

Abstract
The plasma and tissue binding properties of two corticosteroids, dexamethasone (DEX) and methylprednisolone (MPL), were assessed in the rat in anticipation of developing physiologically based pharmacokinetic and pharmacokinetic/pharmacodynamic models. The tissue-to-plasma partition coefficients (KP) of DEX and MPL were measured in liver, muscle, and lung in vivo after steady-state infusion and bolus injection in rats. Since KP is often governed by reversible binding to macromolecules in blood and tissue, an attempt was made to assess KP values of DEX and MPL by in vitro binding studies using rat tissue homogenates and to compare these estimates to those obtained from in vivo kinetics after dosing. The KP values of both steroids were also calculated in rat tissues using mechanistic tissue composition–based equations. The plasma binding of DEX and MPL was linear with moderate binding (60.5% and 82.5%) in male and female rats. In vivo estimates of steroid uptake appeared linear across the tested concentrations and KP was highest in liver and lowest in muscle for both steroids. Assessment of hepatic binding of MPL in vitro was severely affected by drug loss at 37°C in male liver homogenates, whereas DEX was stable in both male and female liver homogenates. With the exception of MPL in liver, in vitro–derived KP estimates reasonably agreed with in vivo values. The mechanistic equations modestly underpredicted KP for both drugs. Tissue metabolism, saturable tissue binding, and active uptake are possible factors that can complicate assessments of in vivo tissue binding of steroids when using tissue homogenates.
SIGNIFICANCE STATEMENT
Assuming the free hormone hypothesis, the ratio of the unbound drug fraction in plasma and in tissues defines the tissue-to-plasma partition coefficient (KP), an important parameter in physiologically based pharmacokinetic modeling that determines total drug concentrations within tissues and the steady-state volume of distribution. This study assessed the plasma and tissue binding properties of the synthetic corticosteroids, dexamethasone and methylprednisolone, in rats using ultrafiltration and tissue homogenate techniques. In vitro–in vivo and in silico–in vivo extrapolation of KP was assessed for both drugs in liver, muscle, and lung. Although the extrapolation was fairly successful across the tissues, in vitro homogenate studies severely underpredicted the KP of methylprednisolone in liver, partly attributable to the extensive hepatic metabolism.
Introduction
Drugs interact with proteins and other macromolecules in body fluids and tissues. Such binding interactions influence the pharmacokinetics and pharmacodynamics of drugs. Binding to specific carrier proteins, transporters, and enzymes can influence tissue uptake and metabolism, whereas high-affinity binding to receptors influences tissue uptake in some cases (Levy, 1994) and is critical for evoking drug responses. An appreciable portion of drug in the body may interact reversibly with proteins and cellular constituents in a nonspecific manner. Such nonspecific binding is a major determinant of drug distribution in the body and thus plays an important role in pharmacokinetics. The physiologically based Gillette equation (Gillette, 1971) characterizes the effects of body size and plasma and tissue binding on the steady-state volume of distribution (Vss):
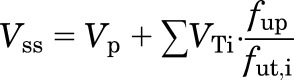 |
where Vp is plasma volume, VTi represents volumes of specific tissues, and fup and fut,i are the fractions of unbound drug in plasma and in specific tissues (assumed as concentration-independent parameters). The Vss, along with clearance, influences important aspects of pharmacokinetics, including the mean residence time and total tissue concentrations. Simulations and limited experimental evidence suggest a stronger influence of tissue binding on half-life compared with plasma binding (Craig and Welling, 1977; Kurz and Fichtl, 1983). The ratio of fup and fut defines the tissue-to-plasma partition coefficient (KP), an important parameter used in physiologically based pharmacokinetic (PBPK) modeling.
Although unbound drug concentrations in plasma can be readily assessed, the study of tissue binding is complicated by methodological factors such as tissue isolation, sample processing, experimental system, and separation of bound versus free drug. In vivo, in vitro, and tissue composition–based computational approaches have been developed. In vivo methods measure drug uptake in tissues once steady state has been established between plasma and equilibrating tissue spaces. Drug in tissue, in excess of that diffused into tissue water, is considered bound to cellular constituents in the absence of metabolism, active transport, and ionization effects (Khalafallah and Jusko, 1984a).
As reviewed extensively (Fichtl et al., 1991; Pacifici and Viani, 1992), tissue binding can be studied ex vivo using perfused organs, tissue slices, and tissue homogenates. Organs such as the liver, kidney, and lungs can be isolated and tissue binding assessed by measuring the steady-state concentration in tissue and the free concentration in the perfusate. Limited duration of organ viability can, however, limit this approach in binding studies. Incubated tissue slices offer the advantage of being technically simple and can be applied to most tissues. However, similar to perfused organs, net drug uptake into slices may represent the sum of several processes (transport, metabolism, and lysosomal trapping), including binding. Tissue homogenates have been successfully applied for generating binding data for numerous drugs in various tissues (Lin et al., 1982; Fichtl et al., 1991; Kalvass et al., 2007; Berry et al., 2010). The advantages of this method include its simplicity and its applicability in most tissues, and results obtained by this technique are not distorted by possible active uptake into cells (Fichtl et al., 1991). However, limitations include the need for tissue dilution and homogenization, which disrupts cellular architecture and may alter the binding characteristics of the tissue.
Mechanistic approaches to predict tissue concentrations and Vss attempt to estimate, based on plasma protein and red blood cell binding, lipophilicity, and pKa, the potential extent of drug partitioning into tissue components such as tissue water, albumin, neutral/charged lipids, and neutral/charged phospholipids (Poulin and Theil, 2000; Berezhkovskiy, 2004; Rodgers et al., 2005; Rodgers and Rowland, 2006). These methods have been fairly successful in predicting Vss for several drugs (Graham et al., 2012), although factors such as active transport, binding to specific proteins, and lysosomal trapping can limit their applicability in some cases.
The synthetic corticosteroids, dexamethasone (DEX) and methylprednisolone (MPL), are frequently prescribed with varying dosage regimens, thus exposing body tissues to a wide range of concentrations. Classic theory holds that, in accordance with the “free hormone hypothesis” (Mendel, 1989), unbound steroids diffuse into intracellular spaces. However, limited experimental evidence indicates a role for active membrane uptake and transporters in steroid disposition (Schinkel et al., 1995; Lackner et al., 1998; Crowe and Tan, 2012). The ubiquitous presence of the glucocorticoid receptor in various tissues may influence steroid uptake in tissues due to high-affinity binding in the cytosol. Although the plasma pharmacokinetics of both steroids have been investigated in rats and humans (Dunn et al., 1991; Hochhaus et al., 2001; Samtani and Jusko, 2005; Hazra et al., 2007), limited information is available concerning their distribution into tissues. Studies in rabbits showed that prednisolone is taken up by several tissues in amounts exceeding the diffusion of free drug into tissue water (Khalafallah and Jusko, 1984a,b).
