

Abstract
Our previous report examined the pharmacokinetics (PK) of methylprednisolone (MPL) and adrenal suppression after a 50 mg/kg IM bolus in male and female rats, and we described in detail the development of a minimal physiologically based pharmacokinetic/pharmacodynamic (mPBPK/PD) model. In continuation of such assessments, we investigated sex differences in genomic MPL responses (PD). Message expression of the glucocorticoid-induced leucine zipper (GILZ) was chosen as a multitissue biomarker of glucocorticoid receptor (GR)-mediated drug response. Potential time-dependent interplay between sex hormone and glucocorticoid signaling in vivo was assessed by comparing the enhancement of GILZ by MPL in the uterus [high estrogen receptor (ER) density] and in liver (lower ER density) from male and female rats dosed within the proestrus (high estradiol/progesterone) and estrus (low estradiol/progesterone) phases of the rodent estrous cycle. An expanded-systems PD model of MPL considering circadian rhythms, multireceptor (ER and GR) control, and estrous variations delineated the determinants controlling receptor/gene-mediated steroid responses. Hepatic GILZ response was ∼3-fold greater in females, regardless of estrous stage, compared with males, driven predominantly by increased MPL exposure in females and a negligible influence of estrogen interaction. In contrast, GILZ response in the uterus during proestrus in females was 60% of that observed in estrus-phased females, despite no PK or receptor differences, providing in vivo support to the hypothesis of estrogen-mediated antagonism of glucocorticoid signaling. The developed model offers a mechanistic platform to assess the determinants of sex and tissue specificity in corticosteroid actions and, in turn, reveals a unique PD drug-hormone interaction occurring in vivo.
Significance Statement
Mechanisms relating to sex-based pharmacodynamic variability in genomic responses to corticosteroids have been unclear. Using combined experimental and systems pharmacology modeling approaches, sex differences in both pharmacokinetic and pharmacodynamic mechanisms controlling the enhancement of a sensitive corticosteroid-regulated biomarker, the glucocorticoid-induced leucine zipper (GILZ), were clarified in vivo. The multiscale minimal physiologically based pharmacokinetics/pharmacodynamic model successfully captured the experimental observations and quantitatively discerned the roles of the rodent estrous cycle (hormonal variation) and tissue specificity in mediating the antagonistic coregulation of GILZ gene synthesis. These findings collectively support the hypothesis that estrogens antagonize pharmacodynamic signaling of genomic corticosteroid actions in vivo in a time- and estrogen receptor-dependent manner.
Introduction
The corticosteroids (CSs) exert diverse biochemical effects in numerous tissues. Most adverse and therapeutic actions of CS occur in tissues upon drug binding the glucocorticoid receptors (GR). The CS can cause effects that are rapid in onset, such as cell trafficking and adrenal suppression (Yao et al., 2008). In contrast, pharmacogenomic (PG) regulation by the drug-receptor complex occurs in a delayed manner (Jusko, 1995) owing to a series of intracellular transduction steps, including gene regulation and consequent mRNA and protein synthesis. Several generations of increasingly mechanistic pharmacokinetic (PK) and pharmacodynamic (PD) models of gene-mediated steroid actions have been developed (Nichols et al., 1989; Xu et al., 1995; Sun et al., 1998; Ramakrishnan et al., 2002; Mager et al., 2003; Hazra et al., 2007b; Ayyar et al., 2018). These models have demonstrated that the major determinants of steroid response include 1) disposition characteristics of the steroid, 2) relative receptor affinity of the steroid, 3) availability of cytosolic receptors, and 4) system control of the biomarker(s) (mRNA and protein) that mediate endpoint responses.
Our early studies used the adrenalectomized (ADX) male rat model to examine CS actions. The use of ADX animals obviated complicating factors such as the influence of endogenous glucocorticoids. This experimental paradigm was subsequently evolved to examine the PK/PD effects of MPL in intact (non-ADX) male rats, with consideration of endogenous steroid effects and circadian rhythms (Hazra et al., 2007b). These assessments were, however, confined to males. Sex is a relevant factor influencing the PK/PD of drugs. Despite this recognition, to date, few preclinical PK/PD studies have included sex as a variable. The overarching goal of this work was to expand our mechanistic assessments of MPL PK/PD from intact male rats to female rats with consideration of estrous cycle variations.
Our companion report (Ayyar et al., 2019a) compared the plasma and tissue PK of MPL as well as the time course of corticosterone (CST) suppression by MPL after a 50 mg/kg IM bolus in male and female rats. Prominent sex differences were observed in PK, with higher drug exposures in females, regardless of estrous stage. No sex difference in plasma protein binding of MPL was observed (Ayyar et al., 2019b). Robust suppression of plasma CST occurred in both sexes upon dosing. Sex- and estrous-dependent differences in basal CST rhythms (Atkinson and Waddell, 1997) were considered.
Sex differences in the receptor-/gene-mediated mechanisms of MPL have not been investigated thus far. The glucocorticoid-induced leucine zipper (GILZ) is a sensitive CS-regulated gene that has emerged as an important mediator of anti-inflammatory CS actions (Ayroldi and Riccardi, 2009). The characterization of GILZ as a multitissue biomarker for CS PK/PD/PG effects has been reported in male rats (Ayyar et al., 2015, 2017). Mechanistic experiments in a human uterine epithelial cell line indicated that enhancement of GILZ mRNA and protein by dexamethasone (DEX) was potently antagonized in the presence of 1–10 pM of estradiol (Whirledge and Cidlowski, 2013; Whirledge et al., 2013). The siRNA-mediated knockdown of the estrogen receptor (ER) reversed the antagonism, suggestive of an ER-dependent mechanism. Chromatin immunoprecipitation analysis revealed that GR and ER recruitment to a specific glucocorticoid response element (GRE) site in the GILZ promoter region was decreased in the presence of both DEX and estradiol compared with their respective ligand alone (Whirledge and Cidlowski, 2013), indicative of a competitive antagonistic interaction occurring at the molecular level.
GILZ expression was chosen as a biomarker in our studies to investigate sex-, tissue-, and estrous cycle–dependent variability in the receptor-/gene-mediated PD activity of MPL. The time course of GILZ enhancement after MPL dosing was quantified in liver, a tissue with high GR and relatively low ER, and in uterus, a tissue with lower GR and a high ER content (Izawa et al., 1984; Kuiper et al., 1997; Plowchalk and Teeguarden, 2002). Organs were harvested from male rats and female rats dosed within two distinct phases of their reproductive cycle: when systemic concentrations of estradiol were either low (estrus, E) or elevated (proestrus, PE) (Smith et al., 1975). It was hypothesized that elevated estradiol production during PE in females would antagonize MPL-enhanced GILZ mRNA expression in tissues with ER. This final report of a three-part series focused on 1) characterizing potential sex- and tissue-dependent differences in GILZ mRNA dynamics after MPL and 2) developing a minimal physiologically based pharmacokinetic/PD/PG (mPBPK/PD/PG) systems model of MPL considering circadian rhythms, multireceptor control, and the rodent estrous cycle to delineate in a quantitative manner the multifactorial control of genomic steroid responses in vivo.
Materials and Methods
Animal study designs, care of animals, and procedures for collection of tissue samples are described in Part II (Ayyar et al., 2019a).
Plasma Estradiol Determination
The plasma concentrations of estradiol in normal (i.e., nondosed) E- and PE-phased female rats were quantified using a competitive enzyme-linked immunosorbent assay kit (LifeSpan BioSciences, Seattle, WA). Rat plasma samples were used undiluted. All other procedures followed the manufacturer’s protocol. Four to five rats from each animal group were used, and all samples were run in duplicate. The range of the standard curve was 3–300 pg/ml, and a four-parameter logistic model was applied to fit the standard curve.
Messenger RNA Quantification
RNA Preparation.
Frozen liver and uterus samples were ground into a fine powder under liquid nitrogen. Powdered samples were weighed, added to prechilled TRI reagent (Invitrogen, Carlsbad, CA), and homogenized. Total RNA extractions were carried out using TRI reagent and further purified by passage through RNeasy minicolumns (Qiagen, Valencia, CA) according to the manufacturer’s protocols for RNA cleanup. The RNA concentrations were quantified spectrophotometrically (NanoDrop 2000c; Thermo Fisher Scientific, Waltham, MA), and purity and integrity were assessed by agarose-formaldehyde gel electrophoresis. All samples exhibited 260/280 absorbance ratios of approximately 2.0, and all showed intact ribosomal 28S and 18S RNA bands in an approximate ratio of 2:1 as visualized by ethidium bromide staining. Final total RNA preparations were diluted to desired concentrations (∼25 ng/μl) in nuclease-free water (Ambion, Austin, TX) and stored in nuclease-free tubes at −80°C until use.
