

Abstract
Phosgene (Carbonyl Chloride, COCl2) remains an important chemical intermediate in many industrial processes such as combustion of chlorinated hydrocarbons and synthesis of solvents (degreasers, cleaners). It is a sweet smelling gas, and therefore does not prompt escape by the victim upon exposure. Supplemental oxygen and ventilation are the only available management strategies. This study was aimed to delineate the pathogenesis and identify novel biomarkers of acute lung injury post exposure to COCl2 gas. Adult male and female C57BL/6 mice (20-25 g), exposed to COCl2 gas (10 or 20ppm) for 10 minutes in environmental chambers, had a dose dependent reduction in PaO2 and an increase in PaCO2, 1 day post exposure. However, mortality increased only in mice exposed to 20ppm of COCl2 for 10 minutes. Correspondingly, these mice (20ppm) also had severe acute lung injury as indicated by an increase in lung wet to dry weight ratio, extravasation of plasma proteins and neutrophils into the bronchoalveolar lavage fluid, and an increase in total lung resistance. The increase in acute lung injury parameters in COCl2 (20ppm, 10min) exposed mice correlated with simultaneous increase in oxidation of red blood cells (RBC) membrane, RBC fragility, and plasma levels of cell-free heme. In addition, these mice had decreased plasmalogen levels (plasmenylethanolamine) and elevated levels of their breakdown product, polyunsaturated lysophosphatidylethanolamine, in the circulation suggesting damage to cellular plasma membranes. This study highlights the importance of free heme in the pathogenesis of COCl2 lung injury and identifies plasma membrane breakdown product as potential biomarkers of COCl2 toxicity.
Keywords: Red blood cell fragility, plasmalogens, BAL proteins, inflammation, free heme
Graphical Abstract
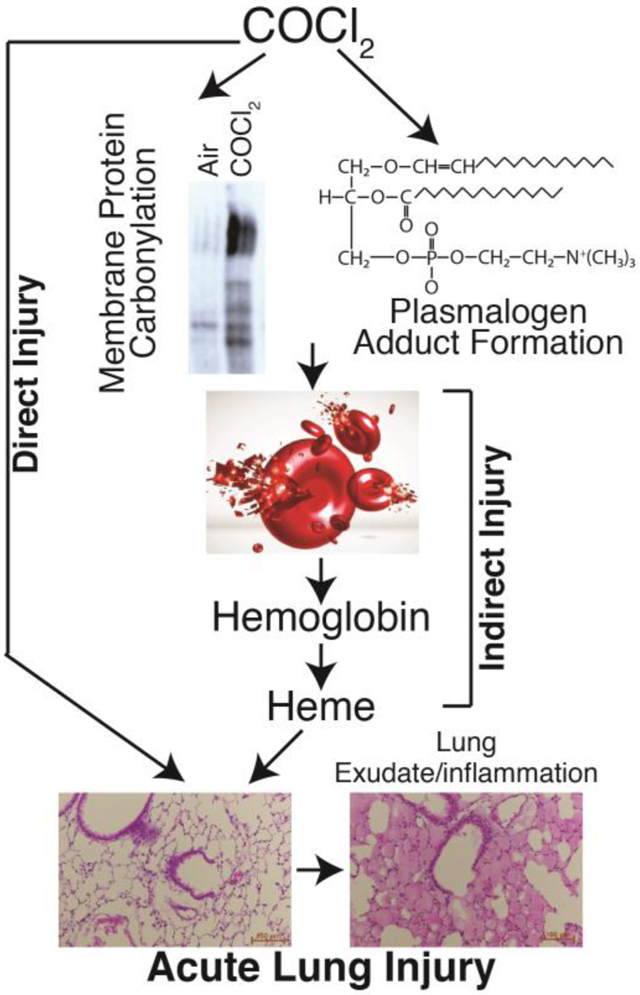
Introduction
Phosgene (COCl2) was first synthesized in 1812 by exposing a mixture of carbon monoxide and chlorine to sunlight. It reacts slowly with water to form carbon dioxide (CO2) and hydrochloric acid (HCl). Phosgene is a widely used in industry for the synthesis of isocyanate-based polymers, carbonic acid esters and acid chlorides. It is also used in the manufacture of dyestuffs, some insecticides and pharmaceuticals and in metallurgy. Although most COCl2 is produced on-site, a fair amount is transported by rail or truck making transportation accidents likely (19; 25; 38). Like the halogens chlorine (Cl2) and bromine (Br2), COCl2 is cheap and produced easily; however, unlike Cl2 and Br2 it cannot be detected by smell in small but dangerous concentrations, increasing the probability that people may be exposed to potentially harmful concentrations before they can react (19). As an example, a severe accident involving release of COCl2 occurred in a DuPont Factory in Belle, West Virginia in 2010 resulting in worker fatalities https://www.csb.gov/dupont-corporation-toxic-chemical-releases/. Phosgene may also be generated during welding and during fires involving plastics and solvents containing Cl2 placing workers and first responders at serious risk (54). Phosgene exposure resulted in more than 80% of all chemical-related deaths in WWI (25) and it may have been used against civilian targets as recently as 2017 and 2018 in Syria http://www.worldinwar.eu/the-gas-used-in-khan-shaykhun-probably-the-phosgene-syria/. Finally, chloroform (CHCL3), a former anesthetic agent, is metabolized to COCl2 in the liver and the kidneys (7; 43), which may account for some of the well-known CHCL3 toxicity COCl2-induced ALI show evidence of persistent apnea periods, bradycardia, and higher pulmonary edema compared to Cl2 toxicity (36). At higher concentrations (>150 ppm·min) COCl2 exposure can cause life-threatening and latent non-cardiogenic pulmonary edema that is seen 6 to 24 hours post-exposure (11; 48). Patients who survive often develop chronic lung disease, with airflow obstruction, fibrosis (44), airway hyper-reactivity, and impaired gas exchange (5; 25; 51). These patients often require hospitalization (42; 53), and are predisposed to bacterial infections (8). A severe accident involving release of COCl2 occurred in a DuPont Factory in Belle, West Virginia in 2010 resulting in worker fatalities. Phosgene may also be generated during fires involving plastics and solvents containing Cl2 placing workers and first responders at serious risk (54). According to the Medical Management Guidelines for Phosgene issued by the Agency for Toxic Substances Disease Registry, there is currently no effective medical countermeasure for phosgene exposure, and emergency medical treatment (like in the case of Cl2 and Br2) consists of support of cardiopulmonary functions via supplemental oxygen and possibly bronchodilators and corticosteroids https://www.atsdr.cdc.gov/mmg/mmg.asp?id=1201&tid=182 Cl2 and Br2, mainly interact with small molecular weight antioxidants present in the epithelial lining fluid as well as lipids and proteins on the surface of airway and epithelial cells (52; 57), as well as with plasmalogens, present in all cell membranes and the epithelial lining fluid, as well as glutathione, to form halogenated adducts (fatty acids and aldehydes). These species were detected in the alveolar space, lung tissues and plasma of Cl2 and Br2 exposed mice (10; 15) and may be responsible for the mediation of injury to distal organs, such as the hearts of halogen-exposed mice (3). Whether COCl2 reacts with plasmalogens is not known but there is evidence regarding its reaction with phospholipids in vivo in CHCl3 hepatotoxicity as well as with phosphatidylcholine in vitro (12; 18). In addition, because of its slow hydrolysis rate and solubility, COCl2 may cross the blood-gas barrier, enter the capillary circulation and damage RBCs directly (45; 49). Interestingly, humans exposed to COCl2 show transient declines in RBC count (41). If this decline is due to hemolysis, free heme/hemoglobin mediated processes may underlie tissue injury after COCl2 exposure. Indeed, one study showed that exposure of rats to COCl2 increased hemolysis (49). Presently, the mechanisms by which COCl2 injures the mammalian blood gas barrier has not been rigorously documented.