The aim of this first report of a three-part series (Ayyar et al., 2019a,b) was to determine the binding of DEX and MPL in vitro using tissue homogenates and to compare results with data obtained in vivo in male and female rats. Further comparison was made with predictions based on three mechanistic tissue composition equations of binding in rat tissues. Findings from the in vitro and in vivo studies were used to support the development of a minimal PBPK model of MPL in rats, presented in a companion article (Ayyar et al., 2019a).
Materials and Methods
Reagents and Chemicals
DEX and 6α-MPL (≥98.5% purity) were purchased from Sigma-Aldrich (St. Louis, MO). High-performance liquid chromatography (HPLC)–grade methylene chloride, heptane, and glacial acetic acid were obtained from Fisher Scientific (Pittsburgh, PA). Rats were dosed with methylprednisolone sodium succinate (MPS) (Solu-Medrol; Pharmacia & Upjohn Company, Kalamazoo, MI) via the intramuscular route. Milli-Q water was used (Millipore Corporation, Bedford, MA). DEX sodium phosphate solution (pharmaceutical grade) was purchased from Bimeda Pharmaceuticals (Dublin, Ireland) and standard DEX (purity >98%) was purchased from Sigma-Aldrich. DEX-D5 (internal standard, purity >98%) was purchased from Toronto Research Chemicals (Toronto, ON, Canada).
Animals
Normal male and female Wistar rats were purchased from Envigo Inc. (Indianapolis, IN). The animals were housed in the State University of New York at Buffalo Laboratory Animal Facility and acclimatized under constant temperature (22°C) and humidity with a controlled 12-hour/12-hour light/dark cycle for 1 to 2 weeks. Three female rats were housed per cage, whereas two males were maintained in each cage. In all studies, rats had free access to rat chow and drinking water. The protocols adhered to the Principles of Laboratory Animal Care (National Institutes of Health publication 85-23, revised 1985) and were approved by the State University of New York at Buffalo Institutional Animal Care and Use Committee.
In Vivo Distribution.
Tissues and plasma harvested from a previously conducted infusion study (Ayyar et al., 2017) were used to assess MPL distribution at steady state. Briefly, male Wistar rats were given a 0.3 mg/kg per hour subcutaneous infusion of MPL for 1 week, and blood and tissues were harvested at various time points. Since steady state in plasma was achieved before 6 hours after the start of infusion, MPL concentrations in plasma, liver, muscle, and lung were measured in male rats (n = 3) infused for 24 hours to determine KP. The in vivo distribution of MPL in liver was also assessed by measuring the plasma and liver concentration-time profiles of MPL in male and female Wistar rats given a 50 mg/kg intramuscular bolus of drug. An in vivo subcutaneous DEX infusion study was conducted in male Wistar rats (n = 3) with the rate of 30 μg/h for 24 hours to achieve steady state, and blood and tissues were harvested for the determination of KP. In addition, another group of male Wistar rats were given a 2.25 mg/kg subcutaneous bolus of DEX and various tissues and blood were harvested to explore the tissue distribution of DEX in male rats (n = 3) at various time points. In all studies, blood was harvested using EDTA as an anticoagulant (4 mM final concentration) to obtain plasma. Harvested tissues were rapidly dissected, frozen in liquid nitrogen, and stored at −80°C until further use.
Tissues for In Vitro Binding.
Normal male and female Wistar rats (n = 2 to 3 per sex per drug) were euthanized by aortic exsanguination under isoflurane anesthesia. Blood samples were collected into syringes precoated with EDTA and centrifuged immediately at 2000g for 15 minutes at 4°C. Plasma samples were collected and pooled for each animal group and used in plasma protein binding studies. Harvested tissues, except for liver, were rapidly dissected, frozen in liquid nitrogen, and stored at −80°C until further use. Assessments of in vitro stability and tissue binding of DEX and MPL in liver homogenates employed fresh tissue.
Experiments
In Vitro Plasma Binding.
Plasma protein binding of MPL and DEX was measured by ultrafiltration using Centrifree micropartition devices (Millipore Corporation) with a 30-kDa molecular mass cutoff filter. Briefly, varying volumes of ethanolic MPL solutions (0.00625, 0.0125, 0.025, 0.005, 0.25, and 1 mg/ml) were added (2.0% volume) to pooled blank plasma samples from each group to yield six plasma samples containing MPL per group (0.125, 0.25, 0.5, 1, 5, and 20 µg/ml). Similar to MPL, different volumes of DEX methanolic stock solutions were spiked into blank plasma samples from individual male and female rats. After incubation at 37°C for 30 minutes, aliquots (500 µl) of plasma of each concentration were transferred into ultrafiltration devices and centrifuged at 2000g for 20 minutes. The filtrates and remaining plasma samples were stored at −20°C until analysis of both free and total steroid concentrations. No degradation was observed upon incubation in plasma samples at 37°C for up to 60 minutes. Preliminary studies indicated negligible nonspecific binding of either steroid to the ultrafiltration device.
In Vitro Tissue Homogenate Binding.
Freshly harvested livers were placed on a chilled dish, crudely minced with a pair of scissors, and mixed. The wet weight of livers was determined and added to prechilled 1× PBS (pH 7.4) to prepare 3, 4, 6, and 10× dilutions of tissue and homogenized using a Polytron homogenizer (Brinkmann Instruments, Westbury, NY) at speed setting 5 with two bursts of 15-second duration with 30-second intervals. Stock solutions of DEX or MPL were added (1.0% volume) to each homogenate dilution to produce final concentrations of 1 μg/ml (DEX) and 10 μg/ml (DEX and MPL). A total volume of 500 µl of each homogenate was loaded into ultrafiltration devices, incubated at 37°C for 15 minutes, and then centrifuged at 2000g for 20 minutes at 37°C. The filtrates and homogenates in the donor chamber were stored at −80°C until further analysis. The volume of ultrafiltrate recovered after centrifugation was kept to <10% of the initial homogenate loaded. Measurement of DEX and MPL stability in rat liver homogenates was performed in parallel to liver binding experiments. Aliquots containing 80 μl of the spiked liver homogenates were incubated at 37°C and the reaction terminated at various times (0, 3, 7, 10, 15, 20, and 30 minutes). The samples were stored at −80°C until further analysis. Tissues harvested from control (untreated) male Wistar rats were used for assessment of MPL and DEX binding in lung and muscle. Tissue binding in both lung and muscle were carried out similar to liver, with the exception that muscle binding was assessed using a 6× dilution only due to technical reasons. The filtrates and homogenates in the donor chamber were stored at −80°C until further analysis. Preliminary experiments confirmed that no depletion occurred in vitro in the lung and muscle homogenates throughout the binding experiment. Total protein content in the tissue homogenates was determined using the Lowry method (Lowry et al., 1951).
