

Abstract
In response to infection, patients with inborn errors of metabolism may develop a functional deterioration termed metabolic decompensation. The biochemical hallmarks of this disruption of metabolic homeostasis are disease specific and may include acidosis, hyperammonemia or hypoglycemia. In a model system previously published by our group, we noted that during influenza infection, mice displayed a depression in hepatic mitochondrial enzymes involved in nitrogen metabolism. Based on these findings, we hypothesized that this normal adaptation may extend to other metabolic pathways, and as such, may impact various inborn errors of metabolism. Since the liver is a critical organ in inborn errors of metabolism, we carried out untargeted metabolomic profiling of livers using mass spectrometry in C57Bl/6 mice infected with influenza to characterize metabolic adaptation. Pathway analysis of metabolomic data revealed reductions in CoA synthesis, and long chain fatty acyl CoA and carnitine species. These metabolic adaptations coincided with a depression in hepatic long chain β-oxidation mRNA and protein. To our surprise, the metabolic changes observed occurred in conjunction with a hepatic innate immune response, as demonstrated by transcriptional profiling and flow cytometry. By employing an immunomodulation strategy to deplete Kupffer cells, we were able to improve the expression of multiple genes involved in β-oxidation. Based on these findings, we are the first to suggest that the role of the liver as an immunologic organ is central in the pathophysiology of hepatic metabolic decompensation in inborn errors of metabolism due to respiratory viral infection.
Keywords: β-oxidation, fatty acids, metabolism, innate immune response, influenza
1. INTRODUCTION
Although individually rare, the inborn errors of metabolism (IEM) constitute a medically important class of disorders that are increasingly recognized through state Newborn Screening programs [1]. Studies of the metabolic disturbances resulting from these monogenic disorders have played an important role in the elucidation of many critical biochemical pathways. Examples of these small molecule diseases include organic acidemias (OA), urea cycle disorders (UCD), primary mitochondrial disorders (pMD), and fatty acid oxidation disorders (FAOD). The untreated clinical course for most of these diseases ranges from mental retardation to coma and death, reinforcing the need for lifelong medical management [2–5]. This ongoing patient care requires judicious monitoring of growth and developmental parameters, as well as nutritional and biochemical status [2, 6, 7].
Despite close monitoring of metabolic and nutritional status, patients with IEM may experience a functional deterioration known as metabolic decompensation [8–10]. These periodic episodes are potentially life threatening, display high rates of morbidity [11], and can result in long-term sequelae [12]. During metabolic decompensation, disease-specific biochemical perturbations occur and include acidosis (e.g. OA, pMD), hyperammonemia (e.g. UCD) and hypoglycemia (e.g. FAOD). Common precipitants include fasting and infection, predominantly, respiratory viral infection. In a cohort of patients with UCD, we previously reported that infection was the most common cause of metabolic decompensation (i.e. hyperammonemia) and was associated with markers of increased morbidity when compared to other precipitants [11]. Based on these clinical data, we constructed a model system of metabolic decompensation due to influenza infection in spf-ash [13], a mouse model of ornithine transcarbamylase deficiency, the most common urea cycle disorder. Influenza infection in spf-ash resulted in a further disruption of urea cycle function. Interestingly, despite having no visible pathologic changes by histology, wild-type (WT) animals also had reduced mitochondrial urea cycle enzyme activities and hepatic urea cycle intermediates arginine and aspartate.
The liver is a critical organ in many IEM. Many of the life-threatening biochemical perturbations seen on clinical testing during metabolic decompensation (i.e. hyperammonemia, acidosis, hypoglycemia) can be traced to impaired intermediary metabolism in the liver, prompting us to reconsider this biochemical instability as hepatic metabolic decompensation. Since WT mice underwent hepatic metabolic adaptations in nitrogen metabolism during influenza infection, we hypothesized that hepatic metabolic decompensation due to infection involves aspects of normal hepatic physiology, to which, IEM are unable to adapt. To expand the phenotype of hepatic metabolic adaptations seen during infection, we carried out untargeted metabolic profiling of livers using a dual mass spectrometry platform in C57Bl/6 mice infected with PR8 influenza. These studies, combined with cellular immunology investigations, suggest that the innate immune system in the liver, particularly Kupffer cells, plays an important role in the modulation of hepatic metabolism during influenza infection and participates in hepatic metabolic decompensation in IEM.
