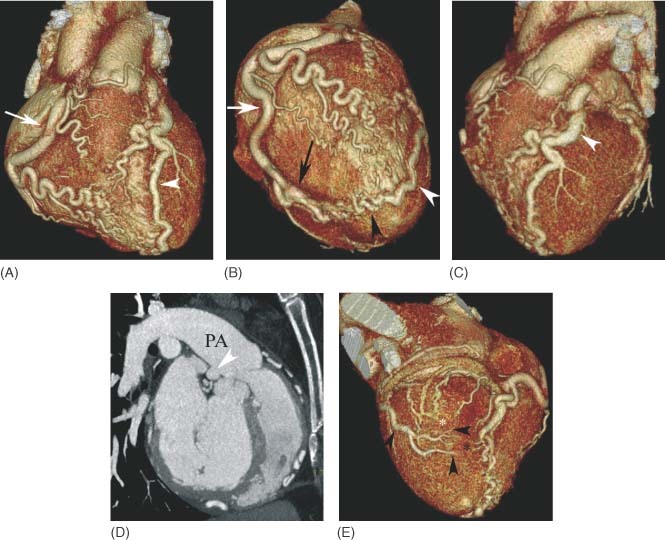
Multislice computed tomographic coronary angiography (Philips Medical Systems, Cleveland, OH). (A) Anterior volumetric rendering demonstrates a dilated right coronary artery (RCA; arrow) originating from the right coronary cusp of the aorta and passing under the right atrial appendage (*). Extensive collateral vessels are present across the free wall of the right ventricle. The left anterior descending artery (LAD) (arrowhead) is also dilated, although not quite as large as the massively dilated RCA. (B) Volumetric rendering of the apex of the heart from the right side demonstrates a tortuous and dilated RCA (white arrow) that supplies a dilated right posterior descending artery (RPDA) (black arrow) with extensive apical collateralization (black arrowhead) to the wrap‐around LAD artery (white arrowhead). (C) Volumetric rendering in a left anterior oblique (LAO) projection demonstrates the dilated LAD artery (arrowhead) coursing around the left side of the pulmonary artery (PA) toward its origination from the posterior aspect of the main PA. (D) Sagittal maximum intensity projection view demonstrates the origin of the dilated left coronary artery (arrowhead) from the posterior aspect of the main PA. (E) Left circumflex artery branches (black arrowheads) are seen crossing over the inferolateral wall of the left ventricle to anastomose with branches of the RPDA (black*) and posterior left ventricular branches from the distal RCA (white*).
Literature Review
One hundred fifty‐one adult cases of ALCAPA were reviewed, of which 12% were diagnosed at autopsy. The average reported age was 41 years; the oldest being 83 years old. There is a greater than 2:1 predominance of females. At the time of presentation, 66% had symptoms of angina, dyspnea, palpitations, or fatigue. Seventeen percent presented with ventricular arrhythmia, syncope, or sudden death. A minority (14%) were asymptomatic (Table 1). Of those with life‐threatening presentations, more than half (62%) had no antecedent symptoms. The average age of life‐threatening presentations was 33 years old ± 14 years. The average age of sudden death was 31 years old ± 11 years.
Table 1.
Population Summary of the 151 Adult Cases
| Age | 40.6 ± 15 years old |
|---|---|
| Sex | 69% female; 31% male |
| Presentation | 68% subacute symptoms; 18% life threatening; 14% asymptomatic |
Four patients had pregnancies complicated by angina, sinus arrest, or congestive heart failure. Twenty‐one patients had a total of 51 uncomplicated pregnancies.
Abnormalities on physical examination were a frequent finding. Eighty‐seven percent of the patients had murmurs. Seventy‐one percent had systolic murmurs, and 28% demonstrated a continuous or “to‐and‐fro” murmur predominantly localized to the left sternal border or apical region.
The ECG frequently demonstrated infarct patterns with Q‐waves noted in 50% of patients. Left ventricular hypertrophy was noted in 28% and left axis deviation in 15%. A normal ECG was found in only 4%.