Thus the purpose of this study was to assess the extent of COCl2 injury to the blood gas barrier and red blood cells of unanesthetized mice. To accomplish these goals, we exposed equal numbers of adult male and female mice to 10 or 20 ppm COCl2 in environmental chambers for 10 min and returned them to room air, and assessed the following parameters at different times postexposure: survival, body weight changes, physiological and histochemical indices of lung function and injury including arterial blood gases, plasmalogen levels, and levels of hemoglobin and free heme in the plasma. We show that mice exposed to COCl2 showed evidence for increased RBC fragility, oxidative injury to RBC membranes, damage to plasma plasmalogens and the onset of delayed but severe lung injury which mimics human Adult Respiratory Distress Syndrome (56). Furthermore, there was considerable oxidation of important RBC structural proteins and band 3 which may contribute to hemolysis. The results of these studies pave the road for additional studies for the development of countermeasures against hemoglobin and heme, such as haptoglobin and hemopexin, as novel countermeasures for COCl2 injury.
Materials and Methods
Reagents.
Ketamine was obtained from Vedco Inc. (St. Joseph MO); Xylazine from Vet One, (Boise, ID); the heme assay kit was from QuantiChrom (Product No. DIHM-250; BioAssay Systems, Hayward, CA); 4mm Pyrex solid glass beads from Sigma-Aldrich (St Louis, MS); 4-20% Tris·HCl Criterion precast gels from Bio-Rad Laboratories (Product number: 567-1094, Hercules, CA); Amido Black from Sigma-Aldrich (St Louis, MS); RIPA buffer from Thermo Fisher Scientific (MA); Oxyblot protein oxidation detection kit from EMD Millipore (Product number: S7150, Billerica, MA)
Animals.
Adult male and female C57BL/6 mice (20-25g) were bought from Charles River (Wilmington, MA). Mice were allowed to acclimatize in the Animal Vivarium located in the basement of the Biomedical Research Building II for at least four days, where they were cared for by personnel from Animal Resources Program. All animal care and experimental procedures were approved by the Institutional Animal Care and Use Committee at the University of Alabama in Birmingham (protocol # 21451). Mice that exhibited respiratory symptoms, or refused to eat and drink, were not included in these studies.
Exposure to Phosgene Gas.
On the day of the exposure, mice of either sex were placed, five at a time, in a 4.5 L glass exposure chamber (1; 2). The exposure chambers were located inside a negative pressure hood, inside a room at the Animal Vivarium, maintained at negative pressure compared to the rest of the Vivarium
The exposure chamber was connected to either compressed air or COCl2 gas (nominal concentrations of 20 or 10 ppm, certified within 2%, purchased from SpecGas, Inc. Warminster, PA). The flow rate was set at 5 L/min. the concentration of COCl2 was monitored by an Analytical Technology, Inc. (Collegeville, PA) A21 Gas Sampling System, F12D Gas Transmitter with Phosgene Sensor (00-1016) Standard Range = 0-100 ppm; Resolution = 0.1 ppm) in a random fashion in 30% of the exposures. In addition, the concentration of CO2 in the chambers was monitored by a UEi C20 combustion meter (Beaverton OR). The gas exiting the chamber was passed through a 10% solution of NaOH to scavenge the COCl2 and vented to the roof of the building.
At the end of each ten min exposure, the gas was turned off, the chamber lid was removed and after a short period of time, the mice were returned to their cages in the Vivarium where they were provided with food and water ad libitum and observed by both laboratory and technicians of the Animal Facility. All personnel involved in the exposure of mice to COCl2 underwent special training by the Occupational and Safety division of the University of Alabama at Birmingham. A COCl2 detector, (purchased from Gas Sensing, Hull IA), equipped with an audible alarm and a strobe light was mounted on the wall of the exposure room and was set to sound an alarm if the COCl2 concentration in the room exceed 0.5 ppm COCl2.
Histological analysis of the mouse lung.
Mice were euthanized with an intra-peritoneal injection of ketamine and xylazine (160 and 80 mg/kg body weight, respectively). The chest was opened and the lungs were removed, fixed in 10% formalin for 24 hours and dehydrated in 70% ethanol before embedding in paraffin. Paraffin-embedded tissues were cut into 4 μm sections then de-paraffinized and rehydrated using Citrisolv (d-limonene based solvent) and isopropanol, respectively. The sections were stained with hematoxylin and eosin (H & E). Images were captured using a Leica DMI 6000 B microscope (Leica Microsystems Inc., Bannockburn, IL) and Leica Application Suite V4.2 software.
Plasma heme assays.
Non-encapsulated heme levels were measured in plasma samples by two different methods: first, by using the QuantiChrom heme assay kit (Product No. DIHM-250; BioAssay Systems, Hayward, CA), according to the manufacturer’s instructions. This method measures total non-encapsulated heme and hemoglobin levels and second, by spectral deconvolution with least square fitting analyses (46), developed by Dr. Patel, which allows for separate measurements of non-encapsulated hemoglobin and heme.
Red Blood Cell Fragility.
Red Blood Cells from air and COCl2 exposed mice were washed thoroughly to remove free heme. RBCs were re-suspended, at 1.0% hematocrit, with 4mm Pyrex solid glass beads (10 beads, 0.4 ml RBC suspension volume in 2 ml round bottom Eppendorf tubes) in DPBS. This solution was rotated 360° for 2 hours at 24 rpm at 37°C. The hemoglobin released from the RBCs during rotation was transferred into a new tube and centrifuged at 13,400g for 4 min and the absorbance of the supernatant were recorded at 540nm. Subsequently, one hundred percent hemolysis of RBCs was achieved by treating them with 1% Titon x-100 solution. The fractional hemolysis of the sample was then obtained by dividing the optical density of the sample by the optical density of the 100% hemolyzed sample.
Respiratory mechanics.
Mice were mechanically ventilated and challenged with increasing concentrations of methacholine as described previously (60). Briefly, mice were anesthetized with diazepam (17.5 mg/kg) and ketamine (450 mg/kg), intubated, connected to a ventilator (flexiVent; SCIREQ, Montreal, PQ, Canada) and ventilated at a rate of 160 breaths per minute at a tidal volume of 0.2 ml with a positive end-expiratory pressure of 3 cm H2O. Total respiratory system resistance (R) and elastance (E) were recorded continuously as previously described (ibid.). Baseline lung volume was set via deep inhalation. Increasing concentrations of methacholine chloride (0–50 mg/ml, Sigma-Aldrich, St Louis, MS) were administered via aerosolization within an administration time of 10 sec. Airway responsiveness was recorded every 15 seconds for 3 minutes after each aerosol challenge. Broadband perturbation was used and impedance was analyzed via constant phase model.
Arterial blood gases.
Mice were anesthetized with Isoflurane (5% for induction, 2% for maintenance) using compressed air as vehicle. The abdomen was opened, the mesentery was externalized to the left side of the mouse to visualize the abdominal aorta. Arterial blood was collected into a heparinized syringe through a 23 gauge needle inserted into the aorta. Blood gas analysis was performed immediately after collection using an Element POC analyzer (Heska, Loveland, CO).
SDS-PAGE and Oxyblots.
