

Abstract
Brain tumors remain a great clinical challenge, in part due to their capacity to invade into eloquent, inoperable regions of the brain. In contrast, inflammation in the central nervous system (CNS) due to injuries activates microglia and astrocytes culminating in an astroglial scar that typically “walls-off” the injury site. Here, the hypothesis is tested that targeting peritumoral cells surrounding tumors to activate them via an inflammatory stimulus that recapitulates the sequelae of a traumatic CNS injury, could generate an environment that would wall-off and contain invasive tumors in the brain. Gold nanoparticles coated with inflammatory polypeptides to target stromal cells in close vicinity to glioblastoma (GBM) tumors, in order to activate these cells and stimulate stromal CNS inflammation, are engineered. It is reported that this approach significantly contains tumors in rodent models of GBM relative to control treatments (reduction in tumor volume by over 300% in comparison to controls), by the activation of the innate and adaptive immune response, and by triggering pathways related to cell clustering. Overall, this report outlines an approach to contain invasive tumors that can complement adjuvant interventions for invasive GBM such as radiation and chemotherapy.
Keywords: glial scars, glioblastoma, inflammation, reactive gliosis, stromal reprogramming
1. Introduction
Primary brain tumors are unique in that they rarely metastasize outside the brain.[1–4] Nevertheless, brain tumors are characterized by high morbidity and mortality partly due to their localization and often locally invasive growth.[3] Gliomas, tumors arising from glial cells, account for almost 30% of all primary brain tumors, and 80% of all malignant ones, and are responsible for the majority of deaths from primary brain tumors.[5] WHO grade IV gliomas–glioblastoma multiforme (GBM), are the most malignant and frequently occurring gliomas. For patients with newly diagnosed GBM, the therapeutic regimen is maximally safe and feasible resection of the tumor mass, followed by concomitant and adjuvant temozolomide (TMZ) plus radiotherapy followed by TMZ alone.[3,5] Despite these measures, survival rates of GBM patients have remained dismal, with little improvement over the last 50 years. An important reason for this is that GBMs are highly invasive and can invade deep and into eloquent regions of the brain, making resections highly risky.[3,5] Therefore, limiting this invasive property may lead to better outcomes for managing GBM.
A critical determinant of brain tumor invasion is its extracellular matrix (ECM).[6–8] In general, injury (physical or chemically induced) to the central nervous system (CNS) leads to inflammation of glial cells, which in turn leads to the formation of a glial scar that contains a remodeled ECM. The remodeled ECM is a key component in the pathophysiology of nervous system injury, e.g., the ECM of the glial scar formed by inflamed glial cells after traumatic brain or spinal cord injury, or bacterial and fungal cell wall exposure, is inhibitory to axonal regeneration.[9–11] Glial scar ECM is rich in chondroitin sulfate proteoglycans (CSPGs), a diverse family of covalently linked protein–chondroitin sulfate (CS) glycosaminoglycan (GAG) polysaccharide complexes. CSPGs have been used as a proxy to study the growth inhibitory properties of the glial scar. The growth promoting or inhibitory/repulsive effects of CSPGs are exerted predominantly by the various sulfated GAGs that form the CSPGs.[11,12] CSPG-rich glial scarring around sites of traumatic brain or spinal cord injury provides a critical barrier, quelling inflammation and preventing wider spread of tissue damage by “walling-off” the injury site.[9,13] In the context of cancer biology, the literature on the role of GAGs and CSPGs in promoting or inhibiting tumors is equivocal and contradictory, but may be unified by the observation that many CS-GAGs cause repulsion of tumor cells.[7,14–19]
Here we hypothesized that the purposeful evocation of inflammation using an inflammatory-stimulus around invasive CNS tumors will contain their spread or size, limiting their ability to form larger tumors. To test this hypothesis, we i) investigated the ability of CSPGs to inhibit invasion of CNS tumors in vitro; ii) designed nanocarriers that generate inflammation induced reactive gliosis around brain tumors in a syngeneic rat model of GBM, and a xenogeneic rat model of human GBM; and iii) investigated this approach’s ability to contain the spread/size of invasive tumors in vivo.
2. Results
2.1. CSPGs Repel GBM Cells
To determine the effects of CSPGs on repelling tumor cells an established in vitro model of glial scarring was used:[16,20] the spot assay using a prototypical inhibitory CSPG, aggrecan. Aggrecan was chosen because it is a constituent of glial scars, and is sulfated with all the families of CS-GAGs.[7,13] Tumor cells were repelled by the boundary posed by the aggrecan spot (Figure 1a). Since CSPGs exert their inhibitory effects primarily via the CS-GAG side chains, chondroitinase ABC (cABC) was used to enzymatically digest and cleave the CS-GAG chains to determine whether the boundary was sustained in the absence of CS-GAG side chains. Chondroitinase digestion of aggrecan abolished the inhibitory effects of intact aggrecan (Figure 1b), confirming that CSPGs—the principal inhibitory components of the glial scar—contributed a biochemical barrier to the invasion of GBM cells in vitro.
Figure 1.
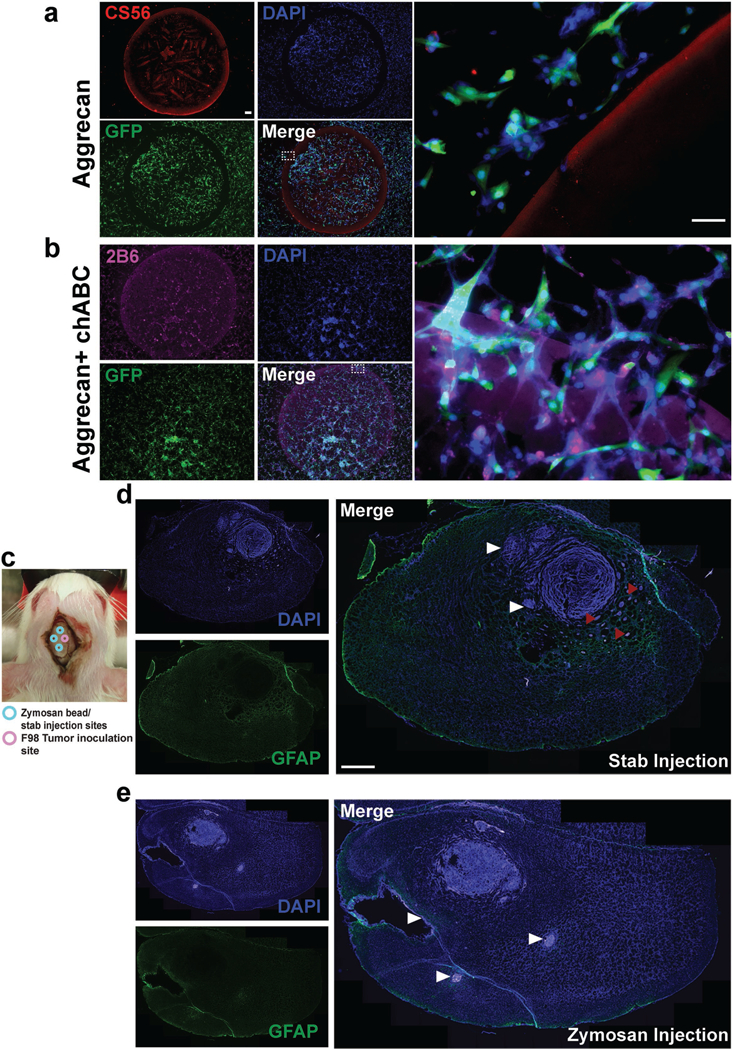
Astroglial scar constituents repel brain tumor cells. a) An in vitro model of glial scar was implemented using a spot assay with the CSPG, aggrecan. U87mg GBM cells expressing green fluorescent protein (GFP) were used. DAPI indicates the nuclear stain 4’,6-diamidino-2-phenylindole, and CS56 is an antibody that stains intact CSPGs. A spot of CS56+ aggrecan (1 mg mL−1, 2 μL spot) repels the tumor cells. Dotted line indicates zoomed in region on right, confirming that tumor cells cannot cross the boundary posed by aggrecan. b) Enzymatic digestion of CSPG GAGs abolishes CSPG-mediated tumor cell invasion inhibition. 2B6 is an antibody that stains the GAG stubs following enzymatic digestion of aggrecan by chondroitinase ABC (chABC). Tumor cells were able to cross the spot boundary posed by aggrecan upon chABC digestion indicating that CSPGs mediate their repulsive effects via CS-GAGs. c) Schematic of coinjection of zymosan beads (or control stab wounds in separate animals) and F98 tumors in Fischer rats. d) Glial fibrillary acidic protein (GFAP) is an immunofluorescent stain for reactive astrocytes. Stab wounds, as indicated by white arrow heads partly repelled tumor growth but were unable to prevent tumor microsatellite migration as indicated by red arrow heads. e) Zymosan beads caused fulminant gliosis and cavitation (white arrow heads) and caused tumors to remain as compact masses. Scale bar in (a) is 50 μm, zoomed in region is 200 μm and in (d) is 200 μm.
To stimulate peritumoral expression of CSPGs, Fischer rats were coinjected with highly motile, syngeneic F98 GBM cells,[21] and zymosan—a fungal pathogen known to cause robust glial inflammation and subsequently, scarring (Figure 1c).[22] Animals were sacrificed 21 d after tumor inoculation. F98 tumor cells were excluded from regions of control stabs (Figure 1d) and of zymosan bead injection (Figure 1e). In control animals, tumors formed microsatellites away from stab sites, and in animals treated with zymosan beads, tumor cells were constrained within the boundaries of the bead injection sites. Zymosan injection caused robust astroglial activation (Figure 1e) indicating that astroglial scarring largely contained and constrained tumor cell migration in vivo.