Several studies have established diagnostic criteria for ALCAPA using echocardiography.11, 12 This includes identification of a dilated RCA, retrograde Doppler flow from the LCA to PA, and prominent septal flow from collateralization. In 72 patients where echocardiograms were performed, 46% were diagnostic or suggestive of ALCAPA, and 44% were abnormal but did not indicate ALCAPA (predominantly mitral regurgitation and segmental ventricular dysfunction). The echocardiogram was normal in 10%. In 26 stress ECG and 38 stress imaging studies, ischemia was detected in 85% and 87% of the cases, respectively. Right heart catheterizations were performed in 69 patients in whom 75% had a left to right shunt, with an average reported shunt of 1.5.
Traditionally, ALCAPA has been diagnosed by angiography or autopsy; however, the development of cardiac CT and MRI has allowed noninvasive evaluation of the coronary anatomy. From 1995 to 2008, 20 of 63 cases had been diagnosed primarily by cardiac CT (9) or MRI (11) (Figure 3).
Figure 3.
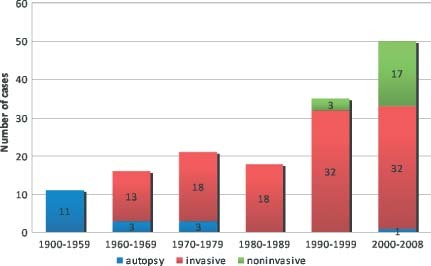
Time periods and modalities of anomalous origin of the left coronary artery from the pulmonary artery (ALCAPA) diagnoses.
CT and MRI findings of ALCAPA in adults include direct visualization of the origin of the LCA from the posterior aspect of the PA, dilated and tortuous RCA, and visualization of dilated intercoronary collateral arteries along the external surface of the heart or within the interventricular septum.13, 14, 15, 16 Both modalities may be used to assess left ventricular (LV) systolic function. MRI has the additional advantage of demonstrating communication and flow from the LCA into the PA, is useful in assessing mitral valvular function, and delayed gadolinium enhancement may help determine myocardial viability.16
Associated congenital abnormalities included 4 coronary arteriovenous fistulas to the PA; 1 LAD artery fistula to the LV; 1 primum atrial septal defect (ASD) with cleft mitral valve; 1 secundum ASD; 1 isolated bicuspid aortic valve; and 1 bicuspid aortic valve in a patient with LV noncompaction and splint. Three patients had reported extracardiac collateral circulation apparent at the time of bypass surgery, including bronchial artery collaterals.
The definitive treatment for ALCAPA is surgical intervention. Surgical correction involves ligation of the anomalous vessel or establishment of dual coronary perfusion.17 Twenty‐one percent of the patients undergoing surgery had ligation, whereas 79% had establishment of some form of dual coronary perfusion. The overall surgical mortality is 1% to 4%. Postoperative stress ECG and imaging studies demonstrated improvement in ischemia in 90% and 93% of cases, respectively.
Of the nonsurgical patients, 14 were unknown and 18 were treated medically, 10 of which had declined surgical intervention, and 8 patients ages 53 to 83 were recommended medical therapy. Of the 7 medically treated patients with reported follow‐up, 5 were described as well up to 5 years after diagnosis; 1 died of cancer after 4 years; and 1 relented to ligation and saphenous vein graft (SVG) bypass surgery after 1 year of ongoing angina. She was alive and well 51//2 years postoperatively despite known graft stenosis.
Discussion
The first reports of anomalous arteries from the PA are attributed to Krause in 186518 and Brooks in 1885.19 In their autopsy accounts, they describe accessory arteries originating from the anterior PA that join a web of mediastinal and thoracic vessels, including branches from normal aortic coronary arteries. In discovering septated venous sinusoids and noting the dilated, vein‐like morphology of the anomalous pulmonary vessels, Brooks first proposed the direction of blood flow to be toward the PA.
The first adult report of ALCAPA came from Maude Abbott's 1908 entry in Osler's Modern Medicine of an asymptomatic 60‐year‐old woman discovered at autopsy following an accidental death.20 Her description of a dilated and aneurysmal RCA with extensive, tortuous branch anastamoses with thin‐walled vein‐like vessels coursing in the normal distribution of the LCA territory, but originating from the posterior sinus of the PA, became the classic autopsy finding in adult ALCAPA.