Mice were anesthetized and euthanized. Their lungs were then lavagd with one ml of saline which was instilled and withdrawn three times. The cells were pelleted by centrifugation (10 min at × 200g). Cleared supernatants were used to measure the protein concentration by the BCA assay; equal volume of BAL (2μl) were loaded on 4-20% Tris·HCl Criterion precast gels (Product number: 567-1094, Bio-Rad Laboratories, Hercules, CA). In addition, RBCs were separated from the plasma and hemolyzed with 20 mM hypotonic HEPES (4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid) buffer. The mixture was centrifuged at 14000xg for 20 min and the RBC pellet was dissolved in RIPA (Radioimmunoprecipitation assay) buffer. The protein was quantified by the BCA method and equal amounts of proteins (10μg) were loaded into a 4-20% gradient gel and proteins were separated and stained with Amido Black.
The presence of protein carbonyl groups was assessed using the Oxyblot protein oxidation detection kit (Product number: S7150, EMD Millipore, Billerica, MA), according to the manufacturer’s protocol (2). Briefly, the carbonyl groups in the protein side chains were derivatized to 2,4-dinitrophenylhydrazone by reacting with 2,4-dinitrophenylhydrazine. Precisely, 10 μg of protein was used for each sample, and the 2,4-dinitrophenol-derivatized protein samples were separated by polyacrylamide gel electrophoresis, as described previously. Polyvinylidene fluoride membranes were incubated for 1 hour in the stock primary antibody (1:150 in 1% PBS/TBST buffer), and after washing, for 1 hour in the stock secondary antibody (1:300 in % PBS/TBST buffer). Membranes were washed 3× in TBST and visualized, as described previously. The abundance of protein carbonylation was assessed by densitometry of each lane and normalization for each lane protein loading was done by SDS PAGE gel quantification.
Quantification of plasmalogens in plasma.
Equal number of male and female mice were sacrificed 24 h post exposure to either air or COCl2. Plasma was subjected to a modified Bligh-Dyer lipid extraction (6) in the presence of lipid class internal standards including 1-0-tetradecanoyl-sn-glycero-3-phosphoethanolamine and 1,2-ditetradecanoyl-sn-glycero-3-phosphoethanolamine (9). Lipid extracts were diluted in methanol/chloroform (1/1, v/v) and molecular species were quantified using electrospray ionization mass spectrometry on a triple quadrupole instrument (Themo Fisher Quantum Ultra) employing shotgun lipidomics methodologies (22). Ethanolamine glycerophospholipid and lysophosphatidylethanolamine molecular species were first converted to 9-fluorenylmethoxycarbonyl (fMOC) derivatives and then quantified in the negative ion mode using neutral loss scanning for 222.2 amu (collision energy = 30eV).
Statistical Analysis.
Statistical analysis was performed using GraphPad Prism version 4.01 for Windows (GraphPad Software, San Diego, CA). The mean ± SEM was calculated in all experiments, and statistical significance was determined by either the one-way or the two-way ANOVA. For one-way ANOVA analyses, Tukey’s post-test post-hoc testing was employed, while for two-way ANOVA analyses, Bonferroni post-tests were used. Overall survival was analyzed by the Kaplan-Meier method. Differences in survival were tested for statistical significance by the log-rank test. A value of P < 0.05 was considered significant.
Results
Whole body exposure to COCl2 in glass chamber.
As shown in Figure 1, COCl2 concentrations in the exposure chamber rose after onset of COCl2 flow and reached a steady state after approximately five min. There were no differences in the concentration profiles whether or not mice were present in the chambers, indicating that absorption of COCl2 by the fur was negligible. The differences in steady state concentrations recorded by the COCl2 detector and the certified nominal COCl2 concentrations were within the stated accuracy of the detector. Based on the profiles shown in Figure 1, the areas under the concentration profiles were 57.5 and 156 ppm × min. For simplicity we are referring to the exposures as 10 and 20 ppm. CO2 concentrations in the exposure chamber were undetectable during the exposure period.
Figure 1. Continuous measurement of COCl2 concentrations inside the exposure chambers.

COCl2 gas (nominal concentrations of 20 ppm or 10 ppm flowed into the exposure chambers (volume=4.5 L) at 4.4 L/min starting at zero time. The concentration of COCl2 was monitored by a. Analytical Technology, Inc. (Collegeville, PA) A21 Gas Sampling System, F12D Gas Transmitter with Phosgene Sensor. Measurements were conducted with five mice in the chamber (dotted lines) or with empty chambers (solid lines). Measurements were repeated twice with similar results.
Exposure to COCl2 results in hypoxemia and respiratory acidosis.
Adult C57BL/6 mice were exposed to air to COCl2 (10ppm or 20ppm) for 10 minutes. Mice appeared alert and unaffected by COCl2 both during the exposure and within the first 12 hours of return to air. After that time till they were sacrificed 24 hours later they exhibited reduced activity levels but they did not exhibit overt respiratory distress, i.e., lacked labored breathing, flaring of the nostrils and expiratory grunting. Measurements of arterial blood gases (Figure 2), PaO2 (2A), PaCO2 (2B), pH (2C), and HCO3− (2D) was performed in anesthetized mice using an Element POC analyzer (Heska, Loveland, CO) as described previously (27). Twenty four hours post COCl2, mice developed dose-dependent hypoxemia [PaO2 (torr)=124±2.6 (14); 82 ±6 (8); 69±4 (8); X ±SEM; (number of mice) for air, 10 ppm and 20 ppm COCL2 respectively] and hypercapnia [PaCO2 (torr)= 26±1.5 (14); 42.3±2 (8); 56±6 (8)] and respiratory acidosis (Fig. 2C), which was compensated by an increases in plasma bicarbonate [HCO3− (mM)= 15±0.7(14); 23±0.9 (8); 26±1.3 (8)] The increase in HCO3− was consistent with a decrease in plasma chloride (Cl−) concentration (data not shown).
Figure 2. Measurements of arterial blood gases in COCl2 exposed mice.

Equal number of male and female mice were exposed to either 10 or 20 ppm COCl2 for 10 min and returned to room air; At 24 h post exposure they were anesthetized with isoflurane, and one ml blood sample was withdrawn from the aorta, following opening of the chest cavity. PO2, PCO2, pH and HCO3− were measured with an Element POC analyzer (Heska, Loveland, CO). Values are means ± 1 SEM. Each point represents a different mouse. Statistical analysis was performed by one way analysis of variance followed by Tukey’s post-hoc testing.
Exposure to COCl2 increases mortality in mice.
As shown in Figure 3, air breathing mice continued to gain weight, eat and drink for the next 21 days (3A). Mice exposed to 10 ppm COCl2 experience a transient 15% decrease of body weight during the first 24 h post exposure, but resumed eating and drinking and gain weight as the air controls (3B). All of these mice were alive at 21 d post exposure. On the other hand, mice exposed to 20 ppm COCl2 for 10 min and returned to room air, continued to lose weight during the first 5 days post exposure (3C). Two of these mice died during the first 24 h post exposure and three mice were sacrificed because their body weights dropped below 30% of their pre-exposure weights. Kaplan-Meyer curves for mice exposed to 10 or 20 ppm COCl2 are shown in Figure 3D. Mice exposed to 20 ppm for 10 min COCl2 exhibited approximately 50% survival following return to room air; all deaths occurring in the first six days post exposure (Figure 3D). Therefore, we opted to perform more detailed studies in mice exposed to 20 ppm COCL2 for 10 min and returned to room air
Figure 3. Measurements of body weight and mortality in mice exposed to COCl2.