2.2. Zymosan-Derived Polypeptides Activate Toll-Like Receptor 2
Direct injection of zymosan into brain tumors is not practical for multiple reasons, e.g., risk of injury due to intracranial injections, incomplete coverage of tumor periphery, etc. Nanoparticles bearing zymosan are ideal to obviate these issues, since nanoparticles leverage the enhanced permeability and retention (EPR) effect[23] to target vascularized tumors such as GBM, and accumulate in the tumor periphery[24]—a location ideal for constraining tumors. Fitch et al.[22] have previously shown both in vitro and in vivo that zymosan causes reactive gliosis by inflaming and activating astrocytes and microglia. However, zymosan beads are insoluble in water and are roughly 13 μm in diameter,[22] posing critical challenges for nanoparticle-based delivery. To circumvent these issues, we extracted a water-soluble polypeptide mixture[25] from zymosan beads (Table S1, Supporting Information). As expected, the mixture contained multiple peptides of the yeast outer membrane (as reported by Laland et al., ref. [25]), and other yeast cell wall enzymes. Addition of this zymosan polypeptide (Zpep) to EOC microglial cells caused nitric oxide production (assessed by a nitrite assay[26]), which increased with time (Figure 2a), and tumor necrosis factor alpha (TNF-α) production (Figure 2b). Since zymosan is a toll-like receptor 2 (TLR2) stimulus,[27,28] Zpep activation of TLR2-related pathways was determined. Zymosan recognition by mammalian cells is mediated by TLR2 and the β-glucan receptor dectin.[27] EOC cells were treated with laminarin (a soluble β-glucan that can block dectin-mediated recognition) or TLR2-blocking antibody prior to exposure to Zpep.[27,29] The blocking of TLR2, but not dectin, led to a decrease in nitric oxide production by EOC cells, indicating that Zpep retains the TLR2-activating properties of zymosan (Figure 2c), and that the Zpep extract did not contain any water-insoluble β-glucans. Addition of Zpep to EOC cells caused robust upregulation of genes associated with microglial activation (Figure 2d).[30] Additionally, we confirmed that Zpep mimics the expected response of zymosan on astrocytes—that zymosan cannot activate astrocytes directly but through secretory factors of microglia exposed to zymosan.[22] These properties were confirmed, as direct addition of Zpep was not able to classically activate C8D1A astrocytes (Figure 2e,f), yet conditioned media from EOC cells exposed to Zpep, induced robust astrocyte activation, as assessed by nitric oxide production and inflammatory gene upregulation (Figure 2e,f).[27,31,32] Taken together, these data indicate that Zpep displays biological properties similar to zymosan and causes robust inflammation in glial cells.
Figure 2.
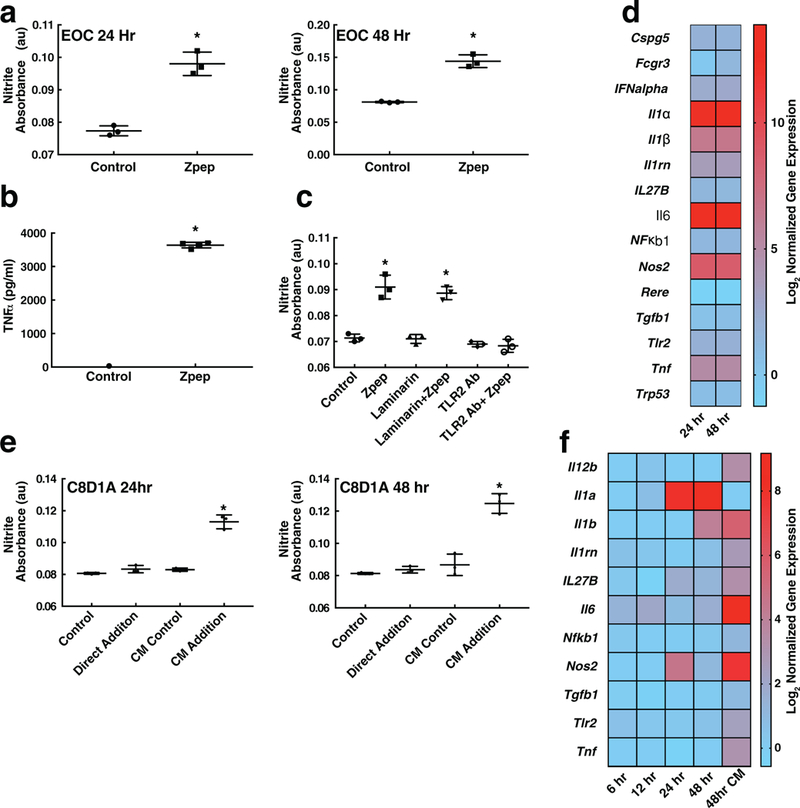
Zymosan peptide (Zpep) causes glial cell activation. Addition of Zpep to EOC mouse microglial cell line causes increased nitric oxide production over time, as assessed by a) the Griess assay and b) the secretion of TNF-α. c) Zpep activates glial cells via toll-like receptor 2 (TLR2). EOC cells were treated with laminarin (to block dectin-mediated recognition) or TLR2-blocking antibody prior to exposure to Zpep. Only TLR2 blocking led to a decrease in nitric oxide production by EOC cells, indicating that Zpep retains the TLR2 activating properties of zymosan. d) Zpep addition led to the upregulation of inflammatory gene transcripts. e) EOC-conditioned medium, but not direct addition of Zpep, activates astrocytes. CM indicates EOC-conditioned medium. Direct addition of Zpep to C8D1A astrocytes failed to induce nitric oxide production even after 48 h of exposure. Conversely, addition of EOC-CM to C8D1A cells induced e) nitrite production and f) upregulated inflammatory gene transcripts at both 24 and 48 h. *p < 0.05; Student’s t-test in a and one-way ANOVA followed by Tukey’s test in (c), (e), and (f). Data are plotted mean ± standard deviation.
2.3. Zpep-Carrying Nanoparticles Stimulate Peritumoral CSPG Expression In Vivo
It has been previously shown that systemically injected nanoparticles accumulate at the periphery of GBM tumors in rodents.[24,33,34] Gold nanoparticles (AuNPs) offer a facile platform for conjugating peptides to the AuNP surface via physical or chemical adsorption, e.g. dative, covalent bond formation between thiol groups on amino acids and the gold surface.[35–38] We decorated the surface of AuNPs (60 nm diameter) with Zpep and 20 kDa polyethylene glycol (PEG) to create inflammatory nanoparticles (AuNP-Z). Particles decorated with PEG alone (AuNP-P) served as vehicle controls. Conjugation of polypeptides onto AuNP surfaces was assessed by UV–vis spectroscopy. As reported previously for AuNP–peptide conjugates,[39] a slight red-shift and an increase in absorption was observed in the AuNP-Z group (Figure 3a). Addition of PEG increased the particle diameter to ≈100 nm, and the subsequent addition of Zpep to ≈90 nm (Figure 3a), as assessed by dynamic light scattering and transmission electron microscopy (Figure S1, Supporting Information). This marginal reduction in size could be due to steric interactions between the polypeptides and PEG.
Figure 3.
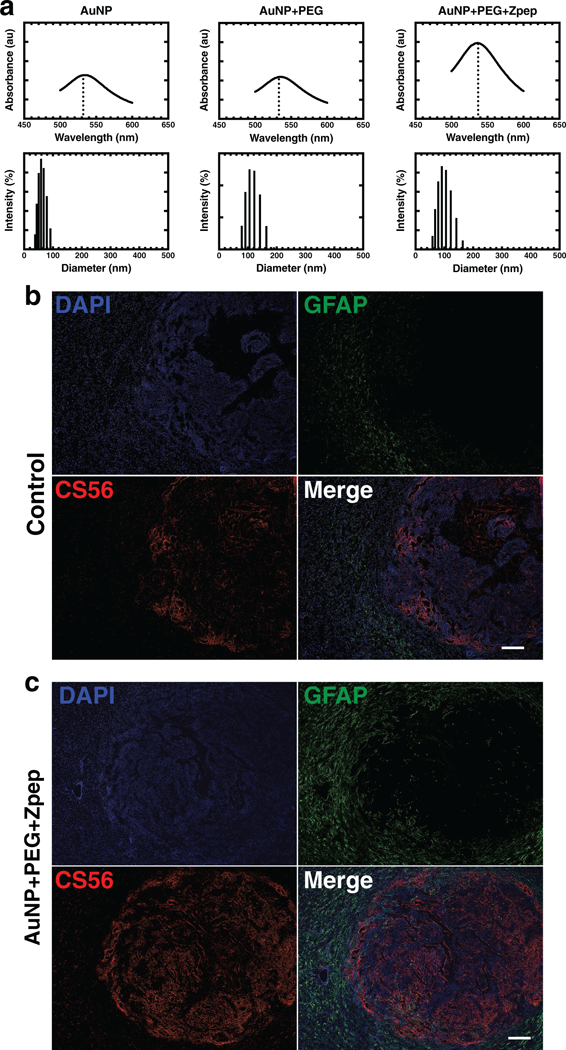
Zpep-bearing gold nanoparticles induce reactive gliosis in vivo. a) UV–vis spectroscopy of AUNPs bearing PEG and Zpep. Addition of PEG alone did not lead to a red-shift (peak absorbance at 535 nm for naked AuNPs and AuNP-P). However, addition of Zpep caused a small red-shift of 4–5 nm (peak absorption at 540 nm for AuNP-Z). Dynamic light scattering of AuNPs (60 nm diameter core) showed an increase in hydrodynamic radius of AuNPs upon addition of PEG (100 nm diameter) and Zpep (90 nm diameter). b,c) In vivo administration of AuNP-Z. F98 gliomabearing Fischer rats were injected intravenously with Zpep-bearing AuNPs (100 μg of Zpep) at 6 d post-tumor inoculation (DPI) and sacrificed at 20 DPI. (b) Control tissue sections from animals bearing F98 tumors, but not receiving any treatment, displayed low astrocyte activation (GFAP staining) and low CSPG production (CS56 staining). Conversely, sections from (c) Zpep-treated animals showed robust astrocyte activation and CSPG production. Scale bar: 50 μm.
