
Figure 4.
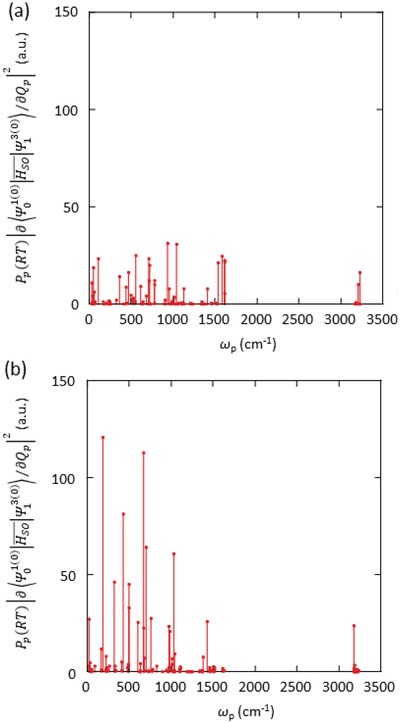
Relationship between and ωp for a) Si(C6H5)4 and b) Ge(C6H5)4 monomers. Conformations including normal vibration modes were optimized at T1 using density functional theory (Gaussian09/B3LYP/6‐31G(d)). SOC data were treated as perturbations based on the scalar relativistic orbitals. Hybrid‐B3LYP and TZP were used as exchange‐correlation functionals and the Slater‐type all‐electron basis set, respectively.