Plasma and Tissue Steroid Analysis.
MPL was measured in plasma, tissues, and plasma and tissue filtrates using normal-phase HPLC (Haughey and Jusko, 1988) with minor modifications in the extraction procedure for each matrix. A detailed description of the assay is provided in a companion article (Ayyar et al., 2019a). DEX was used as the internal standard for all experiments. Chromatography conditions involved a mobile phase of 585 ml MeCl2, 350 ml heptane, 10 ml glacial acetic acid, and 55 ml ethanol and a Zorbax silica column, a Waters model 1515 isocratic pump (Waters Corporation, Milford, MA), and a Waters model 2487 dual wavelength absorbance detector. The lower limit of quantification of MPL was 10 ng/ml in plasma and filtrates and 50 ng/g in tissue with an intra- and interday coefficient of variation of less than 10%. The quantification of DEX in plasma and filtrates from both tissue and plasma was described previously with minor modifications (Li et al., 2017b). Dried residue was reconstituted in 200 μl of acetonitrile/water [30:70 (v/v)] and formic acid was excluded from the mobile phase. For tissue samples, only the steps preceding solid phase extraction of DEX differed compared with plasma samples. Briefly, powdered tissue was homogenized in PBS at 6× dilution. Appropriate volumes of samples were added into 990 μl methanol. Where necessary, blank tissue homogenate was also added to achieve a total volume of 100 μl homogenate. A volume of 10 μl D5-DEX as the internal standard was added to tubes and vortexed, and the sample was centrifuged at 4°C at 14,000g for 20 minutes. The supernatant was transferred to glass tubes and dried under nitrogen. Methanol (50 μl) was used for reconstitution and vortexed twice for 30 seconds. A total of 450 μl water was added and mixed. Samples (450 μl) were transferred to microfuge tubes containing equal volumes of 4% phosphoric acid and subjected to solid phase extraction using Oasis Prime HLB 1-cm3 30-mg cartridges (Waters Corporation). The following steps were identical as reported for plasma samples. The lower limit of quantification of DEX was 0.2 ng/ml (or ng/g) in plasma, filtrates, and tissues.
Tissue Data Correction for Residual Blood.
Drug concentrations obtained in each tissue were converted from nanograms per milliliter to nanograms per gram of tissue by assuming a tissue density of 1 g/ml. Tissue concentrations were corrected for residual trapped blood as follows:
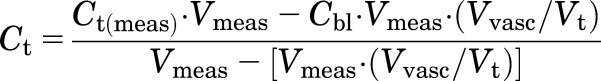 |
(1) |
where Ct and Ct(meas) are the corrected and measured tissue concentrations, Cbl is the measured concentration in blood, Vmeas is the measured volume of collected tissue, and Vvasc/Vt is the fractional vascular volume of blood trapped in tissues as obtained from the literature (Kawai et al., 1998). The corrected tissue concentrations were used for further analysis.
Theoretical and Data Analysis
Calculation of In Vivo KP.
At steady state, the KP for noneliminating tissues is given by:
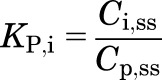 |
(2) |
where C is the measured drug concentration, and the subscripts i, p, and ss denote the tissue, plasma, and steady state.
Because the liver is an eliminating organ, KP,hep at steady state is given by :
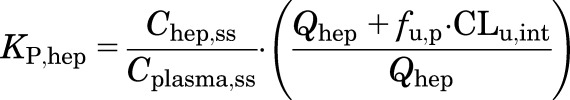 |
(3) |
utilizing the hepatic blood flow rate (Qhep), intrinsic clearance (CLu,int), and fraction of drug unbound in plasma (fu,p), where hep denotes liver.
The in vivo distribution of MPL in liver was also compared between both sexes by measuring by the plasma and liver concentration-time profiles of MPL in male and female Wistar rats given a 50 mg/kg intramuscular bolus of drug. Using these data, KP,hep was calculated as :
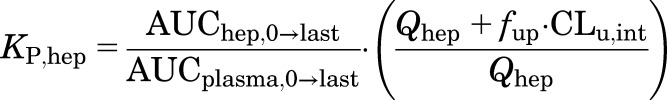 |
(4) |
where AUChep,0→last and AUCplasma,0→last represent the area under the curve for MPL from time 0 until the last measured time point. The values used for CLu,int in male rats were 2987 ml/h for MPL (Ayyar et al., 2019a) and 61.8 ml/h for DEX (calculated).
Assessment of KP from In Vitro Binding.
The binding of a drug to protein can be described by a Langmuir-type equation:
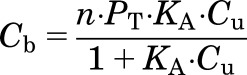 |
(5) |
where Cb is the bound drug concentration, Cu is the unbound drug concentration, KA is the association constant, and n ∙ PT is the maximum binding capacity. When KA ∙ Cu < < 1, then eq. 5 can be simplified to
| (6) |
Equation 6 can be rearranged to calculate the binding factor (BF), defined as the ratio of Cb and Cu:
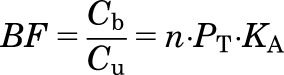 |
(7) |
Since in vitro assessment of drug binding in tissues necessitates preparation of homogenates in buffer, a dilution step is introduced, which results in a reduction of protein concentrations. If the drug binding characteristics are independent of protein concentration, then eq. 7 can be expressed as:
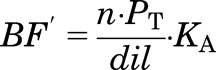 |
(8) |
where dil is the fold dilution and n ∙ PT ∙ KA is:
| (9) |
The fraction of unbound drug in an in vitro homogenate (fu,t) can be expressed as:
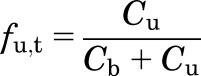 |
(10) |
Dividing by Cu and from eq. 9,
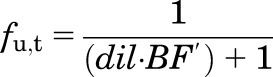 |
(11) |
and
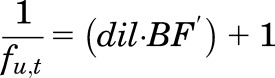 |
(12) |
It will be shown that eq. 12 for estimating in vitro binding in tissue homogenates is equivalent to the definition of in vivo partitioning of unbound drug into a tissue (KP,u).
For liver and lung, BF′ was measured across four tissue dilutions and extrapolated to an undiluted state to compute fu,t using eq. 12. Since binding in muscle was assessed only at a single dilution, fu,t was back-extrapolated to account for dilution as described previously (Kalvass et al., 2007):
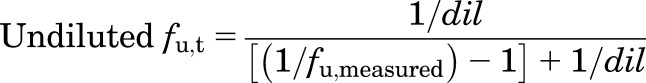 |
(13) |
Under linear, time-invariant conditions and assumption of the free hormone hypothesis (Mendel, 1989), the in vivo partitioning into a tissue KP is defined as follows:
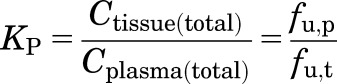 |
(14) |
KP,u represents the partitioning of free drug within a tissue:
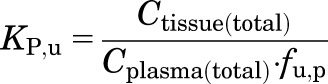 |
(15) |
Based on eqs. 14 and 15, KP,u can be simplified to:
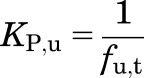 |
(16) |
which is equivalent to eq. 12. Finally, in vivo KP can be predicted as the product of fu,p and 1/fu,t measured in vitro (eq. 14).