To test the capacity of AuNP conjugates to cause peritumoral CSPG expression in vivo, Fischer rats were inoculated with syngeneic F98 tumor cells, and either AuNP-Z (100 μg Zpep total dose) or AuNP-P were injected intravenously at 7 d posttumor inoculation (DPI). Animals were sacrificed at 21 DPI and prepared for histology. We observed that tumors were large and fragmented in the control and AuNP-P groups. In stark contrast animals given AuNP-Z showed smaller, compact tumors (Figure S2b, Supporting Information), similar to that observed with direct injection of zymosan beads. Immunofluorescent staining of glial fibrillary acidic protein (GFAP), a marker of reactive astrocytes, and CSPGs (CS56 antibody) was weak in control and AuNP-P groups (Figure 3b and Figure S2a, Supporting Information). However, CS56 and GFAP immunoreactivity was strong in the AuNP-Z group (Figure 3c) with high reactivity within and at the tumor periphery— a phenomenon reminiscent of non-migratory gliomas.[7] Taken together, these data show that systemic injection of AuNP-Z particles induced elevated CSPG and GFAP expression around brain tumors in vivo, and thus constraining them.
2.4. AuNP-Z Administration Slows Tumor Growth and Leads to Smaller Tumors
To test the ability of AuNP-Z to retard and contain tumor growth, Rowett nude (RNU) rats[40] bearing xenogeneic U87 tumors[41] were injected intravenously with AuNPs at 9 DPI, having confirmed tumor formation. Tumor volumes were monitored longitudinally using T2-weighted, magnetic resonance imaging. AuNP-Z administration led to slower growing tumors with significantly smaller volumes in animals receiving AuNP-Z (134 ± 100 mm3 in AuNP-Z SD vs 311 ± 109 mm3 in control group) (Figure 4a). Because we observed that a few days after AuNP administration the tumors seemed to pick up in growth rate (Figure 4a), we split the initial dose equally into two halves and injected animals at 9 DPI and 13 DPI. This dosing regimen led to even smaller and slower growing tumors (86 ± 35 mm3 in AuNP-Z DD) in comparison with the single-dose cohort (Figure 4b,c).
Figure 4.
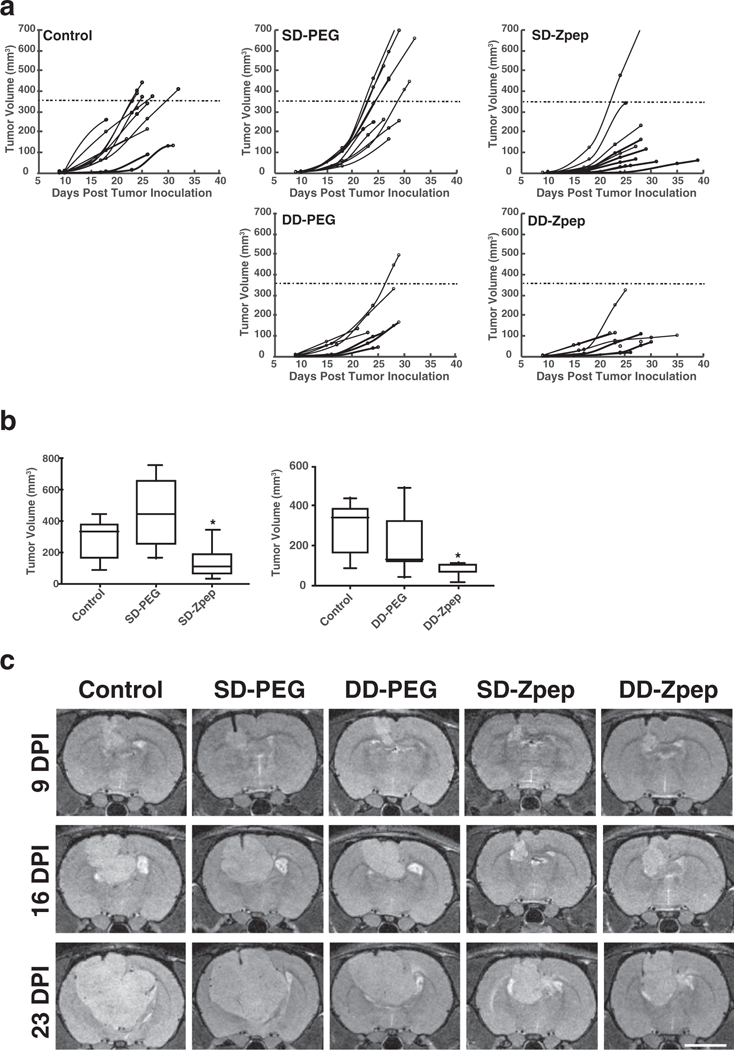
Zpep-bearing gold nanoparticles curtail tumor growth. a) Tumor volume plotted longitudinally for all five experimental groups. Tumor volumes were calculated from MRI images of animals imaged roughly weekly starting at 9 DPI until death. MRI images were analyzed for growth by outlining the tumor on 0.5 mm slices through tumor-bearing rat brains and calculating the volume (mm3). SD refers to single dose and DD to double dose of AuNPs. The timeline of the dosing regime is indicated for the SD and the DD groups. The dashed line indicates median volume at time of death of animals in the control group. Each dot on the growth curve indicates an MRI session from which the tumor volume was calculated. b) Box and whisker plots summarizing terminal volumes of the five experimental groups in (a). Zpep administration curtailed tumor growth significantly compared to other groups. c) Representative T2-weighted MRI images of the experimental groups at the tumor injection site. Images are shown at 9, 16, and 23 DPI for the same animal. *p < 0.05; one-way ANOVA (Kruskal-Wallis test), followed by the Uncorrected Dunn’s post-hoc test.
To confirm the presence of an inflammatory milieu in the tumor, we performed proteomic analyses. Proteomic analyses[41] of tumor biopsy showed that AuNP-Z administration led to an increase in proteins involved in cell adhesion, exocytosis, antigen processing, leukocyte-mediated immunity, and extracellular cell structures (Figure 5 and Tables 1 and 2 and Table S2, Supporting Information). In stark contrast, administration of AuNP-P particles led to the upregulation of proteins involved in the generation of metabolites, energy derivation, cellular transport, and CNS growth (Table S2, Supporting Information) indicating continued tumor progression.[41,42] Taken together, these data indicate that AuNP-Z administration led to smaller malignant gliomas which was in part mediated by regulation of pathways related to inflammation and cell clustering.
Figure 5.
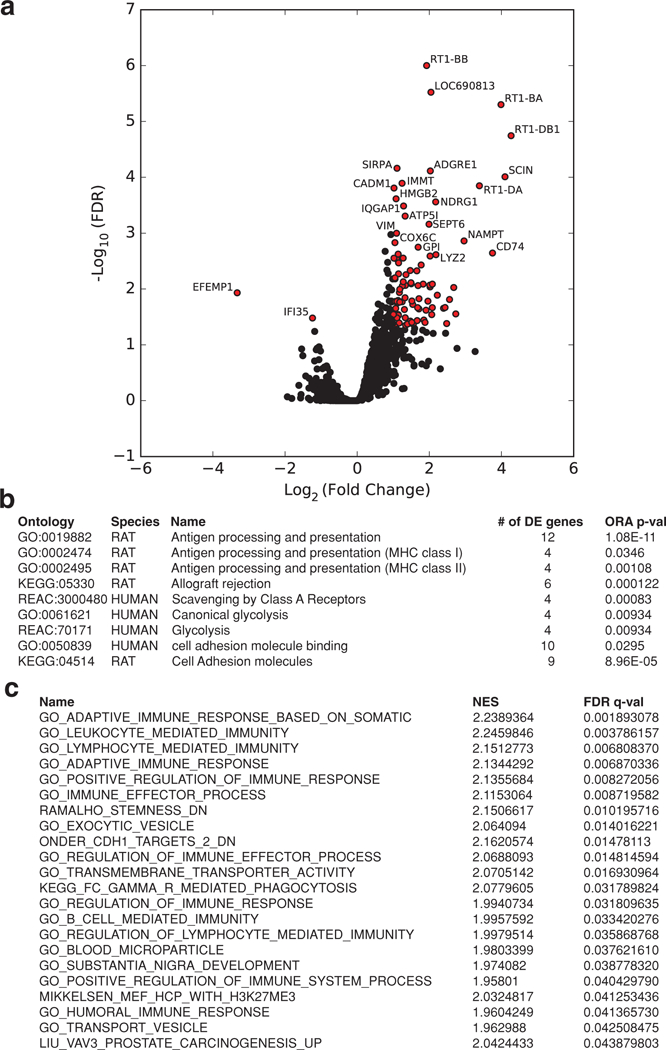
Proteomic analysis of in vivo response to AuNP-Z. a) Volcano plot depicting differential expression of proteins in AuNP-Z treated animals relative to controls (significantly differentially expressed proteins indicated by red dots). Each dot represents a uniquely identified protein accession. Significance threshold was set to a false discovery rate (FDR) of <0.05 and a fold change of greater than ±twofold. These proteins are involved in the pathways listed in (b) and (c). b) Select list of significantly overrepresented pathways in the AuNP-Z group relative to controls. c) List of all the significantly enriched pathways in the AuNP-Z group from GSEA analysis. DE refers to differentially expressed, ORA to over representation analysis, and NES to normalized enrichment score.
Table 1.