This autopsy curiosity became a medical disease with the landmark publication in 1933 of the clinical syndrome of a 3‐month‐old infant with autopsy confirmed ALCAPA by Bland, White, and Garland.21 They described a normal birth, followed by a quiescent period of 2 months, and then the onset of symptoms and associated ischemic ECG changes. Their illustration of an infant with recurrent irritability, pallor, diaphoresis, and dyspnea as a result of cardiac ischemia became known as Bland‐White‐Garland syndrome.
The curious time course of presentation led to the proposal that elevated neonatal pulmonary pressure and oxygenation levels provided for normal early development. After birth, the gradual decline in pulmonary pressure and oxygenation led to venous physiology, providing low perfusion pressure and hypoxic blood to the LCA. By 3 months, closure of the ductus arteriosus led to infant angina, endocardial fibrosis, and ischemic cardiomyopathy. It was presumed that death in the first year was the rule.
In a review of 25 infant and adult cases, Kaunitz confirmed that symptom onset often coincided with closure of the ductus arteriosus 2 months after birth.22 He also noted in the adult cases, RCA dilation and significant development of collateralization from the RCA to the LCA. This became the first distinction proposed between infants who die in the first year and those who survive to adulthood.
Despite Brooks' early postulate of coronary blood flow from anomalous vessels into the PA, initial right heart catheterization studies failed to detect left to right shunting. Moreover, the curious time course of infant presentation focused on the principles of fluctuating pulmonary perfusion pressure and oxygenation in anterograde flow. The increasing association of adult survival with extensive RCA collateral blood supply to LCA territories helped to integrate these concepts.
Edwards subsequently proposed a 3‐phase physiology for ALCAPA rather than distinct infant and adult types.23 Neonatal survival depended on elevated pulmonary pressure and oxygenation to provide adequate anterograde perfusion from the PA to the myocardium. As pulmonary pressure and oxygenation decrease in early infancy, decreased coronary perfusion and subsequent ischemia resulted in the Bland‐White‐Garland syndrome. Survival to adulthood was dependent on the development of collateral circulation from the RCA to the LCA and reversal of flow from the LCA into the PA.
In a series of 11 cases of adult‐type ALCAPA, Jurishica demonstrated a high incidence of sudden death at a young age.8 Only 18% experienced symptoms prior to sudden death, suggesting that aggressive surgical management was appropriate for all adult patients. Nevertheless, early surgical treatment of ALCAPA enjoyed limited success. Among the first techniques was PA banding or ligation to increase coronary perfusion pressure in the LCA,24 under the presumption that perfusion was anterograde. After the development of cardiopulmonary bypass in 1953 by Dr. John Gibbon, Jr., surgical exploration blossomed. Early attempts included left common carotid artery bypass, aorto‐pulmonary window, talcum powder pericardial poudrage and de‐epicardialization to promote collateral circulation, and internal mammary artery bypass.17
Surgical techniques changed with greater appreciation of the physiology of flow reversal. Sabiston reported the first successful ligation of the anomalous LCA at its origin.25 Ligation raised the LCA pressure from 30 mm Hg preoperatively to 75 mm Hg postoperatively. However, the hemodynamic implications of ligation appeared unpredictable as evident by early operative deaths in infants and intraoperative ischemia upon ligation in teens.26, 27 This led to a proposed fourth phase in ALCAPA pathophysiology in which myocardial perfusion of LCA territory from RCA collaterals progressed to excessive shunting into the PA to produce a coronary steal syndrome.28, 29, 30 This would mark the point at which adult patients became symptomatic.