Quantification of mRNA Expression.
The GILZ- and GR-specific quantitative real-time polymerase chain reaction (qRT-PCR) assays were developed and validated according to the Minimum Information for Publication of Quantitative Real-Time PCR Experiments (MIQE) guidelines (Bustin et al., 2009). Both qRT-PCR assays involved the use of in vitro transcribed cRNA standards, gene-specific TaqMan-based probes, and a single-step assay. An extensive description of the subcloning and construction of the in vitro transcribed cRNA standards has been reported previously (Ayyar et al., 2015). Primer and probe sequences were designed and custom-synthesized (Biosearch Technologies, Novato, CA). The qRT-PCR was performed using a Brilliant qRT-PCR Core reagent kit, 1-Step (Stratagene, La Jolla, CA), in a Stratagene MX3005P thermocycler according to the manufacturer’s instructions. Primer and probe sequences were:
GR, forward primer 5′-AACATGTTAGGTGGGCGTCAA-3′; GR, reverse primer 5′-GGTGTAAGTTTCTCAAGCCT AGTATCG-3′; GR, FAM-labeled probe, 5′ TGATTGCAGCAGTGAAATGGGCAAAG-3′; GILZ, forward primer 5′- GGAGGTCCTAAAGGAGCAGATTC- 3′; GILZ, reverse primer 5′- GCGTCTTCAGGAGGG TATTCTC- 3′; and GILZ, FAM-labeled probe, 5′-TGAGCTGGTTGAGAAGAACTCGCA- 3′. The concentrations of the primers, probes, and magnesium chloride were optimized and the reaction conditions reported (Ayyar et al., 2015). A volume of 2.5 μl of total RNA (∼62.5 ng/μl) was used for each sample. Standards were run in duplicate and samples in triplicate; intraassay and interassay CV were less than 20%. Additional reverse-transcriptase minus controls were included to ensure lack of DNA contamination in each sample.
Additional Data Source
The time course of total plasma concentrations of 17β-estradiol across the 4-day rat estrous cycle under 12-hour daily light/dark cycles was obtained from Smith et al. (1975). The data in the report were obtained from female Sprague Dawley Rats weighing between 200 and 250 g, which was comparable to the body weights of the female Wistar rats used in our studies. Mean plasma concentrations of estradiol were extracted from published graphs in the report by computer digitization (WebPlotDigitizer, version 4.1, https://automeris.io/WebPlotDigitizer).
Systems PK/PD Modeling and Data Analysis
Pharmacokinetics.
The concentration-time profiles of MPL in rats as modeled in Part II was incorporated into the current systems model. The mPBPK model equations and parameters describing the plasma and tissue concentrations of MPL in male and female rats have been described in Ayyar et al., 2019a.
Mechanistic Basis for Pharmacodynamics.
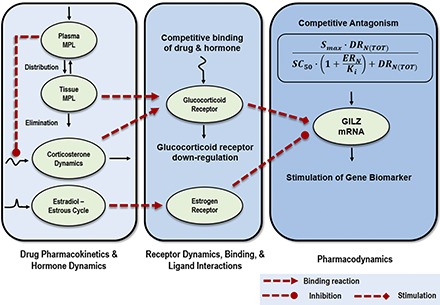
The series of steps controlling the signal transduction events for genomic CS actions, along with the mechanism for the genome-level interaction of estrogens and CS, are depicted in Fig. 1. Free (unbound) plasma concentrations of glucocorticoid hormones and synthetic CS rapidly distribute into intracellular spaces within tissues. Furthermore, systemic administration of CS results in an inhibition of endogenous CST secretion via a negative feedback loop on the hypothalamic-pituitary-adrenal axis, resulting in time-dependent decreases in plasma CST concentrations. The unbound fractions of endogenous and exogenous steroids in tissues interact in a competitive manner with the cytosolic GR based on their relative receptor affinity. Binding of the steroids to its receptor produces an activated complex, which rapidly translocates into the nucleus and binds specific GRE sites on target DNA. This process transcriptionally enhances or downregulates the turnover rates of numerous target genes within a tissue, including GILZ. Circadian-driven production of endogenous CST and GR mRNA in rats produces nonstationarities in the receptor-mediated control of tissue gene expression (Hazra et al., 2007b; Almon et al., 2008). In addition to driving a rapid reduction in free cytosolic receptors by the translocation process, elevated cellular concentrations of CS induce a homologous downregulation of their own receptors via decreased transcription and consequent reductions in mRNA synthesis and free GR densities in the cytosol (Oakley and Cidlowski, 1993). After exerting their transcriptional effects, the steroid-receptor complexes in the nucleus may dissociate from GREs and return to the cytosol. Therefore, the total pool of free cytosolic GR constitutes a composite of recycled and de novo synthesized GR molecules.
Fig. 1.

Genomic mechanism of corticosteroid regulation of GILZ gene expression in tissues and the influence of estrogens on glucocorticoid signaling. FKBP, FK506 binding protein; GR, glucocorticoid receptor; hsp 70/90, heat shock protein 70/90; nGRE, negative glucocorticoid response element; RNAP, RNA polymerase.
Unbound plasma concentrations of E2 in female rats can rapidly equilibrate within tissues (Puig-Duran et al., 1979) and interact with cytoplasmic ER. Activated ER complexes subsequently translocate into the nucleus and modulate its own target genes (Stossi et al., 2004) in a manner similar to that of CS. Cooperative binding of two different steroid hormone receptors at single-target enhancer elements has been demonstrated (Tsai et al., 1989). In the presence of both CS and estrogens, GR and ER complexes in the nucleus can interact within the promoter regions of several CS-regulated target genes, including GILZ (Tynan et al., 2004; Whirledge et al., 2013). This mechanism for CS involves the recruitment of both GR and ER to a specific GRE site, for example, GRE site 1919–1794 in the promoter region of GILZ (Whirledge and Cidlowski, 2013). This interaction results in a competitive antagonism of CS gene regulation by E2.
Mathematical Model.
Figure 2 presents the scheme for the entire mPBPK/PD/PG model for MPL actions in male and female animals. The equations and parameters for modeling the dynamics of CST suppression are described in Part II and used unchanged in the expanded systems model. In the unperturbed system (i.e., no MPL exposure), the model operates under homeostatic (i.e., steady-state) conditions driven by circadian variations in endogenous CST concentrations.
Fig. 2.

Schematic of the mPBPK/PD/PG systems model for corticosteroid actions in male and female rats. Parameters and symbols are defined in the text and tables. Lines with arrows indicate blood flows, binding interactions, conversion of species, or turnover of responses. Dashed lines ending in closed boxes indicate inhibition; dashed lines with open boxes depict a stimulation of turnover exerted by the connected factors.
Model Assumptions.
Certain assumptions were made during the development of this mathematical model: 1) The endogenous time course of E2 across the 4-day estrous cycle in female rats is unaltered upon bolus dosing of MPL; 2) ligand-induced downregulation of the ER in tissues is negligible at physiologic concentrations of E2 and equilibrium binding conditions are operable; 3) the influence of other sex hormones (e.g., progesterone), which are also elevated during PE, is negligible (if existent) compared with E2; 4) only the free fraction of E2 (Montano et al., 1995; Plowchalk and Teeguarden, 2002) can interact with ER in tissues; and 5) the kinetic rate constants controlling GR mRNA and protein dynamics are independent of sex and tissue type.
Mathematical Description of Model
Glucocorticoid Receptor Dynamics.