Gene set enrichment analysis of AuNP-Z treated animals relative to controls. Pathways and significantly enriched core genes are shown
| Name | Core enrichment genes |
|---|---|
| GO_LEUKOCYTE_MEDIATED_IMMUNITY | CADM1,CD74,C2,CTSC,ANXA3,MYO1G,CORO1A,C4A,C1QB,C3,C1QA,PRDX1,PTPN6,PI4K2A |
| GO_ADAPTIVE_IMMUNE_RESPONSE_BASED_ON_SOMATIC_ RECOMBINATION_OF_IMMUNE_RECEPTORS_BUILT_FROM_ IMMUNOGLOBULIN_SUPERFAMILY_DOMAINS |
CADM1,CD74,C2,CTSC,MYO1G,C4A,C1QB,C3,C1QA |
| ONDER_CDH1_TARGETS_2_DN |
SIRPA,CADM1,CTSC,ANXA3,CORO1A,C3,GPM6B,CNTN1,SNCA,NDRG1,CAMK2B,NRCAM,SLC1 A3,SERPINA1,CA9, L1CAM,ARHGDIB |
| GO_LYMPHOCYTE MEDIATED IMMUNITY | CADM1,CD74,C2,CTSC,MYO1G,CORO1A,C4A,C1QB,C3,C1QA,PRDXIPTPN6 |
| RAMALHO_STEMNESS_DN | SIRPA,CTSS,PTPRC,NCAM1,CORO1A,CST3,CTSB,AP2A2,ITGB2,PACSIN1,BIN1,GRIA2 |
| GO_POSITIVE_REGULATION_OF_IMMUNE_RESPONSE |
CADM1,HMGB2,CD74,C2,CTSS,PTPRC,GRB2,WAS,HLA-DRA,FCGR3A,MYO1G,PSMB10,THY1,C4A, C1QB,PSMA2,C3,C1QA,PSMB9,ARPC1B,PAK1,PTPRJ, PSMB8,ARPC4,ELMO1,ARPC3,PTPN6,CD180, PHB,ARPC2,UBE2K,CTSB,HRG,ITGB2 |
| GO_ADAPTIVE_IMMUNE_RESPONSE | CADM1,CD74,C2,CTSS,CTSC,MYO1G,TAP1,C4A,C1QB,C3,C1QA |
| GO_IMMUNE_EFFECTOR_PROCESS |
CADM1,CD74,C2,PTPRC,GRB2,DHX58,WAS,CTSC,ANXA3,FCGR3A,MYO1G,CORO1A,ITGAL,ST AT2,C4A,C1QB,C3,C1QA,ARPC1B,PAK1,ARPC4,ELMO1,PRDX1,ARPC3,PTPN6,CD18O,PI4K2A, ARPC2,LCP1,STAT1 |
| KEGG_FC_GAMMA_R_MEDIATED_PHAGOCYTOSIS | SCIN,DNM1L,PTPRC,WAS,FCGR3A,VASP,ARPC1B,PAK1,DNM1,ARPC4,ARPC3,ARPC2,MARCKS |
| GO_TRANSMEMBRANE_TRANSPORTER_ACTIVITY |
ATP5I,COX6C,CYB5A,COX5A,TAPBP,SLC4A1,ATP6V0D1,VDAC1,ANXA5,SLC25A12,VDAC2,GPM 6A,TAP1,ATP1B1,SLC25A5,SLC1A2,ATP6V0A1,ATP1A2,ATP8A1,SLC12A5,SLC25A4,SLC1A3,ATP6V1G2, SV2A,ATP2B2,ATP5J2,GRIA2,ATP1B2,ATP1A3,BSG,SLC8A2,ATP6V1B2,TTYH1,CALM1,ATP6V1A,ATP5A1, SLC25A1WL,G3,TF,UQCRB,CLICIATP2BIATP5D,UQCRCISLC44AIATP2B4,ATP6V1EIATP1A1 |
| GO_REGULATION_OF_IMMUNE_EFFECTOR_PROCESS |
CADM1,CD74,C2,PTPRC,DHX58,WAS,ANXA5,TAP1,STAT2,C4A,C3,ATP1B1,PPP3CB,SLC1A3,EL MO1,PTPN6,PHB,PHB2,AP2A2,ARF1,MIF,MDH1,LGALS3,RAC1,AP2B1,HPX,AP2M1,C1QBP, RAC2,C9,VAMP2 |
| GO_EXOCYTIC_VESICLE |
SEPT6,DNM1L,ATP6V0D1,VDAC1,ANXA5,VDAC2,RAB3A,STX1B,SNCA,SYN1,DNM1,GAD2, SYNGR3,PI4K2A,SPTBN2,SV2A |
| LIU_VAV3_PROSTATE_CARCINOGENESIS_UP | CD74,CTSS,GBP2,PSMB10,TAP1,C3,C1QA,PSMB9,PSMB8,ARHGDIB,ITGB2,STAT1,LGALS3 |
| MIKKELSEN_MEF_HCP_WITH_H3K27ME3 |
CADM3,NTM,CAMK2B,SLC12A5,SNCB,CRYM,SNAP25,SPTBN2,BCAN,PACSIN1,ATP2B2,GRIA2, ATP1A3,SLC9A2,BSN,TTYH1 |
| GO_REGULATION_OF_LYMPHOCYTE_MEDIATED_IMMUNITY | CADM1,PTPRC,WAS,TAP1,C3,PPP3CB,PTPN6 |
| GO_B_CELL_MEDIATED_IMMUNITY | CD74,C2,C4A,C1QB,C3,C1QA |
| GO_REGULATION_OF_IMMUNE_RESPONSE |
CADM1,HMGB2,CD74,C2,CTSS,PTPRC,GRB2,DHX58,WAS,HLA-DRA,FCGR3A,MYO1G,PSMB10,ITGAL, TAP1,THY1,STAT2,C4A,C1QB,PSMA2,C3,C1QA,GPX1,PSMB9,ARPC1B,PAK1,PPP3CB,PTPRJ,PSMB8, ARPC4,CD81,ELMO1,ARPC3,PTPN6,CD180,PHB,ARPC2,UBE2K,AMBP,CTSB,PHB2,HRG,ITGB2, STAT1,UBE2N,MIF,LGALS3,RAC1,ICAM5,PSMD13,HPX,CALM1,C1QBP,RAC2,CD14,HRAS,HSP90B1, C9,VAMP2,KRT1 |
| GO_BLOOD_MICROPARTICLE |
APOE,HBD,SLC4A1,ANXA5,HBB,HBA1,C4A,C1QB,C3,CP,HP,AMBP,DNPEP,HRG,FGA,PZP,FGB,PFN1, AHSG,MSN,ITIH1,AGT,HPX,AFM,F2,C9,TF,KRT1,PLG,FGG,CLIC1 |
| GO_SUBSTANTIA_NIGRA_DEVELOPMENT | NDRG2,BASP1,INA,CKB,CNP,SIRT2,SYNGR3,G6PD,MAG,PLP1 |
| GO_TRANSPORT_VESICLE |
SEPTG,YKT6,CD74,DNM1L,ARCN1,CTSC,ATP6V0D1,VDAC1,ANXA5,HLA-DRA,VDAC2,RAB3A,STX1B, CTSZ,SNCA,SYN1,DNM1,GAD2, SERPINA1,SYNGR3,PI4K2A,SPTBN2,CLTB,SV2A,SIPA1 |
| GO_HUMORAL_IMMUNE_RESPONSE | C2,PSMB10,C4A,C1QB,C3,C1QA,FGA,FGB,C1QBP,GPI,C9,KRT1 |
| GO_POSITIVE_REGULATION_OF_IMMUNE_SYSTEM_PROCESS |
SCIN,CADM1,HMGB2,CD74,C2,CTSS,PTPRC,GRB2,DHX58,WAS,HLA-DRA,FCGR3A,MYO1G,PSMB10, CORO1A,TAP1,THY1,C4A,ClQB,PSMA2,C3,C1QA,PSMB9,ARPC1B,PAK1,PTPRJ,PSMB8,ARPC4,CD81, ELMO1,ARPC3,PTPN6,CD180,PHB,ARPC2,UBE2K,CTSB,AIF1,HRG,ITGB2,UBE2N,THBS1,MIF,PNP, LGALS3,RAC1,PSMD13,HPX,HCLS1,C1QBP,RAC2,CD14,HRAS,HSP90B1,C9,CALR,VAMP2,KRT1 |
Table 2.
Overrepresentation analysis of AuNP-Z-treated animals relative to controls. Pathways and significantly differentially expressed core genes are shown.
| Ontology | Species | Name | DE genes |
|---|---|---|---|
| GO:0019882 | RAT | Antigen processing and presentation | RT1-BA,PSMB8,CALR,CD74,CTSS,TAPBP,RT1-A2,WAS,RT1-BB,RT1-DA,RT1-DB1,LOC103689996 |
| GO:0002474 | RAT | Antigen processing and presentation (major histocompatibility complex (MHC) class) | CALR,TAPBP,RT1-A2,LOC103689996 |
| GO:0002495 | RAT | Antigen processing and presentation (MHC class) | RT1-BA,CD74,RT1-BB,RT1-DA |
| KEGG:05330 | RAT | Allograft rejection | RT1-BA,RT1-A2,RT1-CE7,RT1-BB,RT1-DA,RT1-DB1 |
| REAC:300048 | HUMAN | Scavenging by class a receptors | APOE,HSP90B1,FTH1,CALR |
| GO:0061621 | HUMAN | Canonical glycolysis | ENO1,PGK1,GPI,HK3 |
| REAC:70171 | HUMAN | Glycolysis | ENO1,PGK1,GPI,HK3 |
| GO:0050839 | HUMAN | Cell adhesion molecule binding | ENO1,NDRG1.RARS,TMPO,IQGAP1,MSN,CADM3,CALR,CADM1,SPTAN1 |
| KEGG:04514 | RAT | Cell adhesion molecules | RT1-BA,PTPRC,CADM3,CADM1,RT1-A2,RT1-CE7,RT1-BB,RT1-DA,RT1-DB1 |
3. Discussion
The possibility that stromal expression of CSPG and GFAP, as a result of purposefully induced glial inflammation, could contain aggressive brain tumors was hitherto unexplored. However, the following notions are worthy of consideration: i) tumors that are naturally bound by scar or encapsulated are typically benign;[6,43–45] ii) benign, encapsulated tumors can be tolerated for several years; and iii) astroglial scar does not impede function as long as it is not thwarting regeneration (as is the case in spinal cord injuries). This study demonstrates that it is possible to leverage endogenous mechanisms of scar formation to moderate tumor growth by modulating the behavior of cells other than those of the tumor. We have previously demonstrated similar containment by targeting brain tumor cells directly using a small cytostatic molecule,[46] although that strategy is hindered by the typical constraints of drug delivery to the brain,[47] often failing to deliver the therapeutic payload to the entire tumor mass.[33,34] Here, we report the targeting of endogenous inflammatory mechanisms for the induced expressed of inhibitory matrix in the stromal space using the EPR effect via gold nanoparticles. Gold nanoparticles have been used extensively in vivo.[38] Our mechanism of tumor containment relies partly on the activation of astrocytes, microglia, and the production of CSPGs (Figures S2 and S3, Supporting Information; Figures 2 and 3), which are highly inhibitory to regenerating axons in CNS trauma but also serve as boundary forming molecules during development. The role of CSPGs in tumor infiltration and progression is not clear, with various conflicting reports on the subject.[7,11,14,18] Recently, Silver et al. showed that noninvasive lesions are associated with a rich matrix containing substantial amounts of CSPGs, whereas glycosylated CSPGs are essentially absent from diffusely infiltrating tumors.[7] We hypothesize that this apparent contradiction is explained by the location of the expressed GAGs. Dual-sulfated GAGs such as CS-E are repulsive to tumor cells and enhance invasion if they are expressed within the tumor mass. Conversely, when expressed in the periphery of tumors, CSPG boundaries can prevent glioma cells from crossing the boundary.