Skepticism over ligation grew with continued infant mortality and increasing adverse outcomes with congenital single coronary arteries.7, 31 Cooley reported the first creation of a 2‐coronary system by performing the first ligation with SVG to the ALCAPA in 1966.32 Two years later, Meyer reported a successful anastomosis of the left subclavian artery to the origin of the ALCAPA.33
Subsequent surgical outcome data endeavored to compare the techniques. The largest surgical outcome study for adults was reported in 1979 comparing 10 adults and 3 teenagers after ligation, and 13 adults and 3 teenagers after ligation with SVG bypass.34 In the latter group, 1 operative death was reported during bypass surgery with no reports of late sudden death at mean follow‐up of 4.8 years; whereas 3 ligation patients had suffered late sudden death at 2, 5.5, and 6.8 years postoperatively. Though the size of the study did not allow for statistical significance, late sudden death in ligation patients suggested that the establishment of dual coronary blood supply was the treatment of choice. In the modern era of surgical technique, it appears that providing a dual coronary supply is superior to single coronary physiology.
Technologic advances subsequently allowed direct coronary reimplantation to the aorta in both infants and adults.17, 29, 35, 36 Reimplantation presented its own obstacles in adults. Adult patients with ALCAPA developed less elastic, friable vessels that often originated distant from surgical access and the implant target.9, 10 Alternatives included intrapulmonary baffling such as the Takeuchi technique.37 Using a rolled flap of PA wall and an aorto‐pulmonary window, an intrapulmonary tunnel was created to connect the anomalous ostium to the aorta through the PA itself.
Over the past 2 decades, the number of reported patients with ALCAPA over age 50 years has increased. This diagnosis in an older cohort correlates with advances in echocardiography and the introduction of cardiac CT and MRI as new noninvasive techniques for evaluation of coronary anatomy. In contrast to their younger counterparts, older patients experienced less frequent life‐threatening presentations. Although the natural course of the disease results in a high incidence of sudden death during childhood and early adulthood, the risk of sudden death appears to decline after age 50 years despite less frequent surgical correction in this population (Table 2).
Table 2.
Clinical Presentations and Outcomes by Age
| All Cases Reported (151 Adults) | Under Age 50 Years (103 Adults) | Age 50 years and Older (48 Adults) | |
|---|---|---|---|
| Presentation | 68% subacute; 18% life threatening; 14% asymp‐ tomatic | 65% subacute; 22% life threatening; 13% asymp‐ tomatic | 75% subacute; 8% life threatening; 17% asymp‐ tomatic |
| Sudden death | 7% | 9% | 2% |
| Medically managed | 15% | 4% | 37% |
These older patients, in whom cardiac surgery is undesirable or unwarranted, continue to be diagnosed at an increased rate. Cardiac CT and MRI are useful not only in diagnosis, but may also offer prognostic information, allow for risk stratification, and be utilized for long‐term follow‐up imaging. MRI also allows for functional assessment of the heart and evaluation for myocardial ischemia and viability. Imaging has reached such an advanced stage of sophistication and precision that these techniques may eventually allow for the establishment of definite clinical guidelines for managing this congenital condition.
Conclusion
ALCAPA is a rare and life‐threatening condition. The availability of newer diagnostic modalities correlates with an increasing incidence in an older cohort. This comprehensive review of adult ALCAPA suggests that older patients experience less frequent life‐threatening presentations and sudden death. Nevertheless, surgical correction should be considered for all patients. Finally, this review emphasizes the lack of adequate comparative surgical and follow‐up data in reported adults, highlighting the need for this information in future studies. The patient reported here was referred for surgical treatment given demonstrable ischemia on exercise stress testing, but declined. At the time of this writing, she remains asymptomatic more than 2 years after diagnosis.