The fifth-generation model of CS proposed by Ramakrishnan et al. (2002) and modified by Hazra et al. (2007b) was adapted to incorporate the binding of endogenous CST to free cytosolic receptors in both sexes (under circadian homeostasis and in the presence of MPL). Thedifferential equations and initial conditions (IC) for GR dynamics in the tissues are:
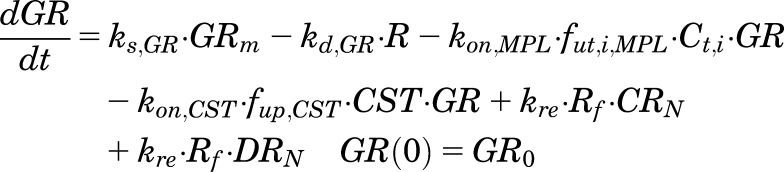 |
(1) |
| (2) |
| (3) |
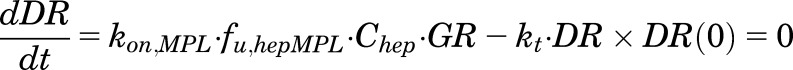 |
(4) |
| (5) |
| (6) |
where GR is the free cytosolic receptor; CR and DR are the cytosolic receptors bound to CST and MPL; CRN and DRN are the nuclear translocated complex concentrations; and NRN_TOT is the total concentration of steroid-receptor complex in the nucleus. The first-order rate constants include receptor synthesis (ks,GR) and degradation (kd,GR), translocation of the receptor complexes into the nucleus (kt), the overall turnover of DRN return receptors to cytosol (kre), as well as the second-order rate constants of hormone- and drug-receptor association (kon,CST and kon,MPL). Part of NRN_TOT may recycle back to the cytosol controlled by the rate constant Rf ∙ kre, and the rest is degraded with a rate constant (1 − Rf) ∙ kre. The total concentrations of MPL in a specific tissue compartment i and CST in plasma are given by Ct,i and CST. The fut,i,MPL and fup,CST are the unbound fractions of MPL in tissue i and CST in plasma. The fut,i,MPL is calculated as the ratio of the plasma unbound fraction of MPL (fup,MPL) and the tissue-to-plasma partition coefficient (KP,i) for tissue i.
The notations used in eqs. 1–6 are important for PD models when multiple steroids are present. When no drug is present, NRN_TOT = CRN. The parameters GR0, CR0, and CRN(0) are the concentrations of the GR, CST bound to receptor, and CST bound to receptor in the nucleus at time zero under baseline conditions at steady state. The GR0 values were fixed to experimentally measured values in liver (476 fmol/mg of protein) and uterus (290 fmol/mg of protein) from untreated rats (Izawa et al., 1984; Hazra et al., 2007b). The steady-state values for CR(0) and CRN(0) at time zero were defined as:
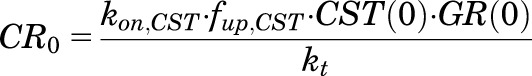 |
(7) |
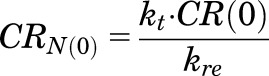 |
(8) |
Eq. 1 was solved under steady-state conditions to yield ks,GR:
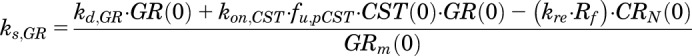 |
(9) |
The GR mRNA (GRm) showed circadian oscillations in livers from male rats (Hazra et al., 2007b), which were described by using an indirect response (IDR) model with the mRNA synthesized by a time-dependent synthesis rate (ks,GRm(t)) and degraded by first-order rate constant (kd,GRm):
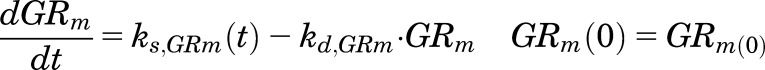 |
(10) |
The time-dependent synthesis rate of hepatic GR mRNA [ks,GRm,liver(t)] was described using a two harmonic function (Hazra et al., 2007b):
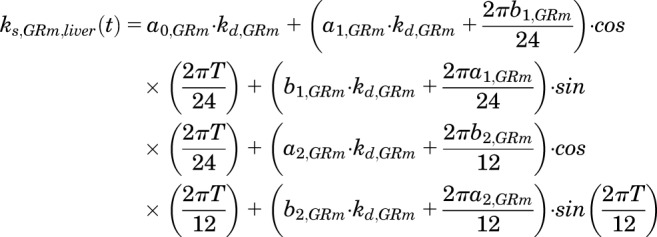 |
(11) |
where ai and bi are Fourier coefficients associated with the harmonic oscillations.
Suppression of GR mRNA expression by MPL in liver was described by a DRN - mediated inhibition of ks,GRm,liver(t) and subsequent inhibition of kd,GRm by a transduction signal generated from DRN to capture a liver-specific rebound phenomenon:
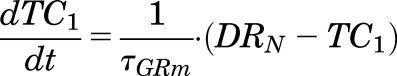 |
(12) |
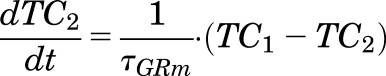 |
(13) |
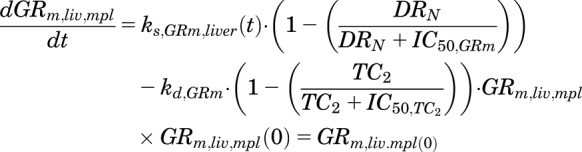 |
(14) |
where TC1 and TC2 are two transit compartments, τGRm is the mean transit time for signal transduction from DRN, IC50,GRm is the concentration of DRN at which the synthesis rate of GR mRNA is reduced to 50% of its baseline, and IC50,TC2 is the concentration of TC2 responsible for 50% inhibition of the loss rate for GR mRNA.
Suppression of GR mRNA expression by MPL in uterus is given by:
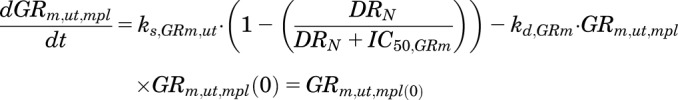 |
(16) |
Because stationarity is assumed, ks,GRm,ut was defined as:
| (17) |
The dynamics of GR mRNA has been previously assessed in liver, adipose, lung, and muscle from intact male rats (Hazra et al., 2007b; Sukumaran et al., 2011; Ayyar et al., 2017) and free cytosolic receptor densities in livers of male rats (Hazra et al., 2007b). Whereas time-dependent down-regulation of receptor mRNA was observed in all tissues after MPL, a rebound in GR mRNA was observed specifically in male livers (Hazra et al., 2007b). It was assumed that hepatic GR mRNA followed a similar temporal pattern in female rat livers. Parameter values for hepatic receptor dynamics were used to simulate receptor dynamics in the rat uterus (no rebound process was incorporated). Corrections for sex and/or tissue differences in baseline receptor mRNA [GRm(0)] were made using measurements in control animals from each group. In addition, free receptor concentrations in the uterus of untreated rats (GR0) were obtained from a previous study (Izawa et al., 1984).
Plasma Estradiol Concentrations.
Endogenous concentrations of plasma E2 vary significantly across the 4-day estrous cycle in rodents. The profile for plasma E2 in control (nondosed) female rats was obtained from Smith et al. (1975). An empirical function (mimicking two joint Gaussian normal distribution curves) was constructed to approximate the time course of plasma E2 [E2(t)]. The equation was fit to the experimental data and subsequently extended to repeat for a second cycle:
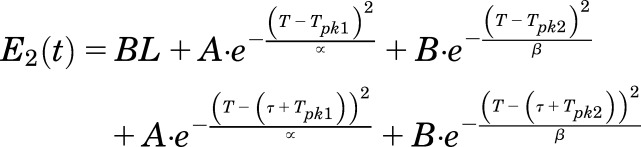 |
(18) |
where BL is the baseline concentration of plasma E2, T is the actual time, the terms Tpk1 and Tpk2 represent the times of peak concentrations within the metestrus or diestrus and PE phases, A and B are the amplitudes of the first and second peak, α and β are fitting coefficients for the two peaks, and τ is the duration of one estrous cycle (96 hours). Plasma concentrations of E2 as described by eq. 18 was used as an input to drive subsequent ER-binding dynamics in the liver and uterus.
Estrogen Receptor Binding.
Plasma E2 concentrations (eq. 18) were converted to nanomolar units, and the free concentrations of E2 in plasma (Cf,E2) were assumed to equilibrate with the intracellular spaces (i.e., diffusion is not rate-limiting) where the cytosolic ER is present. The concentration of cytosolic drug-receptor complex (ERB) is given by the following equation:
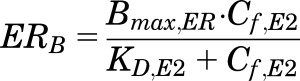 |
(19) |
where Cf,E2 = fup,E2 ∙ E2(t), Bmax,ER is the total concentration of estrogen receptors, and KD,E2 is the equilibrium dissociation constant. The values of Bmax,ER and KD,E2 in rat liver and uterus were obtained from the literature (Notides, 1970; Aten et al., 1978; Branham et al., 2002; Plowchalk and Teeguarden, 2002). The bound hormone-receptor complex translocates into the cell nucleus and the rate of change of activated nuclear complex concentrations (ERN) can be described as:
| (20) |
The value of kt was assumed to be the same as used for describing nuclear translocation of the GR complex.
GILZ mRNA Dynamics.