We used a polypeptide extracted from zymosan[25] to inflame glial cells in vivo and in vitro. Zymosan, a yeast cell wall preparation, was used because it specifically activates microglia and astrocytes when injected directly into the brain, recapitulating the sequelae of a physical injury in its absence. Laland et al. extracted a polypeptide from zymosan and showed that the polypeptide elicited immunogenic reactions similar to that by intact zymosan.[25] Similar to results observed by Fitch et al.[22] and Laland et al.,[25] Zpep was indeed able to activate microglial cells and caused elevated immunogenic reactions and glial scarring in vivo. Interestingly, microglial-conditioned medium, and not the direct addition of Zpep, activated astrocytes, indicating that Zpep exerted its inflammatory effects via macrophages and perhaps other myeloid cells in vivo. Similar results were also observed by Liddelow, et al.,[31] who showed that it was not the direct addition of lipopolysaccharide from bacterial cell walls, but the addition of microglial conditioned medium that activated astrocytes to an inflammatory state.
Tumor-stromal crosstalk leads to immunosuppression of stromal cells, and tumors often co-opt the activities of the stromal cells to increase their growth or invasiveness.[48,49] Therefore, targeting stromal cells of the tumor to reverse their immunosuppressive phenotype is an attractive strategy that has recently gained attention and has yielded favorable outcomes in multiple studies.[50,51] The use of TLR agonists and small molecules[52–57] that can activate microglia and macrophages, or alter their phenotype, has been shown to curb tumor growth by altering the stromal microenvironment into a growth suppressive environment, among other mechanisms. Our results are in line with these studies.
We observed that in vivo administration of Zpep-bearing gold nanoparticles led to a slow-down of tumor growth and smaller tumor volumes. To further characterize this growth suppression, we performed exploratory proteomic analyses. Proteomic analyses was also important because Zpep administration stopped at day 9 DPI, whereas animals were sacrificed a few weeks later. During the phase between injection of Zpep and sacrificing, the tumor did grow, and while early effects of Zpep induced inflammation may have dampened, an overall inflammatory signature would still be present. Pro-teomic analyses showed that the biological response elicited by Zpep-bearing particles was markedly different than that evoked by particles bearing PEG alone. Zpep administration led to increased expression of proteins involved in activation of the immune response, antigen processing, extracellular cellular components, and pathways related to glycolysis and cell adhesion molecule binding (Figure 5).
Gene set enrichment analysis (GSEA) of proteomic data indicated enrichment of pathways related to cytostatic, nonmigratory behavior (Tables 1 and 2). For example, the E-cadherin and MHC class II pathways were upregulated upon Zpep administration in vivo. Onder et al.[58] showed that the loss of the epithelial adhesion molecule E-cadherin results in metastatic dissemination of tumors by inducing wide-ranging transcriptional and functional changes. Zpep administration caused an enrichment of proteins associated with elevated E-cadherin expression. Zagzag et al.[59] showed that the process of brain tumor invasion is associated with decreased expression of MHC antigens allowing glioma cells to invade the surrounding brain in a “stealth”-like manner. Antigen presentation is crucial for the generation of a specific anti-tumor response by the adaptive immune system. Zpep administration led to enrichment of pathways related to upregulation of MHC class I and II, as well as those related to complement signaling, and Fc-receptor-mediated phagocytosis. Pathways related to cytotoxic and phagocytic functions of microglia require microglia to upregulate the expression of complement and Fc-gamma receptors. Activation of phagocytes, including microglia, is the most common function of Fc receptors, indicating that Zpep was acting in part via microglial cells in vivo.[60–62] In stark contrast, animals dosed with AuNP-P showed enrichment of pathways related with neuronal cell communication and synaptic transmission, reminiscent of a quiescent microenvironment that allows for tumor growth. Overall, proteomic analyses point towards a mode of action of Zpep that involves regulation of cell adhesion, migration, and immune activation.
While it was not the primary focus of this work, it was interesting to note that even the robust and significant reduction in tumor volume did not lead to an improvement in survival. In fact, tumor volume did not correlate with survival in our dataset (Figure S4, Supporting Information). Tumor volume is a confounding variable in retrospective studies. Multiple studies have shown that tumor volume is not a reliable prognostic indicator and that gross total resection volume is a better prognostic indicator of survival.[63–65] But this is confounding because if a tumor can be resected, it is likely not located in an eloquent region. Furthermore, if tumors are located in eloquent regions, they may be detected early and lead to earlier treatment due to early presentation of clinical and neurological symptoms.
Clinically, the precise cause of lethality due to brain tumors appears to be highly nuanced and is still unclear.[5] Multiple retrospective studies assign higher importance to tumor location, rather than volume, as a reliable prognostic factor.[63,66] We implanted genetically identical tumor cells in the same location across multiple animals and saw no difference in survival whether the tumors were larger or smaller. We posit that our observed lethality may be due to: i) the tumor being inoculated into an eloquent region; ii) some critical volume threshold already being reached before therapeutic intervention, even by the smaller, constrained tumors, thereby preventing prolonged survival. Conversely, the dosage and timing of the treatment not being adequate; iii) or portions of the tumors escaping, prior to being adequately contained, to other eloquent regions of the brain or body.
Because GBMs are extremely hard to treat, and the exact reason for their lethality is unknown, unorthodox, and orthogonal technologies such as the approach described here may present useful ways to manage GBMs. The methods outlined in this study are one way of doing this. In this study, we used sterile inflammation as a means to recapitulate CNS-injury induced inflammatory sequelae that form a growth inhibitory barrier to tumor invasion. This approach has its limitations because inflammation is a complex process—one that tumors are adept at manipulating and using to their advantage. Proteomics analyses indicated upregulation of pathways related to innate and adaptive immunity and inflammation upon Zpep administration. Recent evidence shows that tumors can down-regulate TLR expression to become anergic to inflammatory stimuli, and wither the toxic effects of inflammation.[67] This said, we suggest that containing the growth of tumors and limiting their volume may be necessary but not sufficient to prolong survival and we anticipate that containing tumor spread may represent an excellent adjuvant strategy in combination with chemotherapy or radiation therapy.
4. Conclusions
It is an established fact that the diffuse infiltrative propensities of GBMs render most therapies ineffective, and a means to prevent tumors from migrating might be useful. Given this criterion, an approach such as the one described in this study allows for control over both the dispersion of tumor microsatellites and the spread of metastatic tumors, provided a leaky vasculature (EPR) is established at tumor sites. Here, tumor growth and tumor volumes were constrained by targeted inflammation of peritumoral stromal cells including astrocytes and microglia. Constraining tumor volume can significantly limit metastatic spread and potentially increase the effectiveness of combination adjuvant therapies.
5. Experimental Section
Cell Lines and Culture Procedures:
U87 cells (human glioma, HTB-14), F98 cells (Fischer rat glioma, CRL-2397), EOC cells (mouse microglia, CRL-2469), LADMAC cells (mouse macrophages/monocytes, CRL-2420), and C8D1A cells (mouse cerebellum astrocytes, CRL-2541) were purchased from ATCC and maintained according to instructions specific to each cell line. The CRL-2469 cell line was cultured in Dulbecco’s modified Eagle medium (DMEM; Corning) and 10% FBS with either 30 ng mL−1 recombinant mouse CSF-1 (416-ML-010, R&D Systems)[56] or with conditioned medium from LADMAC cells. All cells were grown at 37 °C with 5% CO2, passaged with trypsin-EDTA 0.05% and maintained in respective complete cell culture medium (ATCC recommended) and 1% penicillin–streptomycin (Gibco) unless otherwise noted. As noted for some experiments, U87 cells were made to stably express enhanced green fluorescent protein (eGFP), via transfection with an eGFP expression plasmid using the Effectene Transfection Reagent (Qiagen) and further selection of stable transfectants with G418 Sulfate (Gemini).[68]
Spot Assays:
Preparation of 14 mm glass-bottomed Petri-dishes (In vitro Scientific, Sunnyvale, CA) was done according to methods previously described[16,20] with few modifications. Briefly, surfaces were coated with poly-L- lysine (PLL, 1:10 dilution in ultrapure water) (Sigma-Aldrich) and incubated overnight at 37 °C. The following day, the surfaces were rinsed thrice with sterile water and allowed to dry completely. Various concentrations of aggrecan, bovine serum albumin (BSA), and fibronectin with Texas Red (Invitrogen) were spotted onto the prepared surfaces in 2 μL amounts and allowed to dry completely. Next, 80000 U87mg cells (stably expressing eGFP) suspended in standard growth medium were carefully added to the chambers. After 24 h, the cells were fixed with 4% paraformaldehyde and 0.4 M sucrose solution for 15 min and immunostained for CS-56 (Sigma C8035, 1:250). For determining the effect of GAG side chains, aggrecan spots were treated extensively with cABC (1 U mL−1; 3–5 h, 37 °C; Sigma). Next, after aspirating the cABC and rinsing with PBS, 80000 eGFP+U87mg cells were plated as described above. After 24 h, cells were fixed and immunostained for 2B6 (Seikagaku, 1:250) and CS-56 (1:200).
Zpep Extraction:
Zymosan (250 mg, Sigma–Aldrich) was snap frozen in liquid nitrogen and crushed into a fine powder using a mortar and pestle. The crushed zymosan was added to an extraction buffer (10 mL) containing TRIS–HCl (0.5 M), CHAPS (1%), DTT (1%), and PMSF. The extraction process was allowed to go on overnight at room temperature with gentle agitation. The next day, the samples were centrifuged at 5000× g for 10 min and the supernatant was collected following which it was washed, concentrated and dialyzed (3×) using a 10 kDa centrifugal spin column (EMD Millipore, Amicon Ultra-15) according to manufacturer’s instructions. Protein concentrations were determined using a NanoDrop device. Zpep aliquots were stored at −20 °C until further use.