2. MATERIALS AND METHODS
2.1. Infection with A/PR/8/34 (PR8)
The experiments outlined were performed on B6 (C57Bl/6, The Jackson Laboratory, Bar Harbor, ME) mice. Mice were housed in a pathogen-free facility, caged individually, had access to a 24% protein mush-based feed, Nutragel (Bio-Serv, Frenchtown, NJ), and autoclaved reverse osmosis water. Mice were kept in a temperature (22 ± 2˚C) and humidity (30–70%) controlled environment with a 12-hour light cycle. Mouse adapted human influenza virus A/PR/8/34 (PR8) for infection was produced as described previously [14]. Eight-week-old mice were exposed to an infective dose of PR8 (500 TCID50) in an aerosolization chamber (Glas-Col, Terre Haute, IN) [15]. Uninfected control mice were matched for food intake. Mice were sacrificed on Day 5 of infection by 5% Isoflurane inhalation with cervical dislocation. All tissues were isolated and stored at −80˚C until use. Single dose liposomal clodronate (200 μL/animal i.p., Encapsula NanoSciences LLC, Brentwood, TN) was used to deplete Kupffer cells. All animal care and procedures were carried out according to the criteria outlined in the “Guide for the Care and Use of Laboratory Animals” prepared by the National Academy of Sciences and published by the National Institutes of Health (NIH publication 86–23 revised 1985) and were authorized by the Animal Care and Use Committees of the National Human Genome Research Institute.
2.2. Viral titers
Viral titers in lungs and liver were determined using published assay based on the infection of MDCK cells [16]. The inverse of the dilution at which 50% of the wells showed cytopathic effect was recorded as the 50% tissue culture infectious dose (TCID50).
2.3. Kupffer cell isolation and stimulation
Kupffer cell isolation was performed as previously published [17]. Briefly, liver was dissected and incubated on 37°C with collagenase for 30 minutes. Digested tissue was passed through a mesh filter, resuspended in 33% percoll and centrifuged at 500 X g for 20 minutes. Red blood cells were lysed, and cells were washed and plated at 3 × 106 cell/mL. Adherent cells were stimulated with 1 μg/ml LPS (Sigma Aldrich, St. Louis, MO) for 24 hours. Cytokines were analyzed using a Cytometric Bead Array for mouse inflammatory markers on a FACSCalibur flow cytometer according to the manufacturers instructions (BD Biosciences, San Jose, CA).
2.4. Biochemical parameters
Glucose testing was performed on retro-orbital collected whole blood using standard handheld glucometer (AccuCheck, Roche Diagnostics, Indianapolis, Indiana). Serum aspartate aminotransferase and alanine aminotransferase were determined via dry-slide technology (VetTest 8008, IDEXX, Columbia, MO).
2.5. Histology
All histologic procedures were performed by an outside vendor (Histoserv, Germantown, MD) and included hematoxylin, eosin, and oil red o staining. Antibody for 3-nitrotyrosine was used for IHC nitrosylation studies (Cayman Chemical, Ann Arbor, MI) and was performed according to a standard protocol (http://www.abcam.com/tag/ihc%20protocols).
2.6. Electron microscopy
Mouse livers (1 mm3) were fixed overnight at 40C in 2% glutaraldehyde in 0.1M cacodylate buffer (pH 7.4) and washed with cacodylate buffer three times. The tissues were fixed with 2% OsO4 for two hours, washed again with 0.1M cacodylate buffer three times, washed with water and placed in 1% uranyl acetate for one hour. The tissues were subsequently serially dehydrated in ethanol and propylene oxide and embedded in EMBed 812 resin (Electron Microscopy Sciences, Hatfield, PA, USA). Thin sections, approx. 80 nm, were obtained by utilizing the Leica ultracut-UCT ultramicrotome (Leica, Deerfield, IL, USA) and placed onto 300 mesh copper grids and stained with saturated uranyl acetate in 50% methanol and then with lead citrate. The grids were viewed in the JEM-1200EXII electron microscope (JEOL Ltd, Tokyo, Japan) at 80kV and images were recorded on the XR611M, mid mounted, 10.5Mpixel, CCD camera (Advanced Microscopy Techniques Corp, Danvers, MA, USA).