The transcriptional enhancement of GILZ mRNA synthesis by MPL has been described in various tissues from male rats (Ayyar et al., 2017). The expression of GILZ shows nonstationary baselines over time in multiple tissues with a pattern entrained to endogenous CST production in male rats (Ayyar et al., 2017). The dynamics of GILZ mRNA in both sexes was driven by NRN_TOT, where circadian rhythms in gene expression would be expected to follow a pattern of CRN in the absence of MPL, whereas the receptor-mediated enhancement of GILZ would occur in a competitive manner in the presence of endogenous and exogenous agonists (Ariëns et al., 1957). Based on the genomic mechanism of interaction of nuclear steroid-receptor complexes (Whirledge and Cidlowski, 2013), it is assumed that the ERN in tissues interact with NRN_TOT by competing for binding at the same site of the GILZ promoter region, resulting in competitive antagonism of GILZ enhancement. Consistent with this mechanism, the proposed equation accounts for antagonist pharmacology (Gaddum, 1957) via a nuclear ER concentration-dependent increase in the apparent SC50 of the nuclear GR complex:
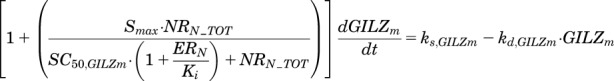 |
(21) |
where ks,GILZm is the zero-order synthesis rate and kd,GILZm the first-order degradation rate constant for GILZ mRNA; Smax is the maximal stimulatory capacity for GILZ synthesis by NRN_TOT; SC50,GILZm is the concentration of NRN_TOT producing half-maximal stimulation of GILZ mRNA; and Ki (inhibition constant) is the nuclear ER complex (ERN) concentration producing 50% inhibition of NRN_TOT-mediated enhancement of GILZ. The ks,GILZm at steady state is given by:
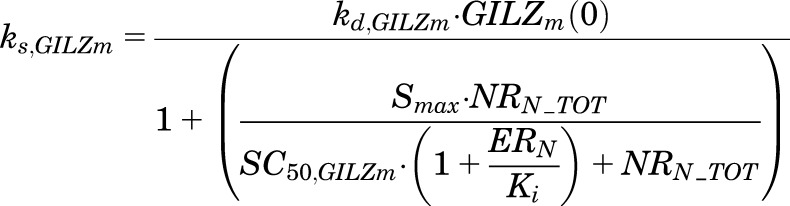 |
(22) |
Data Analysis
Statistical Analysis.
Data were analyzed statistically by Student’s t tests using SigmaPlot 14.0 (Systat Inc., San Jose, CA), and P < 0.05 was considered statistically significant.
PK/PD Data Analysis
The ADAPT 5 software was used for implementing the systems model, including data fitting and simulation of model equations (D’Argenio et al., 2009). The maximum likelihood method was applied for fitting the data. Replicate data at each time point from animals in each experiment were pooled. The goodness of fit was assessed by system convergence, visual inspection of the fitted curves, improved likelihood, examination of residuals, and precision (CV%) of the estimated parameters. The following variance model was specified for the PD outputs:
| (23) |
where Vi is the variance of the ith data point, σ1 and σ2 are the variance parameters, and Yi is the model predicted response. Variance parameters σ1 and σ2 were estimated (or σ2 was fixed to 2) along with model parameters during fittings. Area-under-the-effect-curve (AUEC) values for observed and model-fitted data were calculated using the linear-up log-down trapezoidal method and compared using Phoenix 8.1 (Certara Corporation, Princeton, NJ).
All circadian as well as PK/PD data were interpreted and modeled across the time scale of the rodent reproductive cycle (i.e., 4 days), starting at 12:00 AM on day 1 (E phase) with female rats. Drug was administered between 1.5 and 3 hours after lights on in for males (day 1), females within the E phase (day 1), and females within the PE phase (day 4). For simplicity, the dosing time was assumed to be at circadian time 2.5 hours for all rats. In the females, dosing therefore occurred at 8.5 hours (6 hours of dark + 2.5 hours after lights on) after entering a new phase of the cycle. The same was assumed for males to ensure consistency across groups (although reproductive cycles are irrelevant in males; stable baseline E2 concentrations were assumed). Time-course data from males and from E-, and PE-phased female rats were fitted jointly using a single-systems model. To ensure that a steady state in all PD profiles was achieved after dosing within E on day 1 and before modeling PE, MPL was reinputted into the system on day 8 (i.e., second PE) for the PE female group. Hence, all PD profiles are plotted and modeled with respect to estrous time, with MPL given at 8.5 hours in males and E females and at 176.5 hours in PE females.
Results
Glucocorticoid Receptor Dynamics
Baseline hepatic GR mRNA was compared between males aged 7 (cohort 4) and 11 weeks (cohort 5); no differences were observed (Supplemental Fig. 1). Hepatic GR mRNA between males (cohort 5) and E females (cohort 1) was compared at baseline and 4 hours after MPL dosing. As expected, ∼50% downregulation in GR mRNA occurred 4 hours after MPL compared with baseline in both sexes (Fig. 3). No statistically significant difference in hepatic GR mRNA was found between that at baseline and that after MPL (Fig. 3) in both groups, consistent with previous findings in liver at the mRNA and protein levels (Duma et al., 2010). Therefore, identical receptor concentrations and kinetic rate constants as obtained from fitting hepatic GR mRNA and free cytosolic receptor data in males were assumed in both sexes (note: sex-specific PK and unbound plasma CST concentrations were used to drive receptor dynamics). This assumption was further confirmed, in part, by comparative time-course measurements of hepatic GR mRNA in males and E females up to 24 hours after MPL (Supplemental Fig. 2). Next, uterine expression of GR mRNA was assessed in E (cohort 1) and PE (cohort 2) females at baseline and 4 hours after dosing. Similar to liver patterns, mRNA expression was ∼50% decreased by 4 hours after dosing compared with baseline within both groups, with no significant differences in baseline or suppression across both groups.
Fig. 3.

GR mRNA expression at baseline (black bars) and at 4 hours after MPL (gray bars) in livers from 11-week-old male rats (cohort 5) and estrus-phased female (cohort 1) rats (top panel) and in uterus from proestrus (cohort 2) and estrus (cohort 1) female rats (bottom panel) determined by qRT-PCR. Error bars reflect 1 S.D. from the mean (n = 3 rats/group). *P < 0.05, significant difference between baseline and after dosing at 4 hours; nd, no significant difference.
The dynamics of hepatic GR mRNA in male rats (Hazra et al., 2007b) after MPL dosing along with the model fittings are shown in Fig. 4. MPL caused significant perturbation in GR mRNA, which unlike the profiles for CST (Ayyar et al., 2019), displayed a rebound phenomenon in this tissue before slowly returning to its regular daily rhythm beyond 72 hours. The downregulation, as well as the rebound phase, was captured suitably well. The time course of free hepatic cytosolic receptor density in male rats (Hazra et al., 2007b) and the model fittings are shown in Fig. 4. The developed model, which extended the model of Hazra et al. (2007) to include competitive CST binding, captured the free receptor profiles reasonably well. All parameter values governing GR mRNA and receptor dynamics controlled by MPL were assumed to be the same for CST and were fixed based on Hazra et al. (2007). Estimation of the parameter kon,CST yielded values with low precision (high % CV), likely because of model overparameterization in the absence of nonstationary baseline receptor data. Using the obtained kinetic parameters in liver, simulations of GR mRNA and receptors in uterus from E and PE females were performed after correction for tissue differences in GR mRNA (from Fig. 3) and cytosolic receptor concentrations (Izawa et al., 1984) (Supplemental Fig. 3). The uterine GR mRNA data at 4 hours was well predicted by the model in both groups. In the absence of extended time-course and circadian GR data in uterus, no rebound in GR mRNA as well as a stable baseline were assumed.
Fig. 4.

Hepatic GR mRNA (left panel) and free cytosolic GR density (right panel) in normal male rats (cohort 4) after 50 mg/kg IM MPL. The symbols represent the mean ± S.D., and the solid lines depict the model fit (eqs. 1–14). Parameter values are listed in Table 1. Dark (shaded) and light (unshaded) time periods are indicated.