In Vitro Experiments:
Zpep was used at a concentration of 25 μg mL−1 in vitro. Nitrite production was assessed at time points (indicated in figure legends) using the Griess reagent system (G2930, Promega). TLR2 blocking experiments were performed as described elsewhere.[27–29] TNF-α levels were assessed using anti-TNF- α enzyme-linked immunosorbent assay (ELISA) kit as per the manufacturer’s instructions (BD Biosciences, RayBiotech). Quantitative real-time PCR was performed on a multiplexed Fluidigm system and data were analyzed according to methods described previously.[69] Primers were purchased from Fluidigm Inc.
Gold Nanoparticles:
Gold nanoparticles were purchased from Ted Pella (60 nm diameter, 15 708–6). PEG-thiol (MPEG-SH-20K-1 g) was purchased from Laysan Bio. All reactions were performed in ultrapure DI water (18 MΩ cm−1). Zpep and PEG-SH were conjugated to AuNPs as described elsewhere.[37,70,71] Briefly, the amount of gold nanoparticles each animal received per dose was 57 μg( =1 mL of 60 nm gold nanoparticles, at a concentration of 2.6 × 10e10 particles mL−1). PEG-SH concentration was calculated such that each nanoparticle received 30000 PEG-SH molecules (Qian et al.). PEG was dissolved in ultrapure DDI water. Zymosan peptide was reconstituted at 1 mg mL−1, such that each animal would receive 100 mg final dose (based on initial concentration of zymosan and assuming 100% conjugation efficiency). Zymosan was suspended in 3 × 10−3 m Tris base buffer (Yeh et al., and Paciotti et al.). Nanoparticles were centrifuged (12 000× g, 20 min), and resuspended in ultrapure DDI water. The particles, peptide, and zymosan were combined and left on a rotating test-tube holder overnight at room temperature. Following this, particles were aliquoted into individual doses, and washed twice by centrifugation (12 000× g, 20 min), before being resuspended in sterile saline for in vivo tail vein injections, or ultrapure DDI water for physical characterization. Dynamic light scattering and UV–vis spectroscopy were performed as described previously.[37]
Tumor Inoculation:
All experiments were approved by the Institutional Animal Care and Use Committee at the Georgia Institute of Technology and Duke University. Rowett nude rats or Fischer rats (175–200 g, male, Charles River Laboratories) were inoculated with U87 mg (80 000 cells)[41] or F98 tumor cells (10 000 cells)[21] respectively. The animals were anesthetized using 5% isoflurane and maintained with 2–3% isoflurane during surgical procedures. The animals were placed in a stereotaxic device. A 1 cm incision was made on the head. The periosteum was cut and removed from the skull. A hole was made in the skull 2 mm lateral and 2 mm posterior from bregma. A 26-gauge needle mounted on a 10 μL Hamilton syringe was inserted 2 mm deep from the surface of the brain and retracted 0.5 mm. Tumor cells in 5 μL of DMEM (serum free) were injected using an automated syringe pump at a rate of 1 μL min−1. The needle was held in place an additional 2 min before removal and closing. Animals displaying symptoms of distress from the glioblastoma were anesthetized with ketamine (1 mL kg−1), xylazine (0.17 mL kg−1), and acepromazine (0.37 mL kg−1), transcardially perfused with physiological PBS followed by 4% paraformaldehyde; the brains were dissected and incubated in 4% paraformaldehyde overnight and then stored in 30% sucrose containing 0.01% sodium azide. For cohorts designated to proteomics and histology, anesthetized animals were transcardially perfused with physiological PBS followed by 10% formalin, and stored in 10% formalin at room temperature.
MRI Imaging:
Rats were anesthetized and placed in a Bruker Pharmascan 7-T (Bruker BioSpin MRI) operating with the ParaVision software with a 38 mm quadrature-detection volume coil as head coil.[41,46] The animal was anaesthetized using 2% isoflurane and placed in a home-built cradle, allowing the easy placement of the animal’s head within the MRI coil. The rapid acquisition of high quality T2 weighted images was achieved using the rapid imaging with refocused echoes (RARE) sequence (RARE factor, 6; effective echo time, 36 ms; repetition time [TR], 4200 s; two averages per scan; total acquisition time, 6 min). A slab of 40 transversal slices was recorded using a field of view of 40 mm × 40 mm with a 256 × 256 matrix and a slice thickness of 0. 5 mm. This slab was aligned to cover the injection site of the tumor cells using a pilot scan, which was recorded immediately before the acquisition of the RARE images. MR images were acquired roughly every week following tumor implant to check for tumor growth or regression. ImageJ software (FIJI, version 2.0) was used for further image processing, and for tumor volume calculations. A region of interest (ROI) following the tumor borders was drawn manually in the T2-weighted images. The whole tumor volume was calculated by adding up the voxel volumes within the ROIs of all image slices. In the case that MRI was not possible on days of euthanasia, tumor volume at time of death was extrapolated assuming a linear tumor growth rate.
Proteomics at Duke Proteomics and Metabolomics Shared Resource:
U87mg tumors that had previously been fixed in formalin were macrodissected from the surrounding normal tissue from the brains of nine rats: three different animals per group, three groups total. The wet weight of each was noted. 1 mL of 50 × 10−3 M ammonium bicarbonate (AmBic) was added to each tumor, and heated at 80 °C for 55 min while shaking at 750 rpm. The AmBic was removed, and another 1 mL of AmBic was added for a second rinse. The second rinse was then pipetted off, and the tumor was allowed to cool completely at room temperature (<5 min). Each tumor was transferred to a centrifuge tube, and 8 M urea in 50 × 10−3 m AmBic was added at 10 μL per mg of wet weight. The tissue was then taken through tissue tearing until no tissue pieces were visible, and the samples were homogenized. The samples were then probe sonicated at power level 3 for 5 s bursts, 3 bursts each while on ice. A concentration was determined for each homogenate by Bradford assay. 50 μg from each sample was taken out and concentration normalized in 8 M urea in 50 × 10−3 M AmBic. Then, enough AmBic was added to each to get to 1.8 M urea for subsequent in-solution tryptic digestion. Briefly, the samples were reduced in 10 × 10−3 M dithiothreitol (DTT) at 32 °C for 45 min, alkylated in 25 × 10–3 M iodoacetamide (IAA) at room temperature in the dark for 30 min, and trypsin was added to each at a 1:50 ratio of enzyme to protein for digestion at 32 °C overnight while shaking at 750 rpm. The following morning, the samples were acidified with trifluoroacetic acid to give 0.5% TFA final, and taken through a C18 SPE cleanup (Waters Sep-Pak Vac, 50 mg cartridges, Product # WAT054955). After some of the acetonitrile was evaporated via Speed Vac, the remaining extracts were taken to dryness by lyophilization overnight. The samples were then reconstituted in 200 μL of 1% TFA/2% ACN containing 25 fmol μL−1 yeast alcohol dehydrogenase surrogate standard. A QC pool was prepared by mixing equal volumes of all samples.
Quantitative Mass Spectrometry:
Quantitative 1D liquid chromatography, tandem mass spectrometry (1D LC-MS/MS) was performed on the peptide digests per sample, with additional analyses of conditioning runs and QC pools. Samples were analyzed using a nanoACQUITY UPLC system (Waters) coupled to a QExactive Plus high resolution accurate mass tandem mass spectrometer (Thermo) via a nanoelectrospray ionization source. Briefly, the sample was first trapped on a Symmetry C18 180 μm × 20 mm trapping column (5 μL min−1 at 99.9/0.1 v/v H2O/MeCN) followed by an analytical separation using a 1.7 μm Acquity HSS T3 C18 75 μm × 250 mm column (Waters) with a 90 min gradient of 5% to 40% MeCN/H2O with 0.1% formic acid at a flow rate of 400 nL min−1 and column temperature of 55 °C. Data collection on the QExactive Plus MS was performed in data-dependent acquisition mode with a 70 000 resolution (@ m/z 200) full MS scan from m/z 375 to 1600 with a target AGC value of 1e6 ions followed by 10 MS/MS scans at 17 500 resolution (@ m/z 200) at a target AGC value of 5e4 ions. A 20 s dynamic exclusion was employed. The total analysis cycle time per sample injection was ≈2 h. Following 12 total UPLC-MS/MS analyses (including three replicate QC injections), data were imported into Rosetta Elucidator v 4.0 (Rosetta Biosoftware, Inc.), and analyses were aligned based on the accurate mass and retention time of detected ions (“features”) using PeakTeller algorithm in Elucidator. Relative peptide abundance was calculated based on area-under-the-curve (AUC) of the selected ion chromatograms of the aligned features across all runs. Elucidator was utilized to produce fragment ion spectra, and Mascot Server (v 2.5, Matrix Sciences) performed the database searches. The MS/MS data were searched against two databases: a Swissprot database with Homo sapiens taxonomy and a NCBI refseq database with Rattus norvegicus taxonomy (both downloaded in August 2016) with additional proteins commonly used as internal controls including yeast ADH1, bovine serum albumin, and bovine alpha casein, as well as an equal number of reversed-sequences (“decoys”) for false discovery rate determination. Database search parameters included fixed modification on Cys (carbamidomethyl) and variable modifications on Asn and Gln (deamidation) and Met (oxidation), 2 missed cleavages, precursor tolerance of 5 ppm and product tolerance at 0.2 Da, and trypsin as the enzyme specificity. After individual peptide scoring using the PeptideProphet algorithm in Elucidator, the data were annotated at a 0.9% peptide false discovery rate.
Proteomic Differential Expression:
For proteomics, comparisons between fold changes of the protein-level intensities (PLIs) were used to determine proteins that were differentially expressed (DE) between the conditions (Z: AuNP-Z; P: AuNP-P; C: control, no AuNP; n = 3). The statistical comparison tool QPROT (v1.3.3)[72] (nburnin: 2000; niters: 10 000; normalized: true) was used to compute a z-statistic and FDR for each identified protein compared pairwise between conditions. The p values were obtained from the z-statistic using Python (Version 2.7.11, Anaconda 2.2.0; https://www.python.org/). Proteins with a FDR < 0.05 for a particular fold change comparison between conditions were considered statistically significant and treated as DE for that condition-pair.