Prediction of KP Using In Silico Methods
For comparison, KP was also calculated according to the tissue composition equations proposed by Poulin and Theil (2002) (referred to as method 1 hereafter) and corrected by Berezhkovskiy (2004) (method 2) and the Rodgers and Rowland method for neutral compounds (Rodgers and Rowland, 2006) (method 3) as programmed in the GastroPlus PBPK Simulator (version 9.6.2; Simulations Plus Inc., Lancaster, CA). These publications provide a detailed description of the approaches.
Results
Plasma Protein Binding
Protein binding studies of DEX and MPL were carried out using plasma from normal male and female Wistar rats. Plasma binding of MPL was assessed over total concentrations ranging from 163 to 22,230 ng/ml (∼0.4–60 μM). Figure 1 (left) demonstrates the lack of concentration dependence of MPL binding in plasma. Plasma binding of MPL in both sexes was essentially identical, with 60.5% ± 1.6% bound in males and 60.4% ± 2.3% bound in females. This percentage of bound MPL is comparable to a previously reported estimate in male rats of 63.1% ± 0.8%, obtained using equilibrium dialysis (Haughey and Jusko, 1991). The linearity of binding indicates a nonsaturable, low-affinity binding to plasma proteins. This finding is consistent with the plasma protein binding of MPL observed in humans and rabbits (Ebling et al., 1986). The percent bound in rats, however, was lower than in humans (77.6%) and rabbits (78.5%), indicating a species difference. The linear binding of these steroids allows calculation of only composite binding affinity constants NA ⋅ KA. Assuming that albumin is the only nonspecific binding protein (550 μM in female rats and 510 μM in male rats) (Rose and Klemcke, 2015; Li et al., 2017a), NA ⋅ KA ranges between 3.0 × 103 and 3.2 × 103 M. The estimated NA ⋅ KA for MPL is in close agreement to a previously reported value obtained in a hepatocyte incubation medium (inactivated horse serum; 3.15 × 103 M) (Morais and Wagner, 1985). This is also similar to values for prednisolone (∼2.0 × 103 M) in male rat plasma (Rocci et al., 1980). Plasma binding of DEX was assessed (Fig. 1, right) over total concentrations ranging from 11 to 12,712 ng/ml (∼0.03–32.4 μM) in male, estrus female, and proestrus female rat plasma. No differences were observed across the three groups. Plasma protein binding of DEX was concentration independent (linear) across the range tested. The extent of plasma binding of DEX was higher compared with MPL at 82.5%, which is comparable to a value reported previously for DEX in male rat plasma (84.7% ± 0.7%) (Peets et al., 1969).
Fig. 1.

Percent binding vs. total plasma steroid concentration for MPL (left) and DEX (right) in male (black squares), pooled female (green diamonds), estrus-phased female (orange diamonds), and proestrus-phased female (blue triangles) rat plasma. The inset depicts bound vs. free steroid concentrations of MPL in male and pooled female rat plasma.
In Vivo Tissue Distribution
Steady-state tissue-to-plasma distribution of MPL and DEX was measured in liver, muscle, and lung obtained from male Wistar rats. The KP values for each drug after steady-state infusion were calculated using eq. 1 for muscle and lung and using eq. 2 for liver. The KP values of DEX in these tissues were also assessed upon subcutaneous bolus injection (2.25 mg/kg) in male rats. The concentration-time profiles of MPL after a 50 mg/kg intramuscular bolus were measured in plasma and liver from both male and female rats, and the resultant KP values were calculated using eq. 3. The calculated KP values of MPL and DEX in rat liver, muscle, and lung after bolus injection and/or steady-state infusion are shown in Fig. 2. For MPL, KP was highest in liver (12.8 ± 1.2) and lowest in muscle (1.8 ± 0.4). The KP values for MPL obtained in male livers via intramuscular bolus and subcutaneous infusion and in female livers after intramuscular bolus were similar (13.4 ± 0.9 vs. 13.5 ± 1.4 vs. 11.4 ± 1.4) after correction for organ extraction using eq. 3. Similar to MPL, the in vivo KP of DEX was comparable using either subcutaneous bolus or infusion for each of the tissues. The KP was highest in liver (bolus vs. infusion: 5.5 ± 0.3 vs. 4.8 ± 0.5) but unlike MPL, DEX showed similar accumulation in both lung (bolus vs. infusion: 0.53 ± 0.02 vs. 0.43 ± 0.05) and muscle (bolus vs. infusion: 0.50 ± 0.03 vs. 0.49 ± 0.04).
Fig. 2.

MPL (top) and DEX (bottom) tissue-to-plasma partition coefficients (KP) obtained in vivo by bolus injection in male rats (black bars), infusion in male rats (gray bars), or bolus injection in female rats (dashed gray bars). Error bars represent 1 S.D. from the mean.
Stability of Drug in Tissue Homogenates
Stability of MPL and DEX at 37°C was assessed over time in plasma and in tissue homogenates. Depletion of either steroid was minimal throughout the course of the experiment in plasma, muscle, and lung homogenates (data not shown) and did not appear to present any problem in analyzing the binding data. Robust depletion occurred for MPL in male liver homogenates, with the rate and extent of substrate loss inversely proportional to the degree of homogenate dilution. The rate of MPL depletion in the male liver homogenates obeyed first-order loss kinetics for up to 10 minutes after incubation at 37°C (Fig. 3, left), followed by a plateau in concentrations beyond 15 minutes. Of interest, negligible depletion of MPL occurred in female rat liver homogenates, and concentrations remained stable for the duration of the experiment (Fig. 3, right). Preparation of liver homogenates using cryopreserved powdered liver only modestly decreased the extent of MPL loss at 37°C (Supplemental Fig. 1). No significant depletion of DEX occurred in either male or female rat liver homogenates (Supplemental Fig. 2).
Fig. 3.

Time course of in vitro stability of MPL in liver homogenates prepared at 3× (red), 4× (blue), 6× (orange), and 10× (green) dilutions from freshly harvested male (left) and female (right) livers. Symbols represent the mean ± S.D. (n = 3 per time point), the solid line depicts the first-order loss kinetics of MPL in male liver homogenates, and the dashed lines connect the data points.