References
- 1. Keith JD. The anomalous origin of the left coronary artery from the pulmonary artery. Br Heart J. 1959;21:149–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Yamanaka O, Hobbs RE. Coronary artery anomalies in 126,595 patients undergoing coronary arteriography. Cathet Cardiovasc Diagn. 1990;21:28–40. [DOI] [PubMed] [Google Scholar]
- 3. Hauser M. Congenital anomalies of the coronary arteries. Heart 2005;91:1240–1245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Frescura C, Basso C, Thiene G, et al. Anomalous origin of coronary arteries and risk of sudden death: a study based on an autopsy population of congenital heart disease. Hum Pathol. 1998;29: 689–695. [DOI] [PubMed] [Google Scholar]
- 5. Konstantinowitsch W. Ein seltener Fall von Herzmissbildung. Prager med Wchnschr. 1906;31:657–660. [Google Scholar]
- 6. Abrikossoff A. Aneurysma des linken Herzventrikels mit abnormer Abgangstelle der linken Koronararterie von der Pulmonalis bei einem funfmonatlichen Kinde. Virchows Arch Pathol Anat. 1911;303:413–420. [Google Scholar]
- 7. Wesselhoeft H, Fawcett JS, Johnson AL. Anomalous origin of the left coronary artery from the pulmonary trunk. Its clinical spectrum, pathology, and pathophysiology, based on a review of 140 cases with seven further cases. Circulation. 1968;38:403–425. [DOI] [PubMed] [Google Scholar]
- 8. Jurishica AJ. Anomalous left coronary artery; adult type. Am Heart J. 1957;54:429–436. [DOI] [PubMed] [Google Scholar]
- 9. Moodie DS, Fyfe D, Gill CC, et al. Anomalous origin of the left coronary artery from the pulmonary artery (Bland‐White‐Garland syndrome) in adult patients: long‐term follow‐up after surgery. Am Heart J. 1983;106:381–388. [DOI] [PubMed] [Google Scholar]
- 10. Alexi‐Meskishvili V, Berger F, Weng Y, et al. Anomalous origin of the left coronary artery from the pulmonary artery in adults. J Card Surg. 1995;10:309–315. [DOI] [PubMed] [Google Scholar]
- 11. Yang YL, Nanda NC, Wang XF, et al. Echocardiographic diagnosis of anomalous origin of the left coronary artery from the pulmonary artery. Echocardiography. 2007;24:405–411. [DOI] [PubMed] [Google Scholar]
- 12. Frommelt MA, Miller E, Williamson J, et al. Detection of septal coronary collaterals by color flow Doppler mapping is a marker for anomalous origin of a coronary artery from the pulmonary artery. J Am Soc Echocardiogr. 2002;15:259–263. [DOI] [PubMed] [Google Scholar]
- 13. Horisaki T, Yamashita T, Yokoyama H, et al. Three‐dimensional reconstruction of computed tomographic images of anomalous origin of the left main coronary artery from the pulmonary trunk in an adult. Am J Cardiol. 2003;92:898–899. [DOI] [PubMed] [Google Scholar]
- 14. Khanna A, Torigian DA, Ferrari VA, et al. Anomalous origin of the left coronary artery from the pulmonary artery in adulthood on CT and MRI. Am J Roentgenol. 2005;185:326–329. [DOI] [PubMed] [Google Scholar]
- 15. Takenaga M, Matsuda J, Miyamoto N, et al. Magnetic resonance imaging of Bland‐White‐Garland syndrome—A case of anomalous origin of the left coronary artery form the pulmonary trunk in a 22‐year‐old woman. Jpn Circ J. 1998;62:219–221. [DOI] [PubMed] [Google Scholar]
- 16. Pena E, Nguyen ET, Merchant N, et al. ALCAPA syndrome: not just a pediatric disease. Radiographics. 2009;29:553–565. [DOI] [PubMed] [Google Scholar]
- 17. Dodge‐Khatami A, Mavroudis C, Backer CL. Anomalous origin of the left coronary artery from the pulmonary artery: collective review of surgical therapy. Ann Thorac Surg. 2002;74:946–955. [DOI] [PubMed] [Google Scholar]
- 18. Krause W. Cber den Ursprung einer akzessorischen A. coronaria aus der A. pulmonalis. Ztschr rat Med. 1865;24:225–227. [Google Scholar]