2.7. Metabolomics
Sample processing and metabolite identification with relative quantitation was done by Metabolon (Research Triangle Park, NC) using proprietary methods on GC/MS and LC/MS/MS platforms [18]. Raw data is provided in Supplemental Table 1. Acylcarnitine profiles on tissue homogenates were performed Biochemical Genetics and Metabolism Laboratory, Children’s National Medical Center (Washington, DC).
2.8. mRNA expression profiling and qRT-PCR
RNA was extracted from homogenized liver tissue using a kit (Qiagen, Germantown, MD). For RT-PCR, 1 µg of RNA was reverse transcribed to cDNA using a modified MMLV-reverse transcriptase (iScript, Bio-Rad, Hercules, CA). Real-time quantitative PCR reactions were carried using TaqMan commercial primers (Applied Biosciences, Carlsbad, CA). All results were normalized to β-actin. Reactions were cycled and quantitated with an ABI 7500 Fast Real Time PCR System (Applied Biosystems, Foster City, CA).
2.9. Immunoblot
For western blot analysis, 30 μg of protein was loaded on 4–20% Tris-glycine polyacrylamide gels. The gels were transferred to polyvinylidene difluoride membrane using the iBlot Dry Blotting System (Life Technologies, Grand Island, NY). The membranes were blocked and probed with primary antibodies according to the manufacturers suggested dilutions: VLCAD, LCAD, HADHA (Abcam, Cambridge, MA), and β-actin (Sigma-Aldrich, St. Louis, MO). Incubation was done with appropriate secondary antibodies. Image analyses were performed using an Odyssey Imager (LiCor, Lincoln, NE).
2.10. Flow cytometry
Antibodies were purchased from BD Biosciences (San Jose, CA). Data were acquired on a FACSCalibur flow cytometer (BD Biosciences) and analyzed using FlowJo software (Tree Star, Ashland, OR). Hepatic immune cells were isolated by collagenase digestion of the liver as outlined above.
2.11. β-oxidation of palmitate
Palmitate oxidation was performed on Hep3B cells measuring oxygen consumption rates using a Seahorse XF24 Analyzer (Seahorse Bioscience, North Billerica, MA). Briefly, Hep3B cells (30,000/well) were incubated overnight with TNFα (100U/mL). Oxygen consumption rates were determined while using the XF Cell Mito Stress kit and XF Palmitate-BSA FAO Substrate (Seahorse Bioscience, North Billerica, MA) according to the manufacturers instructions.
2.12. Data analysis and statistics
Summary statistics were calculated for all data. Two-sided student’s t-test, Mann-Whitney t-test, Welch’s T-test and survival analysis was used when appropriate. P-values less than 0.05 were considered to indicate statistical significance. Baseline statistical analyses were performed using Prism 5 (GraphPad Software, Ja Jolla, CA) software package. A conservative approach was adopted to analyze Affymetrix gene arrays and metabolomics in order to minimize false positives. All analyses were performed using R v3.1.0 (http://www.R-project.org/.), Bioconductor v3.0, and oneChannelGUI v1.32 [19]. Quality control was performed by inspecting image representations of the microarrays for obvious defects and principal component analysis performed to verify reproducibility among replicates. Quantile normalization and RMA summarization was performed using an IQR filter of 0.25 to filter probesets that did not change significantly among samples. RankProduct with 500 permutations and limma were applied to obtain differentially expressed genes. Differentially expressed genes or metabolites were then selected as those significantly differentially expressed (Benjamini-Hochberg FDR < 0.05) for both methods and with fold change > 0.5. GO term analysis and networks were computed using ClueGO [20]. Plots were created using ggplot2 (http://ggplot2.org ) and the directlables package (http://directlabels.r-forge.r-project.org/).