In Vitro Binding in Tissue Homogenates
The effect of dilution of tissue homogenates on MPL binding was examined across 3- to 10-fold dilutions of liver and lung homogenates. As shown in Fig. 4 and Supplemental Fig. 3, the extent of MPL binding was directly proportional to the total protein concentration in tissue homogenates. The unbound tissue fractions (fu,t) for MPL in liver, muscle, and lung are presented in Table 1, as are the KP values of MPL calculated on the basis of in vitro binding studies. As shown in Fig. 5, DEX binding was proportional to the total protein concentration in tissue homogenates. The unbound tissue fractions (fu,t) for DEX in liver, muscle, and lung are presented in Table 2, as are the KP values of DEX calculated on the basis of in vitro binding studies.
Fig. 4.

Binding of MPL in homogenates prepared from male rat liver (left), female rat liver (middle), and male rat lung (right) at initial concentrations of 10 μg/ml. Symbols depict the mean ± S.D. of the binding factor (Cbound/Cfree) across four dilutions of tissue homogenate. The dashed line represents the best-fit line extrapolated to an undiluted state of tissue (dil = 1).
TABLE 1.
Comparison of tissue KP values for MPL determined by different methods Values are listed as the mean ± S.D.
| Tissue (Sex) | In Vivo KP | fu,t (in vitro) | In Vitro KP | Method 1 | Method 2 | Method 3 |
|---|---|---|---|---|---|---|
| Liver (male) | 13.4 ± 0.9a | 0.162 ± 0.01 | 2.5 ± 0.2 | 1.2 | 0.9 | 0.8 |
| 13.3 ± 1.4b | ||||||
| Liver (female) | 11.4 ± 1.4a | 0.066 ± 0.003 | 6.1 ± 0.4 | |||
| Lung (male) | 2.3 ± 0.0b | 0.192 ± 0.01 | 2.1 ± 0.1 | 1.3 | 1.0 | 0.9 |
| Muscle (male) | 1.8 ± 0.4b | 0.298 ± 0.05 | 1.4 ± 0.2 | 0.9 | 0.8 | 0.6 |
Intramuscular injection.
Subcutaneous infusion.
Fig. 5.

Binding of DEX in homogenates prepared from male rat liver (left), female rat liver (middle), and male rat lung (right) at initial concentrations of 1 μg/ml (gray) and/or 10 μg/ml (black). Symbols depict the mean ± S.D. of the binding factor (Cbound/Cfree) across four dilutions of tissue homogenate. The dashed line represents the best-fit line extrapolated to an undiluted state of tissue (dil = 1).
TABLE 2.
Comparison of tissue KP values for DEX determined by different methods Values are listed as the mean ± S.D.
| Tissue (Sex) | In Vivo KP | fu,t (in vitro) | In Vitro KP | Method 1 | Method 2 | Method 3 |
|---|---|---|---|---|---|---|
| Liver (male) | 5.5 ± 0.3a | 0.050 ± 0.001 | 3.6 ± 0.5c | 1.3 | 0.7 | 0.6 |
| 4.8 ± 0.5b | 0.068 ± 0.001 | 2.6 ± 0.4d | ||||
| Liver (female) | — | 0.059 ± 0.004 | 3.0 ± 0.5c | |||
| 0.070 ± 0.003 | 2.5 ± 0.4d | |||||
| Lung (male) | 0.53 ± 0.02a | 0.115 ± 0.006 | 1.5 ± 0.3d | 1.4 | 0.8 | 0.6 |
| 0.43 ± 0.05b | ||||||
| Muscle (male) | 0.50 ± 0.0a | 0.236 ± 0.03 | 0.74 ± 0.1d | 0.9 | 0.6 | 0.34 |
| 0.49 ± 0.04b |
Subcutaneous injection.
Subcutaneous infusion.
Low DEX concentration (1 μg/ml).
High DEX concentration (10 μg/ml).
The fu,t for MPL in liver was estimated as 0.162 ± 0.01 in males and 0.066 ± 0.003 in females, which correspond to in vitro–derived KP values of 2.5 ± 0.2 and 6.1 ± 0.4. Therefore, the homogenate studies, under the conditions described, significantly underpredicted in vivo KP of MPL in male liver and, to a lesser extent, in female liver. The fu,t values for MPL in lung and muscle from male rats were estimated to be 0.192 ± 0.01 and 0.298 ± 0.05, corresponding to in vitro–derived KP values of 2.1 ± 0.1 and 1.4 ± 0.2, in good agreement with their in vivo values. For DEX, the fu,t values in liver were similar in males and females at the two concentrations tested and were comparable to the value obtained for MPL in liver from female rats (∼0.06). The in vitro–derived KP values for DEX in liver were about 2-fold lower compared with in vivo estimates. The fu,t value for DEX in lung was 0.115 ± 0.006, and the in vivo KP was overpredicted by around 3-fold. In muscle, the fu,t of DEX was 0.236 ± 0.03, with an in vitro–derived KP 0.74 ± 0.1, in relatively good agreement with the in vivo value.
Prediction of Steroid KP in Rat Tissues
KP estimates obtained for MPL and DEX from in vivo and in vitro studies were compared with KP values in rat tissues computed using the tissue composition–based approaches proposed by method 1 (Poulin and Theil, 2002) and corrected by method 2 (Berezhkovskiy, 2004) and method 3 (Rodgers and Rowland, 2006) using the GastroPlus PBPK Simulator. The predicted KP values for MPL and DEX using these approaches are listed in Tables 1 and 2. All methods underpredicted KP values severely for both drugs in liver but were somewhat closer for lung and muscle.
Discussion
This study reports tissue binding data for DEX and MPL in the rat, obtained using different methods. A major goal of this work was to examine whether in vitro assessments of drug partitioning and binding using tissue homogenates (along with in vitro plasma binding information) could be used to reliably predict the in vivo tissue distribution of MPL and DEX in selected tissues. For this to be possible, several conditions must be met: 1) nonspecific binding to components of plasma and tissues dominates the tissue distribution process, 2) steroids distribute and bind relatively uniformly within each organ, 3) kinetics of drug uptake into tissues is not rate limiting (i.e., entry is rapid), 4) contributory binding processes are nonsaturating, and 5) active influx or efflux transport does not contribute significantly to distribution. Such assumptions are also operable with use of in silico methods of prediction of KP.