Pathway Overrepresentation Analysis:
The DE proteins were prefiltered to exclude proteins with an absolute fold change of less than twofold. For each condition-pair, gene ontology was performed using g:profiler version: r1730_e88_eg35 (http://biit.cs.ut.ee/gprofiler/).[73] Both Rattus norvegicus and Homo sapiens datasets were used. Since parsimonious assignment of species is unreliable for closely homologous proteins, each species dataset was run for all proteins (species-indifferent), as well as the subsets of proteins identified as either rat or human. The search included Gene Ontology, KEGG, Reactome, and Regulatory Motif databases and used the built-in g: SCS threshold for significance. Default settings were used. QuickGO (https://www.ebi.ac.uk/QuickGO) web service was used for obtaining additional gene ontology information for visualization with Python.
Pathway Enrichment Analysis:
Gene set enrichment analysis (GSEA) Release 3.0 (http://www.broadinstitute.org/gsea)[74] was used to perform pathway enrichment analysis. Preranked analysis was performed against the curated (c2.all.v6.0), and gene ontology (c5.all.v6.0), datasets using the negative log of the QPROT p-value, signed according to fold-change direction, as the ranking scheme for each condition-pair. The isoform with the lowest p-value was selected in cases of gene symbol collision. Default settings were used except the max and min pathway size exclusion criteria were set to 1000 and 5, respectively.
Immunohistochemistry, Immunofluorescence, and Microscopy:
All antibodies used for IHC and IF are listed in Figure Captions. Fixed, frozen brain tissue was sectioned to 14 μm thickness, and prepared for immunofluorescence using methods described elsewhere.[69] Sections were imaged on a Zeiss Axiovision inverted microscope. For IHC, tissues were processed at the Emory Winship Pathology Core Lab, as described elsewhere.[41] Tissues from paraffin-embedded blocks were sectioned at 5 μm thickness. IHC was performed using DAB chromogenic kit (Wako) following the manufacturer’s protocol. Whole-slide scanning was done using a Hamamatsu Nanozoomer 2.0 HT
Graphing and Statistics:
All graphs were made in Prism 7 (Graphpad Inc.), Python, or MATLAB (Version 9, Mathworks, MA). Layout of figure panels was done with Illustrator (Adobe Inc.). Outliers were omitted when indicated by a Grubb’s test. Where appropriate, Student’s t-tests or one way ANOVA (as described in figure legends), followed by post-hoc tests were run in Prism 7. Survival analysis was also performed in Prism 7, and significance was assessed using the Mantel-Cox log rank test.
Supplementary Material
Acknowledgements
D.P. and J.A. contributed equally to this work. R.V.B. and T.S. conceived the idea. T.S. supervised the study, conducted experiments, and analyzed the data. J.G.L. analyzed and illustrated proteomics data. S.B.P. conceived the Zpep extraction and purification strategy. D.P. and J.A. analyzed the MRI data and assisted with nanoparticle fabrication and in vitro experiments. L.K. assisted with aggrecan spot assays. S.L.C. performed the PCR experiments and analysis. E.G. assisted with surgeries and MRI. T.S., J.G.L., and R.V.B. wrote the manuscript. All authors read and edited the manuscript. The authors thank the Ian’s Friends Foundation for generously supporting this research. Helpful discussions with, and inputs from, Dr. Syed Enam, Dr. Nalini Mehta, Dr. Daniel Brat, and Dr. Peter Fecci are greatly appreciated. Assistance with immunofluorescence by Ms. Ketki Patil and Mr. William Burke is greatly appreciated. The authors thank Laura Dubois, Dr. Arthur Moseley, and Dr. J. Will Thompson at the Duke Proteomics and Metabolomics Shared Resource for their help with proteomics. This work was performed using a Q-Exactive Plus tandem mass spectrometer acquired via the S10 grant NIH S10OD012266-01A1.
Footnotes
Conflict of Interest
The authors declare no conflict of interest.
Supporting Information
Supporting Information is available from the Wiley Online Library or from the author.
Contributor Information
Dr. Tarun Saxena, Department of Biomedical Engineering Pratt School of Engineering Duke University, 101 Science Drive, Durham, NC 27705, USA
Dr. Johnathan G. Lyon, Department of Biomedical Engineering Pratt School of Engineering Duke University, 101 Science Drive, Durham, NC 27705, USA
Dr. S. Balakrishna Pai, Wallace H. Coulter Department of Biomedical Engineering, Georgia Institute of Technology & Emory School of Medicine, UA Whitaker Building, 313 Ferst Drive, Atlanta, GA 30332, USA
Daniel Pare, Wallace H. Coulter Department of Biomedical Engineering, Georgia Institute of Technology & Emory School of Medicine, UA Whitaker Building, 313 Ferst Drive, Atlanta, GA 30332, USA.
Jessica Amero, Wallace H. Coulter Department of Biomedical Engineering, Georgia Institute of Technology & Emory School of Medicine, UA Whitaker Building, 313 Ferst Drive, Atlanta, GA 30332, USA.
Lohitash Karumbaia, Regenerative Bioscience Center, The University of Georgia, 425 River Road, ADS Complex, Athens, GA 30602, USA.
Sheridan L. Carroll, Department of Biomedical Engineering Pratt School of Engineering Duke University, 101 Science Drive, Durham, NC 27705, USA
Eric Gaupp, Wallace H. Coulter Department of Biomedical Engineering, Georgia Institute of Technology & Emory School of Medicine, UA Whitaker Building, 313 Ferst Drive, Atlanta, GA 30332, USA.
Dr. Ravi V. Bellamkonda, Department of Biomedical Engineering Pratt School of Engineering Duke University, 101 Science Drive, Durham, NC 27705, USA, ravi@duke.edu
References
- [1].Beauchesne P, Cancers 2011, 3, 461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Lun M, Lok E, Gautam S, Wu E, Wong ET,J. Neuro-Oncol. 2011, 105, 261. [DOI] [PubMed] [Google Scholar]
- [3].Cuddapah VA, Robel S, Watkins S, Sontheimer H, Nat. Rev Neurosci. 2014, 15, 455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Hamilton JD, Rapp M, Schneiderhan TM, Sabel M, Hayman A, Scherer A, Kröpil P, Budach W, Kretschmar U, Arne Gerber P, J. Clin. Oncol. 2014, 32, e80. [DOI] [PubMed] [Google Scholar]
- [5].Weller M, Wick W, Aldape K, Brada M, Berger M, Pfister SM, Nishikawa R, Rosenthal M, Wen PY, Stupp R, Nat. Rev. Dis. Primers 2015, 1, 15017. [DOI] [PubMed] [Google Scholar]
- [6].Mueller MM, Fusenig NE, Nat. Rev. Cancer 2004, 4, 839. [DOI] [PubMed] [Google Scholar]
- [7].Silver DJ, Siebzehnrubl FA, Schildts MJ, Yachnis AT, Smith GM, Smith AA, Scheffler B, Reynolds BA, Silver J, Steindler DA, J. Neurosci. 2013, 33, 15603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Gu L, Mooney DJ, Nat. Rev. Cancer 2016, 16, 56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Silver J, Miller JH, Nat. Rev Neurosci. 2004, 5, 146. [DOI] [PubMed] [Google Scholar]
- [10].Sofroniew MV, Trends Neurosci. 2009, 32, 638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Silver DJ, Silver J, Curr. Opin. Neurobiol. 2014. 27, 171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Gilbert RJ, McKeon RJ, Darr A, Calabro A, Hascall VC, Bellamkonda RV, Mol. Cell. Neurosci. 2005, 29, 545. [DOI] [PubMed] [Google Scholar]
- [13].Galtrey CM, Fawcett JW, Brain Res. Rev. 2007, 54, 1. [DOI] [PubMed] [Google Scholar]
- [14].Varga I, Hutoczki G, Szemcsak CD, Zahuczky G, Toth J, Adamecz Z, Kenyeres A, Bognar L, Hanzely Z, Klekner A, Pathol. Oncol. Res. 2012, 18, 413. [DOI] [PubMed] [Google Scholar]
- [15].Viapiano MS, Bi WL, Piepmeier J, Hockfield S, Matthews RT, Cancer Res. 2005, 65, 6726. [DOI] [PubMed] [Google Scholar]
- [16].Karumbaiah L, Anand S, Thazhath R, Zhong Y, McKeon RJ, Bellamkonda RV, Glia 2011, 59, 981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Karumbaiah L, Saxena T, Betancur M, Bellamkonda RV, Curr. Med. Chem. 2014, 21, 4257. [DOI] [PubMed] [Google Scholar]
- [18].Wade A, Robinson AE, Engler JR, Petritsch C, James CD, Phillips JJ, FEBS J. 2013, 280, 2399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Belting M, Thromb. Res. 2014, 133, S95. [DOI] [PubMed] [Google Scholar]
- [20].Tom VJ, Steinmetz MP, Miller JH, Doller CM, Silver J, J. Neurosci. 2004, 24, 6531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Mathieu D, Lecomte R, Tsanaclis AM, Larouche A, Fortin D, Can. J. Neurol. Sci. 2007, 34, 296. [DOI] [PubMed] [Google Scholar]