Equal number of male and female C57BL/6 were placed five at a time in the environmental exposure chambers and exposed to air (n=18), 10 ppm COCl2 (n=20) or 20 ppm COCl2 (n=20) for 10 min. At the end of the exposure, the gas flow was stopped, the top lid of the chamber removed and the mice were transferred to their normal holding cages. Their body weights were measured daily and expressed as % of initial (i.e. pre-exposure) weights. Mice exposed to air (A) continued to gain weight; mice exposed to 10 ppm COCL2 for 10 min (B) lost no more than 20% of their initial body weight in the first 24 hours but gained weight thereafter. Mice exposed to 20 ppm for 10 min lost a significant higher levels of their body weights (C). Two male and female mice each were found dead in their cages one day post exposure. The sharp drops of body weight shown in 3C indicate that the mice were either sacrificed due to a loss of more than 30% of body weight or were found dead. Kaplan-Meyer survival curves (D) for 10 ppm COCl2 (n=20; dotted line) or 20 ppm COCl2 (n=20; solid line) for 10 min (equal numbers of male and female mice). All air exposed mice were alive at 21 d post exposure. * p=0.0006 as compared to air or 10 ppm COCl2 according to the Long-rank (Mantel-Cox) test.
Exposure to 20 ppm COCl2 damages the distal lung regions and results in inflammation.
Consistent with the data shown in Figure 3, mice exposed to COCl2 (20 ppm, 10 min) had severe injury to the blood gas barrier 24 hours post exposure (Figure 4). There was a progressive and significant increase of the lung/wet dry weight (4A) (W/D=4.3±0.1 (9); 4.7±0.3 (5); 5.1±0.22(10);X ±SEM; (number of mice) for air, 10 ppm and 20 ppm COCL2 respectively) consistent with the presence of interstitial and alveolar edema, and a 10 fold increase of plasma proteins in the bronchoalveolar lavage fluid (BALF) (4B) [BAL Pr.(mg/ml)=0.2±0.03 (13); 4.45±1.34 (5); 10.3±0.84 (13)] Interestingly, the magnitude of increased lung edema far exceeded (by 5-10fold) that observed in mice exposed to HCl (60), Cl2 or Br2 (1; 2; 29); maximal BALF protein levels in these mice are in the 1-2mg/ml range. SDS PAGE analysis of cell free BALF showed a large increase in a number of protein bands, including a 67 kDA band. Previous Western blotting studies (34) have shown that the 67 kDa band corresponds to plasma albumin (4C). It should be noted that the pattern observed in SDS-PAGE gels was similar to that of plasma (far right two lanes in Figure 4C), indicating complete breakdown of the alveolar barrier.
Figure 4. Measurement of lung edema in mice exposed to COCl2.

Equal number of male and female mice were sacrificed 3 or 24 hours post exposure to either air or 20 ppm COCl2 for 10 min; their chests were opened and lungs removed en bloc, blotted and non-lung tissue was removed. They were then weighted for determination of wet weight and placed in an oven at 70 °C for seven days at which point they were reweighted for measurement of dry weights. Data shown are wet/dry weights (A). Mean±1 SEM; n=9 air; n=5 for 3 hours post COCl2, and n=10 for 24 hours post COCl2; In a separate set of animals mice lungs were lavaged with 1 ml of saline. The cells were pelleted by centrifugation (10 min at x200g) and protein in the cell free lavage was determined by the BCA method (B). Mean±1 SEM; n=9 air; n=5 for 3 hours post COCl2, and n=10 for 24 hours post COCl2. BALF proteins were also separated by SDS-PAGE (2 μl BAL). No significant amount of proteins were detected in the BALF of air breathing mice; however, significant amounts of a protein with a molecular weight of around 70 kDa (most likely albumin) was detected in the BALF of mice harvested 24 hours post COCl2 exposure (C). Large number of other plasma proteins were also seen. Statistical analysis was done by ANOVA with Tukey’s post-test (A, B).
The total cell count remained unchanged at both 3 and 24 h post exposure (data not shown). However at 24 hours post exposure (Figure 5) there was a significant increase of the % neutrophils in the BAL (5A)[% of neutr. =0.14±0.1 (14); 8.5±1.8 (13); X ±SEM; (number of mice) for air and 20 ppm COCL2 respectively] and a concomitant decrease of the % of alveolar macrophages (5B) [% mac. =100±0.1 (14); 91±1.8 (13)]. H&E staining of the lungs of mice sacrificed at 24 hours post COCl2 (20 ppm for 10 min) showed that most alveoli were filled with exudate exhibiting eosin staining, indicative of high protein content (5C), consistent with data shown in Figure 4. In contrast, H&E stains of the distal lung regions of mice exposed to air, were free of exudate and showed normal architecture. Macroscopic examination of the lungs of COCl2 exposed mice showed severe hemorrhage (5C).
Figure 5. Measurements of distal lung injury in mice exposed to COCl2.

Equal number of male and female mice were sacrificed 24 h post exposure to either air or 20 ppm COCl2 for 10 min. Neutrophils (A) and macrophages (B), identified by their characteristic shapes were counted in the BALF with a hemocytometer and expressed as % of total cells. Means±1 SEM; n=14 air; n=13 COCl2; statistical analysis by unpaired t-test. In addition, the lungs were fixed with formalin at a 25 cmH2O constant pressure. Thin sections were cut from all lobes, fixed and stained with hematoxylin and eosin (C). The characteristic pictures show that the lungs in COCl2 mice were filled with fluid containing proteins and cells. A small number of alveoli appear empty of fluid and were responsible for gas exchange. In addition, macroscopic examination of the lungs showed significant hemorrhage in COCl2 exposed mice (C).
Exposure to 20 ppm COCl2 increases airway resistance.
Measurements of airway resistance and elastance with flexi Vent showed a large increase of baseline airway resistance and a concomitant decrease of dynamic elastance at 24 hours post exposure (Figure 6). Following methacholine challenge, airway resistance (6A) and elastance (6B) changed similarly in both COCl2 and air exposed mice; these data indicate that in spite of the large increase of baseline airway resistance, airways did not become hyperreactive following exposure to COCl2, in contrast to what seen in Cl2 and Br2 exposed mice. Surprisingly, Newtonian airway resistance, an index of injury to upper airways, did not change in the COCl2 group indicating that the increase of airway resistance was mainly due to damage to small airways.
Figure 6. Measurements of airway resistance and elastance in mice exposed to COCl2.

Male only C57BL/6 mice were exposed to either air (n=10; black circles) or COCl2 (20 ppm for 10 min; n=5; black diamonds) and returned to room air for 24 hours. They were then anesthetized and connected to a flexiVent for measurements of Airway Resistance (A) or Elastance (B). Numbers are means ± 1 SEM; *p<0.05 (unpaired t-test). * p<0.05 by Student’s t-test for the indicated levels of resistance or elastance
Exposure to COCl2 damages red blood cells and causes hemolysis.
First, RBC membranes were analyzed for the presence of oxidative damage (Figure 7). For these studies, RBC were collected and membranes prepared as outlined in Methods. The measurement of membrane associated carbonyl adducts (7A, B) from RBC collected 24 hours post exposure to COCl2 (20 ppm, 10min) showed significantly higher levels of oxidation (7A, B) of a 90-95 kDa protein as well as a number of higher molecular weight proteins [fold increase over air: 1.9±0.2 (8)]. Due to the limited repertoire of proteins in RBC membrane, the 90-95 kDa protein is likely to be band 3 (21); notice that both 4.1a and 4.1b bands are clearly visible and their ratio is about 1:1 in most of the preparation, as previously reported (17); the high molecular species are likely to be ankyrin and spectrin. SDS-PAGE of RBC ghosts revealed no difference in total levels of these proteins (data not shown).