Plasma Estradiol and Tissue Receptor Occupancy
Plasma concentrations of E2 were assayed in a group of nondosed female rats within either the E (n = 4) or PE (n = 4) stages (blood samples were taken at 3 hours in PE females and between 2 and 6 hours after lights on in E females). As depicted in Fig. 5 (left panel), plasma E2 concentrations were significantly elevated in PE (19.6 ± 5.9 ng/ml) compared with E (2.0 ± 0.48 ng/ml). A similar trend was demonstrated by Smith et al. (1975), but their study monitored plasma concentrations of 17β-E2 more extensively over the entire course of the rat estrous cycle. Figure 5 (middle panel) depicts the profile of plasma E2 over 4 days in the female rat along with its mathematical characterization. Plasma E2 was stable throughout the E phase (∼6.9 ng/ml) but showed a sharp rise during PE to peak concentrations of ∼39 ng/ml. Since the model assumed that the hormonal profiles in females were not perturbed by drug, an empirical function was constructed to approximate the time course of plasma E2 that captured the data reasonably well. Total E2 was corrected for plasma protein binding to albumin (Plowchalk and Teeguarden, 2002), and the free concentrations of E2 were subsequently used to drive ER binding in the cytosol and subsequent ERN concentrations in liver and uterus (Fig. 5; right panel). The simulations indicated higher occupancy and amounts of ERN in uterus compared with liver, largely attributable to a higher ER density in uterus (560 fmol/mg protein) compared with liver (24.5 fmol/mg protein). Parameter values for ER binding are listed in Table 2, along with their literature sources.
Fig. 5.

Top panel: Plasma 17β-estradiol (E2) concentrations in nondosed proestrus and estrus female rats in blood samples taken between 2 and 6 hour after lights on during each phase and determined using enzyme-linked immunosorbent assay. Error bars reflect 1 S.D. from the mean (n = 4 rats/group). **P < 0.001, significant difference. Middle panel: Plasma concentrations of E2 in female rats over the 4-day estrous cycle; symbols are measurements from individual rats [data taken from Smith et al. (1975)], and the solid line depicts the model-fitted profile (eq. 18) shown to repeat for a second cycle. Bottom panel: Simulated profile (eq. 20) of the estrogen complex in the nucleus (ERN) throughout the rodent estrous cycle in the uterus (left axis) and in liver (right axis). Parameter values are listed in Table 2.
TABLE 2.
Model parameters for plasma 17β-estradiol (E2) and estrogen receptor (ER) dynamics
| Parameter | Definition | Estimate (% CV) or Value (Source) |
|---|---|---|
| Plasma estradiol concentrations | ||
| BL (pg/ml) | Baseline concentrations of E2 | 6.85 (5.8) |
| A (pg/ml) | Peak amplitude in met/diestrus | 8.6 (14.8) |
| B (pg/ml) | Peak amplitude in proestrus | 32.3 (10.5) |
| Tpk1 (h) | Time of peak amplitude in met/diestrus | 52.3 (3.5) |
| Tpk2 (h) | Time of peak amplitude in proestrus | 82.2 (0.8) |
| α | Fitting coefficient for first peak | 191 (37.5) |
| β | Fitting coefficient for second peak | 106 (16.7) |
| τ (h) | Duration of estrous cycle | 96 (fixed) |
| ER binding and dynamics | ||
| fup,E2 | Unbound fraction of estradiol in plasma | 0.053 (Plowchalk and Teeguarden, 2002) |
| Bmax,ER (liver) (fmol/mg of protein) | ER content in liver | 24.5 (Aten et al., 1978; Dickson and Eisenfeld, 1979) |
| Bmax,ER (uterus) (fmol/mg of protein) | ER content in uterus | 560 (Notides, 1970) |
| KD,ER(liv) (pM) | ER binding constant in liver | 140 (Dickson and Eisenfeld, 1979) |
| KD,ER(uterus) (pM) | ER binding constant in uterus | 100 (Branham et al., 2002) |
| kt (h−1) | Translocation rate constant | 58.3 (Assumed equal to GR) |
TABLE 1.
Model parameters for glucocorticoid receptor (GR) dynamics
| Parameter | Definition | Estimate (CV%) |
|---|---|---|
| a0,GRm,liver | Fourier coefficient for liver GR mRNAa | 14.3 |
| a1,GRm,liver | −1.53 | |
| a2,GRm,liver | 0.554 | |
| b1,GRm,liver | −3.04 | |
| b2,GRm,liver | 1.18 | |
| kd,GRm (h−1) | Degradation rate constant for GR mRNA | 0.14 (17.0) |
| IC50,GRm (fmol/mg) | Half-maximal inhibition of GR mRNA production | 15.2a |
| τGRm (h) | Transduction delay for mRNA rebound | 15.6a |
| IC50,TC2 (fmol/mg) | Half-maximal inhibition of GR mRNA removal | 60.5a |
| kd,GR (h−1) | Degradation rate constant for receptor | 0.05a |
| kon,MPL (nM−1·h−1) | Association rate constant for methylprednisolone (MPL) | 0.016a |
| kon,CST (nM−1·h−1) | Association rate constant for corticosterone (CST) | 0.001 (fixed) |
| fup,mpl | Unbound fraction of MPL in plasma | 0.4b |
| fu,liv,mpl | Unbound fraction of MPL in liver | 0.032 (calculated) |
| fu,cst | Unbound fraction of CST in plasma | 0.017a |
| kre (h−1) | DRN nuclear loss rate constant | 1.31a |
| Rf | Fraction recycled | 0.93a |
| kT (h−1) | Translocation rate constant | 58.3a |
| GR(0) (fmol/mg protein) | Free cytosolic receptor initial concentration | 476.0 (liver)a 320.0 (uterus)c |
GILZ mRNA Dynamics
Liver.
The PD enhancement profiles of hepatic GILZ mRNA by MPL in males and females, along with the model fitted profiles and simulated driving forces, are shown in Fig. 6. The developed model was able to jointly capture the data quite well. Table 3 lists parameter values that were either fixed or estimated from the fitting. Figure 6 (top left) displays the time course of enhancement of hepatic GILZ mRNA by MPL in male rats along with the model fits. The enhancement profile of GILZ mRNA was nearly identical across both male studies (cohorts 4 and 5; comparison not shown). In males, GILZ increased rapidly from the baseline (1893 ± 423 molecules/ng of RNA) to the peak (7867 ± 1821 molecules/ng of RNA) by 0.75 hour after dosing. The model-fitted profile showed a return to baseline at around 24 hours in males, with GILZ mRNA displaying modest circadian rhythmicity in expression (driven by endogenous CST concentrations). The PD profiles of hepatic GILZ message in E and PE females are shown in Fig. 6 (top right). GILZ, in general, showed higher peak expression, a more prolonged return profile, and a resultant 2.5 to 3-fold increase in AUEC in females, regardless of estrous stage, compared with males (Table 4). The model jointly captured the observed sex differences fairly well, although peak responses were somewhat underpredicted in both groups. The estimated first-order degradation rate constant of GILZ mRNA in liver (kdeg,GILZm) value of 7.5 hours−1 (21.8% CV) was significantly higher compared with estimates in other tissues, including lung, muscle, and adipose (Ayyar et al., 2017), as reflected by the profile of GILZ mRNA in liver. The extent and pattern of upregulation of hepatic GILZ mRNA in all groups (Fig. 6; bottom left) corresponded with that of the nuclear steroid-receptor complex (Fig. 6; bottom right).
Fig. 6.

GILZ mRNA expression in liver from male rats (green; top left), estrus-phased female rats (red; top right), and proestrus-phased female rats (blue; top right) given 50 mg/kg IM MPL. Symbols represent the mean ± S.D., and the solid lines depict the model fit (eq. 21). Parameter values are listed in Table 3. Bottom right: Comparison of the model-fitted profiles of hepatic GILZ mRNA in the three groups. Bottom left: Comparison of the model-simulated profile of hepatic drug/hormone-receptor complex in the nucleus NRN_TOT (eq. 6) in the three groups. Dark (shaded) and light (unshaded) periods are indicated Table 4.
TABLE 3.
Model parameters for glucocorticoid-induced leucine zipper (GILZ) mRNA dynamics
| Parameter | Definition | Estimate (% CV) |
|---|---|---|
| kd,GILZm (h−1) | Degradation rate constant for GILZ | 7.5 (21.8)a; 1.9 (27.5) b |
| Smax | Maximal stimulatory capacity by DRN | 7.5a,b,c |
| SC50,GILZm (fmol/mg) | DRN producing half-maximal stimulation | 558 (5.5)a; 672 (19.2)b |
| Ki (fmol/mg) | ERN producing half-maximal inhibition of SC50 of DRN | 62.1 (68.6)a,b |
| GILZm(0) (molecules/ng of RNA) | GILZ initial concentration in liver | 1893 (M); 2051 (E); 2538 (PE) |
| GILZm(0) (molecules/ng RNA) | GILZ initial concentration in uterus | 3245 (E); 2400 (PE) |
TABLE 4.