Technical Considerations
It has been argued that drug binding to (bovine) serum albumin depends on albumin concentration in a complex manner (Shen and Gibaldi, 1974). However, binding of both MPL and DEX to rat plasma and to tissue (liver and lung) homogenates was found to be linearly related to protein concentration (Figs. 4 and 5; Supplemental Fig. 3). This is consistent with other reports demonstrating that drug binding is independent of homogenate protein concentration (Lin et al., 1982; Kurz and Fichtl, 1983; Schuhmann et al., 1987). Some studies have employed equilibrium dialysis for the separation of bound and free drug in homogenate studies. This technique involves lengthy equilibration times during which decay of tissue (Pacifici and Viani, 1992) as well as volume shifts (Boudinot and Jusko, 1984) can occur. Ultrafiltration also presents certain limitations. First, nonspecific binding of drug to the filter and collection apparatus can confound measurements of binding. Second, filtration of greater than 20% of the sample volume can lead to shifts in binding equilibrium (Shen and Gibaldi, 1974). Preliminary recovery experiments confirmed negligible binding of both DEX and MPL to the apparatus. In addition, filtration conditions were optimized such that the filtrate volume was less than 15% of the initial sample volume. To prevent drug metabolism during equilibrium dialysis in diluted tissue homogenates, Lin et al. (1982) conducted binding studies of ethoxybenzamide at 4°C. Although not the case for ethoxybenzamide, the unbound fractions for several other drugs tend to decrease with an increase in temperature (Ballard, 1974). Therefore, the in vitro homogenate studies for DEX and MPL described in this report were performed using ultrafiltration at 37°C. Total drug was stable at this temperature and thus did not present a problem in analyzing binding data, except for MPL in male liver homogenates, where robust depletion occurred upon incubation. Thus, the time course of total MPL concentrations was followed in liver homogenates during the experiment and used to correct for the drug loss. Despite this apparent correction, MPL binding in male liver homogenates underpredicted in vivo estimates by approximately 4-fold. In vitro binding of MPL was assessed at an initial concentration of 10 μg/ml. Low sensitivity of the HPLC assay in tissues (50 ng/g) coupled with small volumes of filtrate (25–60 μl) precluded the examination of concentration-dependent MPL tissue binding at lower concentrations. However, a single validation experiment (data not shown) performed at 1 μg/ml using 4× diluted liver homogenates with filtered volumes pooled from multiple devices yielded a bound/free ratio comparable to that obtained at 10 μg/ml (0.9 vs. 1.1), suggestive of concentration-independent binding within this range. The degree of MPL loss in homogenates was similar at both concentrations. A salient feature of this study was the investigation of in vitro binding of two steroids (DEX and MPL) in three distinct tissues (liver, lung, and skeletal muscle), for which the in vivo KP values were found to vary across a wide range (0.5–13).
The in vitro homogenate studies examined the tissue binding and stability of MPL in its free alcohol form; therefore, assessments of MPL binding are not confounded by the prodrug conversion. Our in vivo assessments measured total MPL concentrations in rat plasma and tissues after intramuscular and subcutaneous dosing of its water-soluble succinate prodrug, MPS. Since MPS undergoes incomplete conversion (10%–20%) to MPL in rats (Kong and Jusko, 1991), prodrug instability ex vivo can lead to overestimation of MPL concentrations at early time points. Measures taken to minimize possible ex vivo hydrolysis of MPS were as follows: 1) use of EDTA as an anticoagulant when collecting blood, 2) rapid centrifugation of blood after collection at 4°C, 3) storage of plasma and muscle samples at −20°C and −80°C, 4) avoidance of repeated freeze-thaw cycles, and 5) use of a cold temperature (4°C) prior to drug extraction into methylene chloride for measurement by HPLC. After a bolus dose of intramuscular MPS in male rats, a high concentration of MPL at the local injection site (relative to plasma) was measurable within minutes after dosing (Hazra et al., 2007), indicating rapid and significant MPS conversion occurring at the tissue site prior to entry into the circulation. Kong and Jusko (1991) demonstrated using perfused rat livers that only 8%–20% of MPS formed MPL. These findings collectively suggest a minor impact of MPS on the accumulation of MPL in tissues. Pronounced interspecies differences exist in the interconversion of MPS to MPL. The availability of MPL from MPS is incomplete in rats, monkeys, and dogs (10%–20%), whereas it is almost complete (80%–90%) in rabbits and humans (Kong and Jusko, 1991). Multiple studies of MPS in humans indicate a very rapid and extensive conversion of prodrug to active MPL (Derendorf et al., 1985; Al-Habet and Rogers, 1989), which can confound MPL tissue uptake and concentrations at early time points.
Binding to Homogenates Compared with In Vivo Data
There was reasonable agreement between homogenate binding and in vivo distribution in the case of skeletal muscle and lung for both steroids. This permits some speculation concerning the mode of uptake of the drugs into rat tissue. The fact that the extent of in vivo distribution of both steroids in both tissues could be reasonably reproduced in homogenates under the conditions described suggests an absence of active mechanisms at the concentration tested. These observations may be valid for both steroids at lower drug concentrations, since linear uptake of prednisolone was documented in rabbit muscle slices across a wide range of concentrations (Khalafallah and Jusko, 1984a). Therefore, KP values obtained using homogenates can be used to inform this parameter for the development of PBPK models in the case of both steroids.
Corticosteroid Binding in the Liver
Total concentrations of DEX were relatively stable throughout the binding experiments in male and female liver homogenates, and in vitro–derived KP values for DEX in liver from both sexes were in reasonable agreement with in vivo values. On the other hand, in vitro experiments underpredicted the KP of MPL in both male (4-fold) and female (2-fold) liver homogenates compared with in vivo estimates. Robust in vitro loss of MPL in male homogenates was posited as a reason for the underprediction. It was interesting to find that, in the absence of any discernable loss of MPL in female liver homogenates, there was a 2-fold improvement in, but nonetheless a 2-fold underprediction of, the in vivo KP. Based on these observations, it is possible that in vitro metabolism only partially explains the disconnect with in vivo binding data. Lackner et al. (1998) provided evidence for the presence of a glucocorticoid-responsive site in highly purified rat liver plasma membranes (free of transcortin and glucocorticoid receptors), which was shown to mediate high-affinity active uptake of endogenous and some exogenous glucocorticoids. Competitive binding studies indicated that prednisolone, a steroid differing by only a –methyl group at the C6 position compared with MPL, showed approximately 3-fold improved specificity compared with corticosterone, whereas other highly potent steroids such as DEX, betamethasone, and cortivazol, all of them bearing an OH or CH3 group at position C16, exhibited low specificity (Lackner et al., 1998). These observations seem to offer one rational basis for the totality of our findings in regard to in vitro–in vivo prediction of DEX and MPL binding in liver and other tissues using homogenates, a system in which such active import processes would not be operative. Nonlinear tissue uptake of prednisolone observed in vivo and in rabbit liver slices was attributed to either saturable glucocorticoid receptor or transcortin binding (Khalafallah and Jusko, 1984a). The latter mechanism is not applicable because DEX and MPL do not bind to transcortin. The influence of specific high-affinity, low-capacity steroid binding to tissue glucocorticoid receptors is possible in vivo. These receptors are ubiquitous in mammalian tissues and most abundant in liver (Ballard et al., 1974). The argument for hepatic steroid uptake due to receptor binding is further strengthened based on findings of nonlinear prednisolone binding in rabbit liver slices but linear uptake in heart, skeletal muscle, and fat (Khalafallah and Jusko, 1984a). Assessment of factors such as receptor binding and active transport on hepatic uptake will require more focused experimental testing using cytosolic fractions and very low drug concentrations. The three tissue composition–based methods severely underpredicted KP values for both steroids in liver, indicating once again that additional mechanisms of hepatic steroid uptake are likely.