Figure 7. Carbonyl adducts are increased in RBC ghosts post COCl2 exposure.

Mice were exposed to air or COCl2 (20 ppm, 10min). RBCs were separated from the plasma and hemolyzed with 20 mM hypotonic HEPES Buffer 24 hours later. The mixture was centrifuged at 14000 xg for 20 min and the resulting RBC pellet was dissolved in RIPA buffer. The protein was quantified by the BCA method and the presence of protein carbonyl groups was assessed using the OxyBlot Protein Oxidation Detection Kit according to the manufacturer’s protocol. Significant oxidation was noted in a protein around 95 KDa. Oxidation of high molecular weight proteins (around 250 kDa; presumably ankyrin and spectrin) was also observed (A). Each lane represents a different mouse. Additionally equal amounts of proteins were loaded into a 4-20% gradient gel and proteins were separated and stained with Amido Black (data not shown). There was no difference in protein levels between the air and the COCl2 exposed mice. Panel B shows the quantification of carbonyl adducts. Values are means ± 1 SEM; Student’s t-test.
Next, the analysis of RBC fragility, showed that exposure to mechanical stress (rotation of RBCs with glass beads for 2 hours) caused significantly more hemolysis in the RBCs obtained from the mice exposed to COCl2 (20 ppm for 10 min) than the air exposed mice (Figure 8A). Next, we measured total levels of non-encapsulated heme, using an ELISA method which does not discriminate between free heme and heme bound to hemoglobin. There was a doubling of non-encapsulated heme at 24 hours post 20 ppm COCl2 for 10 min (8B), [Total heme (mM)=14±2 (16); 28±2.7 (20); X ±SEM; (number of mice) for air and 20 ppm COCl2 respectively] Measurements of non-encapsulated heme using a spectrophometric technique followed by spectra deconvolution revealed similar increase in of this variable (8C) [Total heme (mM)=20±1.6 (33); 31±3.1 (19); X ±SEM; (number of mice) for air and 20 ppm COCL2 respectively]. Using the latter method, we were able to determine that at 24 h post-exposure to 20 ppm COCl2 for 10 min, approximately 25-30% of the non-encapsulated heme existed as free here, formed most-likely by the oxidation of hemoglobin (8D) (Free Heme (mM)= 5.6±0.5 (33); 9.3±1.8 (19).
Figure 8. COCl2 exposure increases RBC fragility in mice.

Equal number of male and female mice were sacrificed 24 hours post exposure to either air or COCl2 (20 ppm for 10 min). Blood was withdrawn from the left ventricle and plasma separated. RBCs from these mice were washed thoroughly to remove free heme; RBC suspensions of 1.0% hematocrit along with 4mm solid glass beads (Pyrex) in DPBS were rotated 360° for 2 hours at 24 rpm at 37°C. The hemoglobin released from the RBCs during rotation was transferred into a new tube and centrifuged at 13,400g for 4 min and the absorbance of the supernatant were recorded at 540nm. Means ± SEM; n=8 for each group. One hundred percent hemolysis of RBCs was achieved by treating them with 1% Titon x-100 solution. The percent hemolysis of the sample was then obtained by dividing the optical density of the sample from the optical density of the 100% hemolyzed sample. (A). Next, total heme levels was measured in the plasma of air and COCl2 exposed mice using the QuantiChrom heme assay kit, according to the manufacturer’s instructions. Means ± 1 SEM; n=16 air and 20 for COCl2. (B). in the second series of experiments total (C) and free (D) heme levels in plasma were measured using an absorbance spectrum deconvolution (46). Means ± 1 SEM; n=33 air and 19 for COCl2. Total heme levels determined by these two methods were very similar.
Exposure to COCl2 damages plasmalogens.
Our previous data show that the halogensCl2 and Br2 interact with lung plasmalogens resulting in the formation of halogenated lipids (fatty acids and aldehydes), which cause extensive injury to RBCs and distant targets (10; 15; 47). Herein we show that exposure of mice to COCl2 (20 ppm for 10 min) results in significant decrease of plasmenylethanoamine (Figure 9A) [PE (ng/±μl) =1038±89 (7); 783±58 (7)] and an increase of its breakdown product, polyunsaturated lysophosphatidylethanolamine (Figure 9B) [LPE (ng/μl) =16±3.7; 62±13 for air and 20 ppm COCL2 respectively].
Figure 9. Measurement of plasmalogens and their breakdown product in COCl2 exposed mice.

Equal number of male and female mice were sacrificed 24 h post exposure to either air or 20 ppm COCl2 for 10 min. Plasma was subjected to a modified Bligh-Dyer lipid extraction in the presence of lipid class internal standards including 1-0-tetradecanoyl-sn-glycero-3-phosphoethanolamine and 1,2-ditetradecanoyl-sn-glycero-3-phosphoethanolamine. Lipid extracts were diluted in methanol/chloroform (1/1, v/v) and molecular species were quantified using electrospray ionization mass spectrometry on a triple quadrupole instrument employing shotgun lipidomics methodologies (see Material and Methods for more details). Plasmenylethanolamine, (A) and Polyunsaturated Lysophosphatidylethanolamine (B) levels were measured. Values are means ± 1 SEM. Statistical Analysis by the Student’s t-test
Discussion
Herein we demonstrate that male and female unanesthetized mice exposed to 20 ppm of COCl2 for 10 min and returned to room air, exhibited significant mortality with a LC50 of about 5 days. At 24 h post exposure there was significant increase of airway resistance and extensive injury to the blood gas barrier, resulting in the filling of large number of alveoli with proteinaceous exudate, resulting in severe hypoxemia, hypercapnia and partially compensated respiratory acidosis. In addition, we observed significant decreases of plasma plasmalogens, with concomitant increase of plasmalogens degradation products, as well as oxidation of key structural and functional proteins of RBCs which may be responsible at least in part for increased RBC fragility. Furthermore, we documented the presence of free hemoglobin and heme in the plasma which may be responsible for initiation and propagation of injury to distal organs. Exposure to 10 ppm of COCl2 for 10 min did not result in mortality up to 21 days post exposure and the extent of lung injury was milder than observed with exposure to 20 ppm. There were no differences in any of the measured variables among male and female mice.
To this extent, the results of these studies mimic what has been found in humans. COCl2-induced ALI show evidence of persistent apnea periods, bradycardia, and higher pulmonary edema compared to Cl2 toxicity (36). At higher concentrations (>150 ppm·min) COCl2 exposure can cause life-threatening and latent non-cardiogenic pulmonary edema that is seen 6 to 24 hours post-exposure (11; 48). Patients who survive ALI from any source often develop chronic lung disease, with airflow obstruction, fibrosis (44), airway hyper-reactivity, and impaired gas exchange (5; 25; 51). These patients often require hospitalization (42; 53), and are predisposed to bacterial infections(8). A severe accident involving release of COCl2 occurred in a DuPont Factory in Belle, West Virginia in 2010 resulting in worker fatalities. Phosgene may also be generated during fires involving plastics and solvents containing Cl2 placing workers and first responders at serious risk (54). According to the Medical Management Guidelines for Phosgene issued by the Agency for Toxic Substances Disease Registry, there is currently no effective medical countermeasure for phosgene exposure, and emergency medical treatment (like in the case of Cl2 and Br2) consists of support of cardiopulmonary functions via supplemental oxygen and possibly bronchodilators and corticosteroidshttps://www.atsdr.cdc.gov/mmg/mmg.asp?id=1201&tid=182
In this study, we opted to expose mice to COCl2 in a whole body manner, instead of nose only, for various reasons: first we believe that whole body exposure better mimics human exposures; second, we have previously shown that exposure of mice to Cl2 gas causes the upregulation of the unfolded protein response which may result in lung injury, compounding the effects of the toxic gas (35); third, mice exposed in a nose only way are restrained in contrast to our system where they can move around freely. We carefully measured COCl2 concentrations in exposure chambers allowing the accurate calculation of the ppm × time profile which may be of considerable use to compare results of studies between various laboratories.