- [22].Fitch MT, Doller C, Combs CK, Landreth GE, Silver J, J. Neurosci. 1999, 19, 8182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Matsumura Y, Maeda H, Cancer Res. 1986, 46, 6387. [PubMed] [Google Scholar]
- [24].Roller BT, Munson JM, Brahma B, Santangelo PJ, Pai SB, Bellamkonda RV, Drug Delivery Transl. Res. 2015, 5, 116. [DOI] [PubMed] [Google Scholar]
- [25].Laland P, Dedichen J, Laland S, Oftebro R, Thorsdalen N, Voss J, Nature 1963, 199, 465. [DOI] [PubMed] [Google Scholar]
- [26].Griess P, Eur. J. Inorg. Chem. 1879, 12, 426. [Google Scholar]
- [27].Dillon S, Agrawal S, Banerjee K, Letterio J, Denning TL, Oswald-Richter K, Kasprowicz DJ, Kellar K, Pare J, van Dyke T, Ziegler S, Unutmaz D, Pulendran B, J. Clin. Invest. 2006, 116, 916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Oliveira-Nascimento L, Massari P, Wetzler LM, Front. Immunol. 2012, 3, 79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Barbalat R, Lau L, Locksley RM, Barton GM, Nat. Immunol. 2009, 10, 1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Michelucci A, Heurtaux T, Grandbarbe L, Morga E, Heuschling P, J. Neuroimmunol. 2009, 210, 3. [DOI] [PubMed] [Google Scholar]
- [31].Liddelow SA, Guttenplan KA, Clarke LE, Bennett FC, Bohlen CJ, Schirmer L, Bennett ML, Munch AE, Chung WS, Peterson TC, Wilton DK, Frouin A, Napier BA, Panicker N, Kumar M, Buckwalter MS, Rowitch DH, Dawson VL, Dawson TM, Stevens B, Barres BA, Nature 2017, 541, 481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Phulwani NK, Esen N, Syed MM, Kielian T, J. Immunol. 2008, 181, 3841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Blanco E, Shen H, Ferrari M, Nat. Biotechnol. 2015, 33, 941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Wilhelm S, Tavares AJ, Dai Q, Ohta S, Audet J, Dvorak HF, Chan WC, Nat. Rev Mater. 2016, 1, 16014. [Google Scholar]
- [35].Perrault SD, Chan WC,J. Am. Chem. Soc. 2009, 131, 17042. [DOI] [PubMed] [Google Scholar]
- [36].Perrault SD, Walkey C, Jennings T, Fischer HC, Chan WC, Nano Lett. 2009, 9, 1909. [DOI] [PubMed] [Google Scholar]
- [37].Qian X, Peng XH, Ansari DO, Yin-Goen Q, Chen GZ, Shin DM, Yang L, Young AN, Wang MD, Nie S, Nat. Biotechnol. 2008, 26, 83. [DOI] [PubMed] [Google Scholar]
- [38].Yeh YC, Creran B, Rotello VM, Nanoscale 2012, 4, 1871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Thobhani S, Attree S, Boyd R, Kumarswami N, Noble J, Szymanski M, Porter RA, J. Immunol. Methods 2010, 356, 60. [DOI] [PubMed] [Google Scholar]
- [40].Festing MF, Lovell D, Sparrow S, MAY D, Connors T, Nature 1978, 274, 365. [DOI] [PubMed] [Google Scholar]
- [41].Mehta N, Lyon JG, Patil K, Mokarram N, Kim C, Bellamkonda RV, Mol. Ther.–Oncolytics 2017, 4, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Chow RD, Guzman CD, Wang G, Schmidt F, Youngblood MW, Ye L, Errami Y, Dong MB, Martinez MA, Zhang S, Renauer P, Bilguvar K, Gunel M, Sharp PA, Zhang F, Platt RJ, Chen S, Nat. Neurosci. 2017, 20, 1329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Liotta LA, Am. J. Pathol. 1984, 117, 339. [PMC free article] [PubMed] [Google Scholar]
- [44].Tlsty TD, Coussens LM, Annu. Rev. Pathol.: Mech. Dis. 2006, 1, 119. [DOI] [PubMed] [Google Scholar]
- [45].Clark WH, Br.J. Cancer 1991, 64, 631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Munson JM, Fried L, Rowson SA, Bonner MY, Karumbaiah L, Diaz B, Courtneidge SA, Knaus UG, Brat DJ, Arbiser JL, Bellamkonda RV, Sci. Transl. Med. 2012, 4, 127ra36. [DOI] [PubMed] [Google Scholar]
- [47].Lyon JG, Mokarram N, Saxena T, Carroll SL, Bellamkonda RV, Adv. Drug Delivery Rev. 2017, 114, 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Quail DF, Joyce JA, Nat. Med. 2013, 19, 1423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Quail DF, Joyce JA, Cancer Cell 2017, 31, 326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Hwang SY, Yoo BC, Jung JW, Oh ES, Hwang JS, Shin JA, Kim SY, Cha SH, Han IO, Biochim. Biophys. Acta, Mol. Cell Res. 2009, 1793, 1656. [DOI] [PubMed] [Google Scholar]
- [51].Mora R, Abschuetz A, Kees T, Dokic I, Joschko N, Kleber S, Geibig R, Mosconi E, Zentgraf H, Martin-Villalba A, Regnier-Vigouroux A, Clia 2009, 57, 561. [DOI] [PubMed] [Google Scholar]
- [52].Kees T, Lohr J, Noack J, Mora R, Gdynia G, Todt G, Ernst A, Radlwimmer B, Falk CS, Herold-Mende C, Regnier-Vigouroux A, Neuro-Oncology 2012, 14, 64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Chicoine MR, Zahner M, Won EK, Kalra RR, Kitamura T, Perry A, Higashikubo R, Neurosurgery 2007, 60, 372; discussion 381. [DOI] [PubMed] [Google Scholar]
- [54].Sarkar S, Doring A, Zemp FJ, Silva C, Lun X, Wang X, Kelly J, Hader W, Hamilton M, Mercier P, Dunn JF, Kinniburgh D, van Rooijen N, Robbins S, Forsyth P, Cairncross G, Weiss S, Yong VW, Ann. Neurosci. 2013, 20, 154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Sarkar S, Doring A, Zemp FJ, Silva C, Lun X, Wang X, Kelly J, Hader W, Hamilton M, Mercier P, Dunn JF, Kinniburgh D, van Rooijen N, Robbins S, Forsyth P, Cairncross G, Weiss S, Yong VW, Nat. Neurosci. 2014, 17, 46. [DOI] [PubMed] [Google Scholar]
- [56].Pyonteck SM, Akkari L, Schuhmacher AJ, Bowman RL, Sevenich L, Quail DF, Olson OC, Quick ML, Huse JT, Teijeiro V, Setty M, Leslie CS, Oei Y, Pedraza A, Zhang J, Brennan CW, Sutton JC, Holland EC, Daniel D, Joyce JA, Nat. Med. 2013, 19, 1264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Waldmannová E, Caisová V, Fàberová J, Sváčková P, Kovářová M, Sváčková D, Kumžáková Z, Jačková A, Vácová N, Nedbalová P, Int. Immunopharmacol. 2016, 39, 295. [DOI] [PubMed] [Google Scholar]
- [58].Onder TT, Gupta PB, Mani SA, Yang J, Lander ES, Weinberg RA, Cancer Res. 2008, 68, 3645. [DOI] [PubMed] [Google Scholar]
- [59].Zagzag D, Salnikow K, Chiriboga L, Yee H, Lan L, Ali MA, Garcia R, Demaria S, Newcomb EW, Lab. Invest. 2005, 85, 328. [DOI] [PubMed] [Google Scholar]
- [60].Li W, Holsinger RM, Kruse CA, Flugel A, Graeber MB, CNS Neurol. Disord.: Drug Targets 2013, 12, 750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Li W, Graeber MB, Neuro-Oncology 2012, 14, 958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Graeber MB, Li W, Rodriguez ML, FEBS Lett. 2011, 585, 3798. [DOI] [PubMed] [Google Scholar]
- [63].Liu TT, Achrol AS, Mitchell LA, Du WA, Loya JJ, Rodriguez SA, Feroze A, Westbroek EM, Yeom KW, Stuart JM, Chang SD, Harsh G. R. t., Rubin DL, Am.J. Neuroradiol. 2016, 37, 621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Iliadis G, Selviaridis P, Kalogera-Fountzila A, Fragkoulidi A, Baltas D, Tselis N, Chatzisotiriou A, Misailidou D, Zamboglou N, Fountzilas G, Strahlenther. Onkol. 2009, 185, 743. [DOI] [PubMed] [Google Scholar]
- [65].Capelle L, Fontaine D, Mandonnet E, Taillandier L, Golmard JL, Bauchet L, Pallud J, Peruzzi P, Baron MH, Kujas M, Guyotat J, Guillevin R, Frenay M, Taillibert S, Colin P, Rigau V, Vandenbos F, Pinelli C, Duffau H, French Reseau d’Etude des Gliomes (REG),J. Neurosurg. 2013, 118, 1157. [DOI] [PubMed] [Google Scholar]
- [66].Mistry AM, Dewan MC, White-Dzuro GA, Brinson PR, Weaver KD, Thompson RC, Ihrie RA, Chambless LB, J. Neuro-Oncol. 2017, 132, 341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Alvarado AG, Thiagarajan PS, Mulkearns-Hubert EE, Silver DJ, Hale JS, Alban TJ, Turaga SM, Jarrar A, Reizes O, Longworth MS, Cell Stem Cell 2017, 20, 450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Jain A, Betancur M, Patel GD, Valmikinathan CM, Mukhatyar VJ, Vakharia A, Pai SB, Brahma B, MacDonald TJ, Bellamkonda RV, Nat. Mater. 2014, 13, 308. [DOI] [PubMed] [Google Scholar]
- [69].Saxena T, Loomis KH, Pai SB, Karumbaiah L, Gaupp E, Patil K, Patkar R, Bellamkonda RV, ACS Nano 2015, 9, 1492. [DOI] [PubMed] [Google Scholar]
- [70].Powell AC, Paciotti GF, Libutti SK, Methods Mol. Biol. 2010, 624, 375. [DOI] [PubMed] [Google Scholar]
- [71].Paciotti GF, Myer L, Weinreich D, Goia D, Pavel N, McLaughlin RE, Tamarkin L, Drug Delivery 2004, 11, 169. [DOI] [PubMed] [Google Scholar]
- [72].Choi H, Kim S, Fermin D, Tsou C-C, Nesvizhskii AI, J. Proteomics 2015, 129, 121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Reimand J, Arak T, Adler P, Kolberg L, Reisberg S, Peterson H, Vilo J, Nucleic Acids Res. 2016, 44, W83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, Mesirov JP, Proc. Natl. Acad. Sci. USA 2005, 102, 15545. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.