In conclusion, the KP values for MPL and DEX determined from in vitro binding data and calculated from kinetic parameters obtained after administering the drug by two different routes were in reasonable agreement for lung and muscle. This observation suggests that the in vitro binding data obtained can, at least in these two tissues, be used to predict in vivo distribution. Tissue metabolism, saturable tissue binding, and active uptake are possible factors that can complicate assessments of hepatic transport of steroids in vivo when using tissue homogenates. These findings are in general agreement with the statement by Mendel (1989) that, for some steroids, the free hormone hypothesis “is likely to be valid with respect to some tissues, but not with respect to others (in particular, the liver).”
Abbreviations
- DEX
dexamethasone
- HPLC
high-performance liquid chromatography
- MPL
methylprednisolone
- MPS
methylprednisolone sodium succinate
- PBPK
physiologically based pharmacokinetic
Authorship Contributions
Participated in research design: Ayyar, DuBois, Jusko.
Conducted experiments: Ayyar, Song, DuBois.
Performed data analysis: Ayyar, Song, Jusko.
Wrote or contributed to the writing of the manuscript: Ayyar, Song, DuBois, Almon, Jusko.
Footnotes
This work was supported by the National Institutes of Health National Institute of General Medical Sciences [Grants GM24211 and GM131800].
 This article has supplemental material available at jpet.aspetjournals.org.
This article has supplemental material available at jpet.aspetjournals.org.
References
- Al-Habet SM, Rogers HJ. (1989) Methylprednisolone pharmacokinetics after intravenous and oral administration. Br J Clin Pharmacol 27:285–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayyar VS, DuBois DC, Almon RR, Jusko WJ. (2017) Mechanistic multi-tissue modeling of glucocorticoid-induced leucine zipper regulation: integrating circadian gene expression with receptor-mediated corticosteroid pharmacodynamics. J Pharmacol Exp Ther 363:45–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayyar VS, DuBois DC, Almon RR, Jusko WJ. (2019a) Modeling corticosteroid pharmacokinetics and pharmacodynamics, part II: sex differences in methylprednisolone pharmacokinetics and corticosterone suppression. J Pharmacol Exp Ther 370:327–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayyar VS, DuBois DC, Almon RR, Jusko WJ. (2019b) Modeling corticosteroid pharmacokinetics and pharmacodynamics, part III: estrous cycle and estrogen receptor–dependent antagonism of glucocorticoid-induced leucine zipper (GILZ) enhancement by corticosteroids. J Pharmacol Exp Ther 370:337–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballard BE. (1974) Pharmacokinetics and temperature. J Pharm Sci 63:1345–1358. [DOI] [PubMed] [Google Scholar]
- Ballard PL, Baxter JD, Higgins SJ, Rousseau GG, Tomkins GM. (1974) General presence of glucocorticoid receptors in mammalian tissues. Endocrinology 94:998–1002. [DOI] [PubMed] [Google Scholar]
- Berezhkovskiy LM. (2004) Volume of distribution at steady state for a linear pharmacokinetic system with peripheral elimination. J Pharm Sci 93:1628–1640. [DOI] [PubMed] [Google Scholar]
- Berry LM, Roberts J, Be X, Zhao Z, Lin MH. (2010) Prediction of V(ss) from in vitro tissue-binding studies. Drug Metab Dispos 38:115–121. [DOI] [PubMed] [Google Scholar]
- Boudinot FD, Jusko WJ. (1984) Fluid shifts and other factors affecting plasma protein binding of prednisolone by equilibrium dialysis. J Pharm Sci 73:774–780. [DOI] [PubMed] [Google Scholar]
- Craig WA, Welling PG. (1977) Protein binding of antimicrobials: clinical pharmacokinetic and therapeutic implications. Clin Pharmacokinet 2:252–268. [DOI] [PubMed] [Google Scholar]
- Crowe A, Tan AM. (2012) Oral and inhaled corticosteroids: differences in P-glycoprotein (ABCB1) mediated efflux. Toxicol Appl Pharmacol 260:294–302. [DOI] [PubMed] [Google Scholar]
- Derendorf H, Möllmann H, Rohdewald P, Rehder J, Schmidt EW. (1985) Kinetics of methylprednisolone and its hemisuccinate ester. Clin Pharmacol Ther 37:502–507. [DOI] [PubMed] [Google Scholar]
- Dunn TE, Ludwig EA, Slaughter RL, Camara DS, Jusko WJ. (1991) Pharmacokinetics and pharmacodynamics of methylprednisolone in obesity. Clin Pharmacol Ther 49:536–549. [DOI] [PubMed] [Google Scholar]
- Ebling WF, Milsap RL, Szefler SJ, Jusko WJ. (1986) 6 alpha-methylprednisolone and 6 alpha-methylprednisone plasma protein binding in humans and rabbits. J Pharm Sci 75:760–763. [DOI] [PubMed] [Google Scholar]
- Fichtl B, Nieciecki A, Walter K. (1991) Tissue binding versus plasma binding of drugs: general principles and pharmacokinetic consequences, in Advances in Drug Research (Testa B. ed) 20, pp 117–166, Academic Press, London. [Google Scholar]
- Gillette JR. (1971) Factors affecting drug metabolism. Ann N Y Acad Sci 179:43–66. [DOI] [PubMed] [Google Scholar]
- Graham H, Walker M, Jones O, Yates J, Galetin A, Aarons L. (2012) Comparison of in-vivo and in-silico methods used for prediction of tissue: plasma partition coefficients in rat. J Pharm Pharmacol 64:383–396. [DOI] [PubMed] [Google Scholar]
- Haughey DB, Jusko WJ. (1988) Analysis of methylprednisolone, methylprednisone and corticosterone for assessment of methylprednisolone disposition in the rat. J Chromatogr A 430:241–248. [DOI] [PubMed] [Google Scholar]
- Haughey DB, Jusko WJ. (1991) Effect of ketoconazole on methylprednisolone pharmacokinetics and receptor/gene-mediated pharmacodynamics. J Pharmacol Exp Ther 259:826–832. [PubMed] [Google Scholar]