The mechanisms responsible for COCl2 induced injury are poorly understood. COCl2 is a very reactive gas and like Cl2 and Br2 increase the concentration of reactive species in the cytoplasm and mitochondria (4; 13; 31). Recently, we have demonstrated that Cl2 and Br2 react with plasmalogens, present in all cell membranes and the epithelial lining fluid, as well as glutathione to form halogenated adducts (fatty acids and aldehydes), which were detected in the alveolar space, lung tissues, plasma and red blood cell membranes (10; 15; 29). These are potentially damaging species which may destabilize cell membranes and cause extensive injury to the blood gas barrier. Phosgene also reacts with phosphatidylcholine, an important component of cell lipid membranes and pulmonary surfactant (18). Decreased levels of surfactant lipids as well as damage to surfactant proteins may result in severe lung injury, characterized by permeability-type edema and hypoxemia (20; 24). In the present study we found that plasma levels of plasmenylethanolamine, one of the most common plasmalogens, were decreased and plasma levels of lysophosphatidylethanolamine, a partial hydrolysis product of plasmenylethanolamine were increased in COCl2 exposed mice as compared to air exposed mice, suggesting that COCl2 may cross the blood-gas barrier and react with plasma plasmalogens. In addition, HCl, a COCL2 byproduct, has been shown to cause acute lung injury when instilled into the lungs of mice (14; 60). Injury may be compounded by the resulting inflammation due to the egress of neutrophils into the alveolar space. In this respect, in the present study we found that plasma levels of plasmenylethanolamine, one of the most common plasmalogens, were decreased and plasma levels of lysophosphatidylethanolamine, a partial hydrolysis product of plasmenylethanolamine were increased in COCl2 exposed mice as compared to air exposed mice, suggesting that COCl2 reacts with plasmalogens as well.
Exposure of mice to Cl2 or Br2 at 600 ppm for 30 minutes cause approximately 50% mortality within 7 days, similarly to 20 ppm COCl2 for 10 minutes (20/10) in the present study. We typically found BAL protein levels of 1 mg/mL to 1.5 mg/mL in Cl2 and Br2 exposed mice (1; 2; 26-28; 32; 58; 59), as compared to the typical 0.3 to 0.5 mg/mL protein levels in air exposed mice. Strikingly, we observed 10.3 mg/mL mean protein levels in BAL of mice exposed to COCl2, as compared to the 0.5 mg/mL mean protein levels in air exposed mice. Another striking difference is that most alveoli of COCl2 mice at 24 h post exposure were filled with proteinaceous exudate; in contrast Cl2 and Br2 induced injury is patchier. Similarly, mean lung wet/dry ratios in mice exposed to Br2 and Cl2 range from 4.5 to 4.8, as compared to ~4.2-4.3 (ibid) in air exposed mice, but we found mean lung wet/dry ratios of 5.1, with individual values being as high as 6.5 in COCl2 exposed mice. These findings suggest that exposure conditions that lead to similar mortality outcomes are associated a significantly greater lung injury in COCl2 exposed mice, compared to Cl2 or Br2. The reasons for this difference are yet to be determined.
Arterial blood gas measurements in mice are relatively uncommon, due to the difficulty of collecting pure arterial blood in sufficient quantities. PaO2 and PaCO2 values in air-exposed mice, obtained while they were anesthetized, were higher and lower respectively compared to what has been reported in conscious mice (33). Following COCl2 exposure, PaO2 levels were decreased and PaCO2 levels were increased. There was a surprisingly adequate metabolic compensation with increase in HCO3− concentrations which returned the pH towards a normal value. These data indicate that the observed metrics of lung injury correlate well with the observed deficiency in gas exchange.
In addition to direct injury to the lung, halogens cause indirect injury to the lungs and systemic organs by causing red blood cell hemolysis (1; 2). Both heme and hemoglobin (Hb), released post hemolysis, are potent pro-oxidants and pro-inflammatory mediators (16; 23). Heme-induced lipid peroxidation (23) and oxidative stress damages the sugar-phosphate backbone causing strand breaks in the mitochondrial DNA (50). Elevated levels of extracellular hemoglobin and heme have been associated with end organ injury and mortality in critically ill patients and therapies that decrease free heme and hemoglobin are protective in animal models where hemolysis occurs (1; 2; 30; 37; 39; 40; 55)
In summary, these findings characterize in detail the development of lung and systemic injury in mice exposed to COCl2, offer significant new insights into the mechanisms leading to RBC fracture and release of free hemoglobin and heme and set the stage for the development of new countermeasures to limit lung injury and prolong survival.
Acknowledgments
Funding: Supported by the CounterACT Program, National Institutes of Health Office of the Director (NIH OD), the National Institute of Neurological Disorders and Stroke (NINDS), and the National Institute of Environmental Health Sciences (NIEHS), Grant Numbers (5UO1 ES026458 03; 3UO1 ES026458 03S1; 5UO1 ES027697 02) to SM. The authors would like to thank Drs. Wesley W. Holmes, Alfred M. Sciuto and Dana R. Anderson from the Analytical Toxicology Division, USA Army Medical Research Institute of Chemical Defense for many useful discussions on phosgene toxicology and Dr. Adam Molyvdas for the careful review of this manuscript.