Area-under-effect-curve (AUEC) analysis for glucocorticoid-induced leucine zipper (GILZ) responses
| Group | Baseline (molec/ng RNA) | AUECobs (0–24 h) (molec/ng RNA∙h) | AUECpred (0–24 h) (molec/ng RNA∙h) |
|---|---|---|---|
| Liver | |||
| Male | 1893 | 31,262 | 41,952 |
| Female (estrus) | 2051 | 78,657 | 73,927 |
| Female (proestrus) | 2538 | 89,972 | 93,172 |
| Uterus | |||
| Female (estrus) | 3245 | 78,828 | 73,335 |
| Female (proestrus) | 2400 | 47,340 | 44,440 |
Molec, molecules.
Uterus.
The PD enhancement profiles of GILZ mRNA by MPL in uterus from E and PE females, along with model fitted profiles, are shown in Fig. 7. The model captured the dynamics of GILZ in both groups reasonably well. Parameter estimates are listed in Table 3. Baseline expression of uterine GILZ mRNA was significantly lower in PE (2400 ± 727 molecules/ng of RNA) compared with E (3245 ± 548 molecules/ng RNA) and correlated inversely to plasma estradiol concentrations in both estrous phases (Fig. 5), suggestive of antagonism of uterine GILZ mRNA, even in the absence of drug. Uterine GILZ mRNA was enhanced to a greater extent in E compared with PE females, indicated by a higher peak response (13,476 ± 489 vs. 7167 ± 2528 molecules/ng of RNA molecules/ng RNA) and baseline-corrected AUEC values (Table 4) in E compared with PE. The proposed model captured the trends of both groups quite well (Fig. 7; Table 4), although the peak of response was underpredicted. The estimated kdeg,GILZm value in uterus of 1.9 hours−1 (27.5% CV) was considerably lower compared with that of liver, yet 4- to 6-fold greater compared with other tissues (Ayyar et al., 2017), indicating a pronounced intertissue variability for this parameter. The SC50 for GILZ message enhancement by MPL was reasonably similar in liver (558 fmol/mg protein; 5.5%CV) and in uterus (672 fmol/mg protein; 19.2% CV). A common Smax value of 7.5 (Hazra et al., 2008) suitably described the dynamics of GILZ mRNA across all tissues and both sexes. The value for Ki estimated based on fitting of the uterine data were 61.4 fmol/mg protein (68.6% CV). These results collectively support the hypothesis of an estrogen-mediated antagonism of receptor/gene-mediated MPL PD. Joint characterization of both data sets upon removing the antagonistic regulation by ERN from eq. 22 led to a systematic overprediction of uterine GILZ mRNA dynamics in PE females (not shown).
Fig. 7.

GILZ mRNA expression in the uterus from estrus-phased female rats (red) and proestrus-phased female rats (blue) given 50 mg/kg IM MPL. Symbols represent the mean ± S.D., and the solid lines depict the model fit [eq. 21]. Parameter values are listed in Table 3. Dark (shaded) and light (unshaded) periods are indicated.
Discussion
This work represents a continuation of our longstanding efforts to decipher the complex pharmacogenomic mechanisms by which glucocorticoids regulate body functions in vivo. Previous-generation mechanistic PK/PD models were built on experimental studies conducted in ADX male rats and later extended to intact male rats. The current work examined the dynamic effects of CS in female rats, a more physiologically complex, however equally relevant, preclinical animal model. Several factors related to PK and PD needed careful attention owing to complexities arising from 24-hour circadian rhythms, as well as longer 96-hour reproductive rhythms. Despite added complications associated with the use of female rats, our objective was to understand quantitatively the interplay of endogenous glucocorticoid hormones, exogenous CS, and sex hormones using a systems PK/PD modeling approach.
Studies examining the variability in drug response should consider factors influencing variability in drug exposure in relevant biophases (Mager and Kimko, 2016). In vitro properties of MPL PK (Ayyar et al., 2019b), as well as in vivo sex differences in plasma and tissue PK of MPL (Ayyar et al., 2019a), were investigated and accounted for in both sexes using an extended mPBPK modeling approach. Endogenous agonists contribute to PD variability in drug responses (Levy, 1998). The model incorporated sex- and estrous phase–dependent differences in the circadian concentrations of CST (Atkinson and Waddell, 1997), as well as the rapid adrenal suppressive effects of MPL (Ayyar et al., 2019a). Differences (or lack thereof) in cytosolic receptor concentrations in rat livers and uterus were accounted for using experimental measurements from the literature (Izawa et al., 1984; Hazra et al., 2007b; Duma et al., 2010), with kinetic parameters controlling GR dynamics held constant in all systems. In ER-expressing human uterine epithelial cells, the presence of low concentrations (1–10 pM) of E2 produced interactions between the activated ER and GR complexes, resulting in an antagonism of CS-inducible GILZ (Whirledge and Cidlowski, 2013). We estimated that endogenous concentrations of unbound plasma E2 would rise to ∼7.5 pM by the peak of PE, well within the antagonistic range reported in vitro. Of translational and physiologic relevance, we hypothesized that such drug-hormone interactions would contribute to time- and sex-dependent variability in genomic MPL actions as a function of estrous rhythms.
A quantitative systems approach was used to understand the dynamic mechanisms controlling sex- and tissue-specific MPL actions. Previous generation models assumed that the kinetics of drug distribution into tissues was not rate-limiting (i.e., well perfused entry) and that unbound concentrations of MPL in plasma interacted with cytosolic receptors (Hazra et al., 2007a). Here, total MPL concentrations were measured in liver in both sexes (Ayyar et al., 2019a), the unbound tissue fraction was calculated under assumption of the “free hormone” hypothesis (Mendel, 1989), and free drug concentrations in liver were used to drive receptor binding. The time course of hepatic free cytosolic receptor data in males was well captured using this model, despite using the parameter values for receptor kinetics estimated upon assuming free plasma MPL as the driving force (Hazra et al., 2007b), indicating that prior model assumptions regarding the rapid equilibration kinetics of MPL in liver are maintained. It is possible that the unbound liver fraction (fu,hep) represents an “apparent” value since active transport processes may contribute to hepatic uptake of MPL (Lackner et al., 1998; Ayyar et al., 2019).
To realize circadian oscillations in hepatic tyrosine aminotransferase (TAT) dynamics, a previous model (Hazra et al., 2007b) assumed equilibrium binding of CST to receptors as a distinct process independent of MPL kinetics and GR engagement. The present model built on this concept but incorporated competitive binding for free cytosolic receptors by drug and hormone, thus introducing nonstationarity in steroid pharmacogenomics through a coupled process. Estimation of the parameter kon,CST yielded values with low precision (high % CV), likely due to model overparameterization in the absence of nonstationary baseline receptor data. To address this issue, a local sensitivity analysis of this parameter was used during the model-building process. Specifically, kon,CST varied by 100-fold across a physiologically plausible range and fixed to an estimate (0.001 nM−1·h−1) that was ∼16-fold lower compared with that of MPL and provided optimal characterization of the data. Further experimental testing of CST binding kinetics is needed to confirm the accuracy of this model-based value. It was assumed that kinetic rate constants and PD parameters governing the profiles of GR and its mRNA were identical across sex and tissue type. Although more extensive receptor data in tissues in both sexes would ascertain the validity of these assumptions, the model was able to generate hepatic nuclear complex profiles that corresponded reasonably well with the extents and patterns of hepatic GILZ mRNA in both sexes. Following the same assumptions, uterine GILZ mRNA profiles were also captured reasonably well. Nonetheless, one possible reason for the underestimation of peak GILZ responses in females could relate to modest differences in receptor dynamics.
Baseline GILZ mRNA at the time of dosing was higher in PE (2538 molecules/ng of RNA) compared with E and in males (1893 and 2051 molecules/ng of RNA), possibly attributable to elevated plasma CST concentrations in PE (∼143 ng/ml) compared with E and males during that time (∼32 ng/ml) (Ayyar et al., 2019). Model-based simulations revealed that inclusion of CST dynamics produced circadian rhythmicity in NRN_TOT after drug washout (Fig. 6, bottom right), consequently producing a characteristic circadian pattern in tissue GILZ mRNA (Ayyar et al., 2017), one that followed the pattern of plasma CST in a delayed manner. Unlike in liver, baseline GILZ mRNA in uterus was significantly lower in PE (2400 molecules/ng of RNA) compared with E females (3245 molecules/ng RNA), despite higher plasma CST concentrations. This is likely attributable to the antagonistic effects on basal GILZ mRNA produced by an elevated occupancy of ER in uterus during PE (Fig. 5, right panel). Therefore, particular attention was needed to discern whether the lower enhancement of GILZ mRNA by MPL during PE was explained solely by a lower baseline (Sun and Jusko, 1999). Upon modeling uterine GILZ enhancement by MPL in the absence and presence of antagonism, it was evident that inclusion of antagonism by ERN on basal and drug-regulated GILZ enhancement (eq. 22) most suitably described the totality of data. Although the kdeg,GILZm was same in both sexes, it was interesting to find a pronounced intertissue variability in this parameter across liver and uterus, consistent with previous findings (Ayyar et al., 2017). Similar observations of a “systems variability” in the degradation rate constants for some signaling proteins across several lines of multiple myeloma cells was recently reported (Ramakrishnan and Mager, 2019).