- Hazra A, Pyszczynski N, DuBois DC, Almon RR, Jusko WJ. (2007) Pharmacokinetics of methylprednisolone after intravenous and intramuscular administration in rats. Biopharm Drug Dispos 28:263–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hochhaus G, Barth J, al-Fayoumi S, Suarez S, Derendorf H, Hochhaus R, Möllmann H. (2001) Pharmacokinetics and pharmacodynamics of dexamethasone sodium-m-sulfobenzoate (DS) after intravenous and intramuscular administration: a comparison with dexamethasone phosphate (DP). J Clin Pharmacol 41:425–434. [DOI] [PubMed] [Google Scholar]
- Kalvass JC, Maurer TS, Pollack GM. (2007) Use of plasma and brain unbound fractions to assess the extent of brain distribution of 34 drugs: comparison of unbound concentration ratios to in vivo p-glycoprotein efflux ratios. Drug Metab Dispos 35:660–666. [DOI] [PubMed] [Google Scholar]
- Kawai R, Mathew D, Tanaka C, Rowland M. (1998) Physiologically based pharmacokinetics of cyclosporine A: extension to tissue distribution kinetics in rats and scale-up to human. J Pharmacol Exp Ther 287:457–468. [PubMed] [Google Scholar]
- Khalafallah N, Jusko WJ. (1984a) Determination and prediction of tissue binding of prednisolone in the rabbit. J Pharm Sci 73:362–366. [DOI] [PubMed] [Google Scholar]
- Khalafallah N, Jusko WJ. (1984b) Tissue distribution of prednisolone in the rabbit. J Pharmacol Exp Ther 229:719–725. [PubMed] [Google Scholar]
- Kong AN, Jusko WJ. (1991) Disposition of methylprednisolone and its sodium succinate prodrug in vivo and in perfused liver of rats: nonlinear and sequential first-pass elimination. J Pharm Sci 80:409–415. [DOI] [PubMed] [Google Scholar]
- Kurz H, Fichtl B. (1983) Binding of drugs to tissues. Drug Metab Rev 14:467–510. [DOI] [PubMed] [Google Scholar]
- Lackner C, Daufeldt S, Wildt L, Alléra A. (1998) Glucocorticoid-recognizing and -effector sites in rat liver plasma membrane. Kinetics of corticosterone uptake by isolated membrane vesicles. III. Specificity and stereospecificity. J Steroid Biochem Mol Biol 64:69–82. [DOI] [PubMed] [Google Scholar]
- Levy G. (1994) Pharmacologic target-mediated drug disposition. Clin Pharmacol Ther 56:248–252. [DOI] [PubMed] [Google Scholar]
- Li X, DuBois DC, Almon RR, Jusko WJ. (2017a) Effect of disease-related changes in plasma albumin on the pharmacokinetics of naproxen in male and female arthritic rats. Drug Metab Dispos 45:476–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, DuBois DC, Song D, Almon RR, Jusko WJ, Chen X. (2017b) Modeling combined immunosuppressive and anti-inflammatory effects of dexamethasone and naproxen in rats predicts the steroid-sparing potential of naproxen. Drug Metab Dispos 45:834–845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin JH, Sugiyama Y, Awazu S, Hanano M. (1982) In vitro and in vivo evaluation of the tissue-to-blood partition coefficient for physiological pharmacokinetic models. J Pharmacokinet Biopharm 10:637–647. [DOI] [PubMed] [Google Scholar]
- Lowry OH, Rosebrough NJ, Farr AL, Randall RJ. (1951) Protein measurement with the Folin phenol reagent. J Biol Chem 193:265–275. [PubMed] [Google Scholar]
- Mendel CM. (1989) The free hormone hypothesis: a physiologically based mathematical model. Endocr Rev 10:232–274. [DOI] [PubMed] [Google Scholar]
- Morais JA, Wagner JG. (1985) Steroid metabolism in isolated rat hepatocytes. Eur J Drug Metab Pharmacokinet 10:295–307. [DOI] [PubMed] [Google Scholar]
- Pacifici GM, Viani A. (1992) Methods of determining plasma and tissue binding of drugs. Pharmacokinetic consequences. Clin Pharmacokinet 23:449–468. [DOI] [PubMed] [Google Scholar]
- Peets EA, Staub M, Symchowicz S. (1969) Plasma binding of betamethasone-3H, dexamethasone-3H, and cortisol-14C--a comparative study. Biochem Pharmacol 18:1655–1663. [DOI] [PubMed] [Google Scholar]
- Poulin P, Theil FP. (2000) A priori prediction of tissue:plasma partition coefficients of drugs to facilitate the use of physiologically-based pharmacokinetic models in drug discovery. J Pharm Sci 89:16–35. [DOI] [PubMed] [Google Scholar]
- Poulin P, Theil FP. (2002) Prediction of pharmacokinetics prior to in vivo studies. II. Generic physiologically based pharmacokinetic models of drug disposition. J Pharm Sci 91:1358–1370. [DOI] [PubMed] [Google Scholar]
- Rocci ML, Jr, Johnson NF, Jusko WJ. (1980) Serum protein binding of prednisolone in four species. J Pharm Sci 69:977–978. [DOI] [PubMed] [Google Scholar]
- Rodgers T, Leahy D, Rowland M. (2005) Physiologically based pharmacokinetic modeling 1: predicting the tissue distribution of moderate-to-strong bases. J Pharm Sci 94:1259–1276. [DOI] [PubMed] [Google Scholar]
- Rodgers T, Rowland M. (2006) Physiologically based pharmacokinetic modelling 2: predicting the tissue distribution of acids, very weak bases, neutrals and zwitterions. J Pharm Sci 95:1238–1257. [DOI] [PubMed] [Google Scholar]
- Rose R, Klemcke HG. (2015) Relationship between plasma albumin concentration and plasma volume in 5 inbred rat strains. J Am Assoc Lab Anim Sci 54:459–464. [PMC free article] [PubMed] [Google Scholar]
- Samtani MN, Jusko WJ. (2005) Comparison of dexamethasone pharmacokinetics in female rats after intravenous and intramuscular administration. Biopharm Drug Dispos 26:85–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schinkel AH, Wagenaar E, van Deemter L, Mol CA, Borst P. (1995) Absence of the mdr1a P-glycoprotein in mice affects tissue distribution and pharmacokinetics of dexamethasone, digoxin, and cyclosporin A. J Clin Invest 96:1698–1705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuhmann G, Fichtl B, Kurz H. (1987) Prediction of drug distribution in vivo on the basis of in vitro binding data. Biopharm Drug Dispos 8:73–86. [DOI] [PubMed] [Google Scholar]
- Shen D, Gibaldi M. (1974) Critical evaluation of use of effective protein fractions in developing pharmacokinetic models for drug distribution. J Pharm Sci 63:1698–1703. [DOI] [PubMed] [Google Scholar]