Footnotes
Conflict of Interest Statement
None of the authors have any conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Aggarwal S, Ahmad I, Lam A, Carlisle MA, Li C, Wells JM, Raju SV, Athar M, Rowe SM, Dransfield MT and Matalon S. Heme scavenging reduces pulmonary endoplasmic reticulum stress, fibrosis, and emphysema. JCI Insight 3: 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Aggarwal S, Lam A, Bolisetty S, Carlisle MA, Traylor A, Agarwal A and Matalon S. Heme Attenuation Ameliorates Irritant Gas Inhalation-Induced Acute Lung Injury. Antioxid Redox Signal 24: 99–112, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ahmad S, Masjoan Juncos JX, Ahmad A, Zaky A, Wei CC, Bradley WE, Zafar I, Powell P, Mariappan N, Vetal N, Louch WE, Ford DA, Doran SF, Matalon S and Dell'Italia LJ. Bromine inhalation mimics ischemia-reperfusion cardiomyocyte injury and calpain activation in rats. Am J Physiol Heart Circ Physiol 316: H212–H223, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Arroyo CM, Feliciano F, Kolb DL, Keeler JR, Millette SR and Stotts RR. Autoionization reaction of phosgene (OCCl2) studied by electron paramagnetic resonance/spin trapping techniques. J Biochem Toxicol 8: 107–110, 1993. [DOI] [PubMed] [Google Scholar]
- 5.Balakrishna S, Song W, Achanta S, Doran SF, Liu B, Kaelberer MM, Yu Z, Sui A, Cheung M, Leishman E, Eidam HS, Ye G, Willette RN, Thorneloe KS, Bradshaw HB, Matalon S and Jordt SE. TRPV4 inhibition counteracts edema and inflammation and improves pulmonary function and oxygen saturation in chemically induced acute lung injury. Am J Physiol Lung Cell Mol Physiol 307: L158–L172, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Blight EG and DYER WJ. A rapid method of total lipid extraction and purification. Can J Biochem Physiol 37: 911–917, 1959. [DOI] [PubMed] [Google Scholar]
- 7.Branchflower RV, Nunn DS, Highet RJ, Smith JH, Hook JB and Pohl LR. Nephrotoxicity of chloroform: metabolism to phosgene by the mouse kidney. Toxicol Appl Pharmacol 72: 159–168, 1984. [DOI] [PubMed] [Google Scholar]
- 8.Cucinell SA. Review of the toxicity of long-term phosgene exposure. Arch Environ Health 28: 272–275, 1974. [DOI] [PubMed] [Google Scholar]
- 9.Demarco VG, Ford DA, Henriksen EJ, Aroor AR, Johnson MS, Habibi J, Ma L, Yang M, Albert CJ, Lally JW, Ford CA, Prasannarong M, Hayden MR, Whaley-Connell AT and Sowers JR. Obesity-related alterations in cardiac lipid profile and nondipping blood pressure pattern during transition to diastolic dysfunction in male db/db mice. Endocrinology 154: 159–171, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Duerr MA, Palladino END, Hartman CL, Lambert JA, Franke JD, Albert CJ, Matalon S, Patel RP, Slungaard A and Ford DA. Bromofatty aldehyde derived from bromine exposure and myeloperoxidase and eosinophil peroxidase modify GSH and protein. J Lipid Res 59: 696–705, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Duniho SM, Martin J, Forster JS, Cascio MB, Moran TS, Carpin LB and Sciuto AM. Acute changes in lung histopathology and bronchoalveolar lavage parameters in mice exposed to the choking agent gas phosgene. Toxicol Pathol 30: 339–349, 2002. [DOI] [PubMed] [Google Scholar]
- 12.Fan BR, Nguyen T, Waring A and Taeusch W. Staining properties of bovine low molecular weight hydrophobic surfactant proteins after polyacrylamide gel electrophoresis. Anal Biochem 186: 41–45, 1990. [DOI] [PubMed] [Google Scholar]
- 13.Filipczak PT, Senft AP, Seagrave J, Weber W, Kuehl PJ, Fredenburgh LE, McDonald JD and Baron RM. NOS-2 Inhibition in Phosgene-Induced Acute Lung Injury. Toxicol Sci 146: 89–100, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Folkesson HG, Matthay MA, Hebert CA and Broaddus VC. Acid aspiration-induced lung injury in rabbits is mediated by interleukin-8-dependent mechanisms. J Clin Invest 96: 107–116, 1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ford DA, Honavar J, Albert CJ, Duerr MA, Oh JY, Doran S, Matalon S and Patel RP. Formation of chlorinated lipids post-chlorine gas exposure. J Lipid Res 57: 1529–1540, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gaggar A and Patel RP. There is blood in the water: hemolysis, hemoglobin, and heme in acute lung injury. Am J Physiol Lung Cell Mol Physiol 311: L714–L718, 2016. [DOI] [PubMed] [Google Scholar]
- 17.Goodman SR, Yu J, Whitfield CF, Culp EN and Posnak EJ. Erythrocyte membrane skeletal protein bands 4.1 a and b are sequence-related phosphoproteins. J Biol Chem 257: 4564–4569, 1982. [PubMed] [Google Scholar]
- 18.Guastadisegni C, Guidoni L, Balduzzi M, Viti V, Di CE and Vittozzi L. Characterization of a phospholipid adduct formed in Sprague Dawley rats by chloroform metabolism: NMR studies. J Biochem Mol Toxicol 12: 93–102, 1998. [DOI] [PubMed] [Google Scholar]
- 19.Gutch M, Jain N, Agrawal A and Consul S. Acute accidental phosgene poisoning. BMJ Case Rep 2012: 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Haddad IY, Ischiropoulos H, Holm BA, Beckman JS, Baker JR and Matalon S. Mechanisms of peroxynitrite-induced injury to pulmonary surfactants. Am J Physiol 265: L555–64, 1993. [DOI] [PubMed] [Google Scholar]
- 21.Hamasaki N and Okubo K. Band 3 protein: physiology, function and structure. Cell Mol Biol (Noisy -le-grand) 42: 1025–1039, 1996. [PubMed] [Google Scholar]
- 22.Han X, Yang K, Cheng H, Fikes KN and Gross RW. Shotgun lipidomics of phosphoethanolamine-containing lipids in biological samples after one-step in situ derivatization. J Lipid Res 46: 1548–1560, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Higdon AN, Benavides GA, Chacko BK, Ouyang X, Johnson MS, Landar A, Zhang J and Darley-Usmar VM. Hemin causes mitochondrial dysfunction in endothelial cells through promoting lipid peroxidation: The protective role of autophagy. Am J Physiol Heart Circ Physiol 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Holm BA and Matalon S. Role of pulmonary surfactant in the development and treatment of adult respiratory distress syndrome. Anesth Analg 69: 805–818, 1989. [PubMed] [Google Scholar]
- 25.Holmes WW, Keyser BM, Paradiso DC, Ray R, Andres DK, Benton BJ, Rothwell CC, Hoard-Fruchey HM, Dillman JF, Sciuto AM and Anderson DR. Conceptual approaches for treatment of phosgene inhalation-induced lung injury. Toxicol Lett 244: 8–20, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Honavar J, Doran S, Ricart K, Matalon S and Patel RP. Nitrite therapy prevents chlorine gas toxicity in rabbits. Toxicol Lett 271: 20–25, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jilling T, Ren C, Yee A, Aggarwal S, Halloran B, Ambalavanan N and Matalon S. Exposure of neonatal mice to bromine impairs their alveolar development and lung function. Am J Physiol Lung Cell Mol Physiol 314: L137–L143, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jurkuvenaite A, Benavides GA, Komarova S, Doran SF, Johnson M, Aggarwal S, Zhang J, Darley-Usmar VM and Matalon S. Upregulation of autophagy decreases chlorine-induced mitochondrial injury and lung inflammation. Free Radic Biol Med 85: 83–94, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lambert JA, Carlisle MA, Lam A, Aggarwal S, Doran S, Ren C, Bradley WE, Dell'italia L, Ambalavanan N, Ford DA, Patel RP, Jilling T and Matalon S. Mechanisms and Treatment of Halogen Inhalation-Induced Pulmonary and Systemic Injuries in Pregnant Mice. Hypertension 70: 390–400, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Larsen R, Gozzelino R, Jeney V, Tokaji L, Bozza FA, Japiassu AM, Bonaparte D, Cavalcante MM, Chora A, Ferreira A, Marguti I, Cardoso S, Sepulveda N, Smith A and Soares MP. A central role for free heme in the pathogenesis of severe sepsis. Sci Transl Med 2: 51ra71, 2010. [DOI] [PubMed] [Google Scholar]