The AUEC of hepatic GILZ enhancement was significantly greater in females compared with males (Fig. 6; Table 4), controlled by PK-driven increases in hepatic DRN concentrations (Fig. 6) in females. Examination of GILZ response in two distinct tissues (liver and uterus) and two distinct estrous phases (E and PE) in females, however, revealed estrous cycle– and ER-dependence in the postreceptor control of genomic MPL action. In the liver, a tissue of relatively low ER (Table 2), there was a negligible influence of E2-ER signaling, as evidenced by the nearly identical profiles of hepatic GILZ mRNA between E and PE females. In contrast, in uterus, a tissue with high ER content (Table 2), the AUEC of GILZ upregulation was significantly lower in PE compared with E females (Table 4) despite no PK or receptor differences, providing in vivo support to the hypothesis of an ERN-mediated antagonism of MPL-regulated GILZ.
The ability of the proposed systems model to jointly capture genomic GILZ enhancement by MPL across sex, tissues, and (circadian and estrous) time is encouraging and has several implications. First, reasonable prediction of GILZ mRNA dynamics across all the groups justify the assumption of identical kinetic rate constants for GR dynamics across sex and tissues. Second, the development of a systems model platform enabled a separation and systematic examination of drug- and system-specific parameters and their relative contributions in limiting the overall genomic response to MPL dosing. Third, the results suggest that the principal drug-specific determinants of receptor-/gene-mediated MPL response remain its PK and receptor affinity, whereas tissue GR and ER content, endogenous E2 concentrations, and biomarker turnover represent system-specific factors influencing steroid response.
It should be recognized that the estrous cycle regulates not only E2 but also other sex hormones such as progesterone and prolactin (Smith et al., 1975). The role of progesterone as a contributor to sex differences in PD responses (e.g., QT prolongation) has been reviewed (Sedlak et al., 2012). The potential roles of testosterone, progesterone, and other sex hormones in interacting with glucocorticoid signaling requires further examination. The current work examined sex hormone effects within a physiologic range. Our present findings provide a basis for further examination of MPL-regulated pharmacogenomics under exogenous E2 administration using the ovariectomized (OVX) rat model. Such a study design could 1) clarify the disposition kinetics and exposure of E2 at the biophase, 2) permit evaluation of potential PK interactions between MPL and E2, 3) provide a broader characterization of the concentration-dependent antagonistic relationship in PD, and 4) yield robust data to challenge predictions based on the current model.
In conclusion, differences in PK and receptor-mediated PD of MPL were identified based on sex, estrous stage, and tissue type. The time course of MPL actions were interpreted within the context of 24-hour circadian biorhythms as well as 4-day reproductive biorhythms. The developed model offers a mechanistic platform to integrate and evaluate the determinants of sex- and tissue-specificity in CS actions. This mechanistic systems model may also form the basis for explaining the interactions of E2 with other drugs and xenobiotics acting via nuclear receptors.
Abbreviations
- ADX
adrenalectomized
- AUEC
area-under-the-effect curve
- CS
corticosteroid
- CST
corticosterone
- DEX
dexamethasone
- E
estrus
- E2
17β-estradiol
- ER
estrogen receptor
- GILZ
glucocorticoid-induced leucine zipper
- GR
glucocorticoid receptor
- GRE
glucocorticoid response element
- mPBPK
minimal physiologically based pharmacokinetic
- MPL
methylprednisolone
- PE
proestrus
- PD
pharmacodynamic
- PG
pharmacogenomic
- qRT-PCR
quantitative real-time polymerase chain reaction
Authorship Contributions
Participated in research design: Ayyar, DuBois, Almon, Jusko.
Conducted experiments: Ayyar, DuBois.
Performed data analysis: Ayyar, Jusko.
Wrote or contributed to the writing of the manuscript: Ayyar, DuBois, Almon, Jusko.
Footnotes
This work was supported by the National Institute of General Medical Sciences, National Institutes of Health [Grants GM24211 and GM131800].
 This article has supplemental material available at jpet.aspetjournals.org.
This article has supplemental material available at jpet.aspetjournals.org.
References
- Almon RR, Yang E, Lai W, Androulakis IP, Ghimbovschi S, Hoffman EP, Jusko WJ, Dubois DC. (2008) Relationships between circadian rhythms and modulation of gene expression by glucocorticoids in skeletal muscle. Am J Physiol Regul Integr Comp Physiol 295:R1031–R1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ariëns EJ, Van Rossum JM, Simonis AM. (1957) Affinity, intrinsic activity and drug interactions. Pharmacol Rev 9:218–236. [PubMed] [Google Scholar]
- Aten RF, Dickson RB, Eisenfeld AJ. (1978) Estrogen receptor in adult male rat liver. Endocrinology 103:1629–1635. [DOI] [PubMed] [Google Scholar]
- Atkinson HC, Waddell BJ. (1997) Circadian variation in basal plasma corticosterone and adrenocorticotropin in the rat: sexual dimorphism and changes across the estrous cycle. Endocrinology 138:3842–3848. [DOI] [PubMed] [Google Scholar]
- Ayroldi E, Riccardi C. (2009) Glucocorticoid-induced leucine zipper (GILZ): a new important mediator of glucocorticoid action. FASEB J 23:3649–3658. [DOI] [PubMed] [Google Scholar]
- Ayyar VS, Almon RR, Jusko WJ, DuBois DC. (2015) Quantitative tissue-specific dynamics of in vivo GILZ mRNA expression and regulation by endogenous and exogenous glucocorticoids. Physiol Rep 3:e12382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayyar VS, DuBois DC, Almon RR, Jusko WJ. (2017) Mechanistic multi-tissue modeling of glucocorticoid-induced leucine zipper regulation: integrating circadian gene expression with receptor-mediated corticosteroid pharmacodynamics. J Pharmacol Exp Ther 363:45–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayyar VS, DuBois DC, Almon RR, Jusko WJ. (2019a) Modeling corticosteroid pharmacokinetics and pharmacodynamics, part II: sex differences in methylprednisolone pharmacokinetics and corticosterone suppression. J Pharmacol Exp Ther 370:327–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayyar VS, Song D, DuBois DC, Almon RR, Jusko WJ. (2019b) Modeling corticosteroid pharmacokinetics and pharmacodynamics, part I: determination and prediction of dexamethasone and methylprednisolone tissue binding in the rat. J Pharmacol Exp Ther 370:318–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayyar VS, Sukumaran S, DuBois DC, Almon RR, Qu J, Jusko WJ. (2018) Receptor/gene/protein-mediated signaling connects methylprednisolone exposure to metabolic and immune-related pharmacodynamic actions in liver. J Pharmacokinet Pharmacodyn 45:557–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Branham WS, Dial SL, Moland CL, Hass BS, Blair RM, Fang H, Shi L, Tong W, Perkins RG, Sheehan DM. (2002) Phytoestrogens and mycoestrogens bind to the rat uterine estrogen receptor. J Nutr 132:658–664. [DOI] [PubMed] [Google Scholar]
- Bustin SA, Benes V, Garson JA, Hellemans J, Huggett J, Kubista M, Mueller R, Nolan T, Pfaffl MW, Shipley GL, et al. (2009) The MIQE guidelines: minimum information for publication of quantitative real-time PCR experiments. Clin Chem 55:611–622. [DOI] [PubMed] [Google Scholar]
- D’Argenio D, Schumitzky A, Wang X. (2009) ADAPT 5 User’s Guide: Pharmacokinetic/Pharmacodynamic Systems Analysis Software, BioMedical Simulations Resource, Los Angeles, CA. [Google Scholar]
- Dickson RB, Eisenfeld AJ. (1979) Estrogen receptor in liver of male and female rats: endocrine regulation and molecular properties. Biol Reprod 21:1105–1114. [DOI] [PubMed] [Google Scholar]