- 19. Brooks SJ. Two cases of an abnormal coronary artery of the heart arising from the pulmonary artery: with some remarks upon the effect of this anomaly in producing cirsoid dilatation of the vessels. J Anat Physiol. 1886;20:26–32. [PMC free article] [PubMed] [Google Scholar]
- 20. Abbott ME. Congenital heart disease In: Osler W, ed. ModernMedicine: Its Theory and Practice. Philadelphia, PA: Lea & Febiger; 1908:420–421. [Google Scholar]
- 21. Bland EF, White PD, Garland J. Congenital anomalies of the coronary arteries: report of an unusual case associated with cardiac hypertrophy. Am Heart J. 1933;8:787–801. [Google Scholar]
- 22. Kaunitz PE. Origin of left coronary artery from the pulmonary artery: review of the literature and report of two cases. Am Heart J. 1947;33:182–206. [DOI] [PubMed] [Google Scholar]
- 23. Edwards JE. Anomalous coronary arteries with special reference to arteriovenous‐like communications. Circulation. 1958;17: 1001–1006. [DOI] [PubMed] [Google Scholar]
- 24. Case RB, Morrow AG, Stainsby W, et al. Anomalous origin of the left coronary artery. The physiologic defect and suggested surgical treatment. Circulation. 1958;17:1062–1068. [DOI] [PubMed] [Google Scholar]
- 25. Sabiston DC, Neill CA, Taussig HB. The direction of blood flow in anomalous left coronary artery arising from the pulmonary artery. Circulation. 1960;22:591–597. [DOI] [PubMed] [Google Scholar]
- 26. Nadas AS, Gamboa R, Hugenholtz PG. Anomalous left coronary artery originating from the pulmonary artery. Report of two surgically treated cases with a proposal of hemodynamic and therapeutic classification. Circulation. 1964;29:167–175. [DOI] [PubMed] [Google Scholar]
- 27. Lampe CF, Verheugt AP. Anomalous left coronary artery. Adult type. Am Heart J. 1960;59:769–776. [DOI] [PubMed] [Google Scholar]
- 28. Edwards JE. The direction of blood flow in coronary arteries arising from the pulmonary trunk. Circulation. 1964;29:163–166. [DOI] [PubMed] [Google Scholar]
- 29. Backer CL, Stout MJ, Zales VR, et al. Anomalous origin of the left coronary artery. A twenty‐year review of surgical management. J Thorac Cardiovasc Surg. 1992;103:1049–1057. [PubMed] [Google Scholar]
- 30. Baue AE, Baum S, Blakemore WS, et al. A later stage of anomalous coronary circulation with origin of the left coronary artery from the pulmonary artery: coronary artery steal. Circulation. 1967;36: 878–885. [DOI] [PubMed] [Google Scholar]
- 31. Hillestad L, Eie H. Single coronary artery. A report of three cases. Acta Med Scand. 1971;189:409–413. [PubMed] [Google Scholar]
- 32. Cooley DA, Hallman GL, Bloodwell RD. Definitive surgical treatment of anomalous origin of the left coronary artery from pulmonary artery: indications and results. J Thorac Cardiovasc Surg. 1966;52:798–808. [PubMed] [Google Scholar]
- 33. Meyer BW, Stefanik G, Stiles QR, et al. A method of definitive surgical treatment of anomalous origin of the left coronary artery. A case report. J Thorac Cardiovasc Surg. 1968;56:104–107. [PubMed] [Google Scholar]
- 34. Wilson CL, Dlabal PW, McGuire SA. Surgical treatment of anomalous left coronary artery from pulmonary artery: follow‐up in teenagers and adults. Am Heart J. 1979;98:440–446. [DOI] [PubMed] [Google Scholar]
- 35. Neches WH, Mathews RA, Park SC, et al. Anomalous origin of the left coronary artery from the pulmonary artery. A new method of surgical repair. Circulation. 1974;50:582–587. [DOI] [PubMed] [Google Scholar]
- 36. Grace RR, Angelini P, Cooley DA. Aortic implantation of anomalous left coronary artery arising from pulmonary artery. Am J Cardiol. 1977;39:609–613. [PubMed] [Google Scholar]
- 37. Takeuchi S, Imamura H, Katsumoto K, et al. New surgical method for repair of anomalous left coronary artery from pulmonary artery. J Thorac Cardiovasc Surg. 1979;78:7–11. [PubMed] [Google Scholar]