- 31.Lazrak A, Chen L, Jurkuvenaite A, Doran SF, Liu G, Li Q, Lancaster JR Jr. and Matalon S. Regulation of alveolar epithelial Na+ channels by ERK1/2 in chlorine-breathing mice. Am J Respir Cell Mol Biol 46: 342–354, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lazrak A, Creighton J, Yu Z, Komarova S, Doran SF, Aggarwal S, Emala CW Sr., Stober VP, Trempus CS, Garantziotis S and Matalon S. Hyaluronan mediates airway hyperresponsiveness in oxidative lung injury. Am J Physiol Lung Cell Mol Physiol 308: L891–L903, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lee EJ, Woodske ME, Zou B and O'Donnell CP. Dynamic arterial blood gas analysis in conscious, unrestrained C57BL/6J mice during exposure to intermittent hypoxia. J Appl Physiol (1985) 107: 290–294, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Leustik M, Doran S, Bracher A, Williams S, Squadrito GL, Schoeb TR, Postlethwait E and Matalon S. Mitigation of chlorine-induced lung injury by low-molecular-weight antioxidants. Am J Physiol Lung Cell Mol Physiol 295: L733–L743, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li C, Weng Z, Doran SF, Srivastava RK, Afaq F, Matalon S and Athar M. Chlorine induces the unfolded protein response in murine lungs and skin. Am J Respir Cell Mol Biol 49: 197–203, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Li W and Pauluhn J. Phosgene-induced acute lung injury (ALI): differences from chlorine-induced ALI and attempts to translate toxicology to clinical medicine. Clin Transl Med 6: 19, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Liang X, Lin T, Sun G, Beasley-Topliffe L, Cavaillon JM and Warren HS. Hemopexin down-regulates LPS-induced proinflammatory cytokines from macrophages. J Leukoc Biol 86: 229–235, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lim SC, Yang JY, Jang AS, Park YU, Kim YC, Choi IS and Park KO. Acute lung injury after phosgene inhalation. Korean J Intern Med 11: 87–92, 1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lin T, Liu J, Huang F, Van Engelen TS, Thundivalappil SR, Riley FE, Super M, Watters AL, Smith A, Brinkman N, Ingber DE and Warren HS. Purified and recombinant hemopexin: protease activity and effect on neutrophil chemotaxis. Mol Med 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lin T, Sammy F, Yang H, Thundivalappil S, Hellman J, Tracey KJ and Warren HS. Identification of hemopexin as an anti-inflammatory factor that inhibits synergy of hemoglobin with HMGB1 in sterile and infectious inflammation. J Immunol 189: 2017–2022, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lo SH, Chan CC, Chen WC and Wang JD. Grand rounds: outbreak of hematologic abnormalities in a community of people exposed to leakage of fire extinguisher gas. Environ Health Perspect 114: 1713–1717, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mackie E, Svendsen E, Grant S, Michels JE and Richardson WH. Management of Chlorine Gas-Related Injuries From the Graniteville, South Carolina, Train Derailment. Disaster Med Public Health Prep 1–6, 2014. [DOI] [PubMed] [Google Scholar]
- 43.Mansuy D, Beaune P, Cresteil T, Lange M and Leroux JP. Evidence for phosgene formation during liver microsomal oxidation of chloroform. Biochem Biophys Res Commun 79: 513–517, 1977. [DOI] [PubMed] [Google Scholar]
- 44.Mo Y, Chen J, Schlueter CF and Hoyle GW. Differential susceptibility of inbred mouse strains to chlorine-induced airway fibrosis. Am J Physiol Lung Cell Mol Physiol 304: L92–102, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nash T and Pattle RE. The absorption of phosgene by aqueous solutions and its relation to toxicity. Ann Occup Hyg 14: 227–233, 1971. [DOI] [PubMed] [Google Scholar]
- 46.Oh JY, Hamm J, Xu X, Genschmer K, Zhong M, Lebensburger J, Marques MB, Kerby JD, Pittet JF, Gaggar A and Patel RP. Absorbance and redox based approaches for measuring free heme and free hemoglobin in biological matrices. Redox Biol 9: 167–177, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Oh MW, Honavar J, Doran S, Benavides G, Darley-Usmar V, Matalon S, Ford DA, and Patel RP Chlorinated Fatty Acids Are Biomarkers and Potential Mediators of Chlorine Gas Toxicity. Free Radical Biology and Medicine 76(Supplement 1), S165–S166. 11-November-2014. Ref Type: Abstract [Google Scholar]
- 48.Plahovinsak JL, Perry MR, Knostman KA, Segal R and Babin MC. Characterization of a nose-only inhaled phosgene acute lung injury mouse model. Inhal Toxicol 27: 832–840, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sciuto AM, Stotts RR, Chittenden V, Choung E and Heflin MD. Changes in absorbance at 413 nm in plasma from three rodent species exposed to phosgene. Biochem Biophys Res Commun 226: 906–911, 1996. [DOI] [PubMed] [Google Scholar]
- 50.Shokolenko I, Venediktova N, Bochkareva A, Wilson GL and Alexeyev MF. Oxidative stress induces degradation of mitochondrial DNA. Nucleic Acids Res 37: 2539–2548, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Song W, Wei S, Liu G, Yu Z, Estell K, Yadav AK, Schwiebert LM and Matalon S. Postexposure Administration of a {beta}2-Agonist Decreases Chlorine-Induced Airway Hyperreactivity in Mice. Am J Respir Cell Mol Biol 45: 88–94, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Squadrito GL, Postlethwait EM and Matalon S. Elucidating mechanisms of chlorine toxicity: reaction kinetics, thermodynamics, and physiological implications. Am J Physiol Lung Cell Mol Physiol 299: L289–L300, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Summerhill EM, Hoyle GW, Jordt SE, Jugg BJ, Martin JG, Matalon S, Patterson SE, Prezant DJ, Sciuto AM, Svendsen ER, White CW and Veress LA. An Official American Thoracic Society Workshop Report: Chemical Inhalational Disasters. Biology of Lung Injury, Development of Novel Therapeutics, and Medical Preparedness. Ann Am Thorac Soc 14: 1060–1072, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Vaish AK, Consul S, Agrawal A, Chaudhary SC, Gutch M, Jain N and Singh MM. Accidental phosgene gas exposure: A review with background study of 10 cases. J Emerg Trauma Shock 6: 271–275, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wagener BM, Hu PJ, Oh JY, Evans CA, Richter JR, Honavar J, Brandon AP, Creighton J, Stephens SW, Morgan C, Dull RO, Marques MB, Kerby JD, Pittet JF and Patel RP. Role of heme in lung bacterial infection after trauma hemorrhage and stored red blood cell transfusion: A preclinical experimental study. PLoS Med 15: e1002522, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ware LB and Matthay MA. The acute respiratory distress syndrome. N Engl J Med 342: 1334–1349, 2000. [DOI] [PubMed] [Google Scholar]
- 57.Yadav AK, Bracher A, Doran SF, Leustik M, Squadrito GL, Postlethwait EM and Matalon S. Mechanisms and modification of chlorine-induced lung injury in animals. Proc Am Thorac Soc 7: 278–283, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zarogiannis SG, Jurkuvenaite A, Fernandez S, Doran SF, Yadav AK, Squadrito GL, Postlethwait EM, Bowen L and Matalon S. Ascorbate and deferoxamine administration after chlorine exposure decrease mortality and lung injury in mice. Am J Respir Cell Mol Biol 45: 386–392, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zarogiannis SG, Wagener BM, Basappa S, Doran S, Rodriguez CA, Jurkuvenaite A, Pittet JF and Matalon S. Postexposure aerosolized heparin reduces lung injury in chlorine-exposed mice. Am J Physiol Lung Cell Mol Physiol 307: L347–L354, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zhou T, Yu Z, Jian MY, Ahmad I, Trempus C, Wagener BM, Pittet JF, Aggarwal S, Garantziotis S, Song W and Matalon S. Instillation of hyaluronan reverses acid instillation injury to the mammalian blood gas barrier. Am J Physiol Lung Cell Mol Physiol 314: L808–L821, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]