- Duma D, Collins JB, Chou JW, Cidlowski JA. (2010) Sexually dimorphic actions of glucocorticoids provide a link to inflammatory diseases with gender differences in prevalence. Sci Signal 3:ra74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaddum JH. (1957) Theories of drug antagonism. Pharmacol Rev 9:211–218. [PubMed] [Google Scholar]
- Hazra A, DuBois DC, Almon RR, Jusko WJ. (2007a) Assessing the dynamics of nuclear glucocorticoid-receptor complex: adding flexibility to gene expression modeling. J Pharmacokinet Pharmacodyn 34:333–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hazra A, DuBois DC, Almon RR, Snyder GH, Jusko WJ. (2008) Pharmacodynamic modeling of acute and chronic effects of methylprednisolone on hepatic urea cycle genes in rats. Gene Regul Syst Bio 2:1–19. [PMC free article] [PubMed] [Google Scholar]
- Hazra A, Pyszczynski N, DuBois DC, Almon RR, Jusko WJ. (2007b) Modeling receptor/gene-mediated effects of corticosteroids on hepatic tyrosine aminotransferase dynamics in rats: dual regulation by endogenous and exogenous corticosteroids. J Pharmacokinet Pharmacodyn 34:643–667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izawa M, Satoh Y, Iwasaki K, Ichii S. (1984) Glucocorticoid receptor in the rat uterus. Endocrinol Jpn 31:491–500. [DOI] [PubMed] [Google Scholar]
- Jusko WJ. (1995) Pharmacokinetics and receptor-mediated pharmacodynamics of corticosteroids. Toxicology 102:189–196. [DOI] [PubMed] [Google Scholar]
- Kuiper GG, Carlsson B, Grandien K, Enmark E, Häggblad J, Nilsson S, Gustafsson JA. (1997) Comparison of the ligand binding specificity and transcript tissue distribution of estrogen receptors alpha and beta. Endocrinology 138:863–870. [DOI] [PubMed] [Google Scholar]
- Lackner C, Daufeldt S, Wildt L, Alléra A. (1998) Glucocorticoid-recognizing and -effector sites in rat liver plasma membrane: kinetics of corticosterone uptake by isolated membrane vesicles. III. Specificity and stereospecificity. J Steroid Biochem Mol Biol 64:69–82. [DOI] [PubMed] [Google Scholar]
- Levy G. (1998) Predicting effective drug concentrations for individual patients. Determinants of pharmacodynamic variability. Clin Pharmacokinet 34:323–333. [DOI] [PubMed] [Google Scholar]
- Mager DE, Kimko HHC. (2016) Systems pharmacology and pharmacodynamics: an introduction, in Systems Pharmacology and Pharmacodynamics (Mager DE and Kimko HHC eds) pp 3–14, Springer International Publishing, Cham, Switzerland. [Google Scholar]
- Mager DE, Pyszczynski NA, Jusko WJ. (2003) Integrated QSPR--pharmacodynamic model of genomic effects of several corticosteroids. J Pharm Sci 92:881–889. [DOI] [PubMed] [Google Scholar]
- Mendel CM. (1989) The free hormone hypothesis: a physiologically based mathematical model. Endocr Rev 10:232–274. [DOI] [PubMed] [Google Scholar]
- Montano MM, Welshons WV, vom Saal FS. (1995) Free estradiol in serum and brain uptake of estradiol during fetal and neonatal sexual differentiation in female rats. Biol Reprod 53:1198–1207. [DOI] [PubMed] [Google Scholar]
- Nichols AI, Boudinot FD, Jusko WJ. (1989) Second generation model for prednisolone pharmacodynamics in the rat. J Pharmacokinet Biopharm 17:209–227. [DOI] [PubMed] [Google Scholar]
- Notides AC. (1970) The binding affinity and specificity of the estrogen receptor of the rat uterus and anterior pituitary. Endocrinology 87:987–992. [DOI] [PubMed] [Google Scholar]
- Oakley RH, Cidlowski JA. (1993) Homologous down regulation of the glucocorticoid receptor: the molecular machinery. Crit Rev Eukaryot Gene Expr 3:63–88. [PubMed] [Google Scholar]
- Plowchalk DR, Teeguarden J. (2002) Development of a physiologically based pharmacokinetic model for estradiol in rats and humans: a biologically motivated quantitative framework for evaluating responses to estradiol and other endocrine-active compounds. Toxicol Sci 69:60–78. [DOI] [PubMed] [Google Scholar]
- Puig-Duran E, Greenstein BD, MacKinnon PC. (1979) The effects of serum oestrogen-binding components on the unbound oestradiol-17 beta fraction in the serum of developing female rats and on inhibition of [3H]oestradiol uptake by uterine tissue in vitro. J Reprod Fertil 56:707–714. [DOI] [PubMed] [Google Scholar]
- Ramakrishnan R, DuBois DC, Almon RR, Pyszczynski NA, Jusko WJ. (2002) Fifth-generation model for corticosteroid pharmacodynamics: application to steady-state receptor down-regulation and enzyme induction patterns during seven-day continuous infusion of methylprednisolone in rats. J Pharmacokinet Pharmacodyn 29:1–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramakrishnan V, Mager DE. (2019) Pharmacodynamic models of differential bortezomib signaling across several cell lines of multiple myeloma. CPT Pharmacometrics Syst Pharmacol 8:146–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sedlak T, Shufelt C, Iribarren C, Merz CNB. (2012) Sex hormones and the QT interval: a review. J Womens Health (Larchmt) 21:933–941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith MS, Freeman ME, Neill JD. (1975) The control of progesterone secretion during the estrous cycle and early pseudopregnancy in the rat: prolactin, gonadotropin and steroid levels associated with rescue of the corpus luteum of pseudopregnancy. Endocrinology 96:219–226. [DOI] [PubMed] [Google Scholar]
- Stossi F, Barnett DH, Frasor J, Komm B, Lyttle CR, Katzenellenbogen BS. (2004) Transcriptional profiling of estrogen-regulated gene expression via estrogen receptor (ER) α or ERbeta in human osteosarcoma cells: distinct and common target genes for these receptors. Endocrinology 145:3473–3486. [DOI] [PubMed] [Google Scholar]
- Sukumaran S, Jusko WJ, DuBois DC, Almon RR. (2011) Mechanistic modeling of the effects of glucocorticoids and circadian rhythms on adipokine expression. J Pharmacol Exp Ther 337:734–746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun YN, DuBois DC, Almon RR, Jusko WJ. (1998) Fourth-generation model for corticosteroid pharmacodynamics: a model for methylprednisolone effects on receptor/gene-mediated glucocorticoid receptor down-regulation and tyrosine aminotransferase induction in rat liver. J Pharmacokinet Biopharm 26:289–317. [DOI] [PubMed] [Google Scholar]
- Sun YN, Jusko WJ. (1999) Role of baseline parameters in determining indirect pharmacodynamic responses. J Pharm Sci 88:987–990. [DOI] [PubMed] [Google Scholar]
- Tsai SY, Tsai MJ, O’Malley BW. (1989) Cooperative binding of steroid hormone receptors contributes to transcriptional synergism at target enhancer elements. Cell 57:443–448. [DOI] [PubMed] [Google Scholar]
- Tynan SH, Lundeen SG, Allan GF. (2004) Cell type-specific bidirectional regulation of the glucocorticoid-induced leucine zipper (GILZ) gene by estrogen. J Steroid Biochem Mol Biol 91:225–239. [DOI] [PubMed] [Google Scholar]
- Whirledge S, Cidlowski JA. (2013) Estradiol antagonism of glucocorticoid-induced GILZ expression in human uterine epithelial cells and murine uterus. Endocrinology 154:499–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whirledge S, Xu X, Cidlowski JA. (2013) Global gene expression analysis in human uterine epithelial cells defines new targets of glucocorticoid and estradiol antagonism. Biol Reprod 89:66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu ZX, Sun YN, DuBois DC, Almon RR, Jusko WJ. (1995) Third-generation model for corticosteroid pharmacodynamics: roles of glucocorticoid receptor mRNA and tyrosine aminotransferase mRNA in rat liver. J Pharmacokinet Biopharm 23:163–181. [DOI] [PubMed] [Google Scholar]
- Yao Z, DuBois DC, Almon RR, Jusko WJ. (2008) Pharmacokinetic/pharmacodynamic modeling of corticosterone suppression and lymphocytopenia by methylprednisolone in rats. J Pharm Sci 97:2820–2832. [DOI] [PMC free article] [PubMed] [Google Scholar]