3. RESULTS
3.1. Infection with PR8 influenza results in hepatitis in mice
Respiratory viral illness is a common cause of metabolic decompensation [11]. To study the effects of respiratory viral infection on the liver, we employed a model system previously published by our laboratory using the mouse-adapted influenza A/PR/8 (PR8) virus [13]. WT mice were infected on Day 0 with 500 TCID50 using an infection aerosolization apparatus and euthanized on Day 5 (Figure 1A and 1B). To control for nutritional confounders, infected and control animals were matched for food intake (pair-fed) and were euthanized on Day 5 (Figure 1B). Tissues were then collected, snap-frozen and stored at −80°C until analysis. Mice challenged with PR8 displayed signs of infection including reduced oral intake, ruffled fur and decreased social and grooming behaviors beginning at Day 2–3 (Figure 1B, gray shading), consistent with cytokine release [15]. In the lungs of infected animals, increased viral load was detected by qRT-PCR for non-structural protein (NS1) and by MDCK cell dilutions of viral titers (Figure 1C). Weight loss was similar on day 5, regardless of the experimental condition (Figure 1D) suggesting that the animals were well matched for intake. Along with these indicators of infection, significant increases in serum AST (2X, P < 0.01) and ALT (3X, P < 0.05) were seen, indicating hepatitis (Figure 1E). Along with this hepatitis, animals infected with PR8 influenza also dropped their blood glucose by approximately 30 μg/dL compared to pair-fed controls (Figure 1F, P < 0.01) consistent with increased glucose utilization during infection.
Figure 1. PR8 influenza infection in WT mice produces hepatitis.
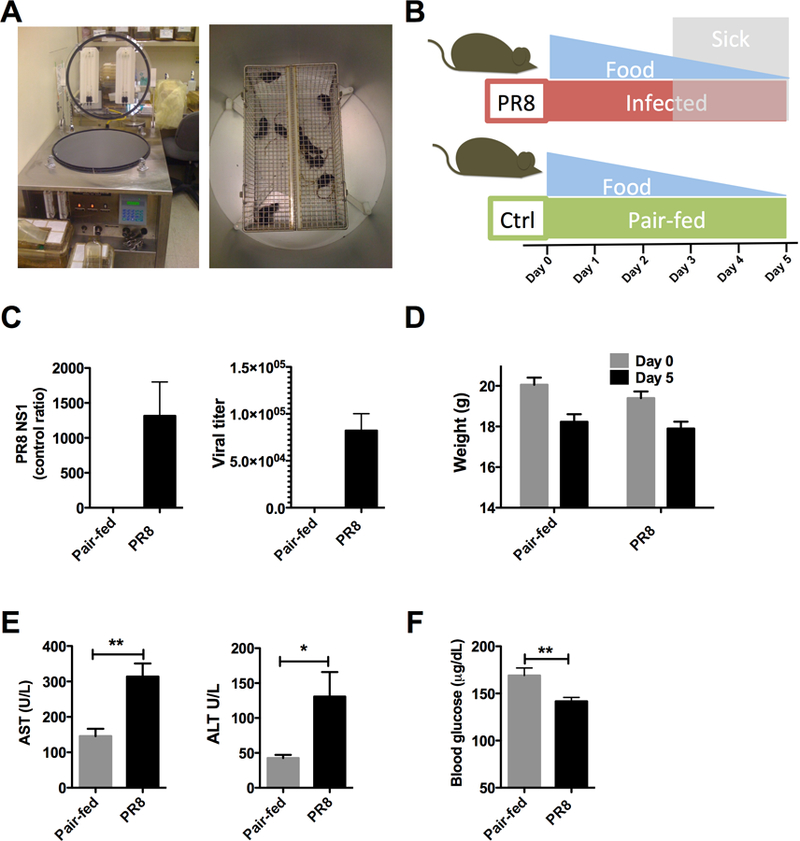
Mice infected with mouse adapted PR8 influenza were euthanized at Day 5 and tissues were harvested. (A) Inhalation infection apparatus (N = 10/group). (B) Study protocol. Shaded areas indicate appearance of sickness behaviors. Uninfected mice were matched for food intake (pair-fed) (C) Viral titers in lungs on Day 5 (N = 5–6 / group). qRT-PCR for influenza PR8 NS1 (left) and lung viral titers (right). (D) Weights for PR8 infection and pair-fed conditions. (F) Blood glucose. PR8 = influenza A/PR/8. Hatched bars indicated statistical significance. * < 0.05. ** < 0.01.
3.2. Liver metabolomics during PR8 infection show changes in fatty acids
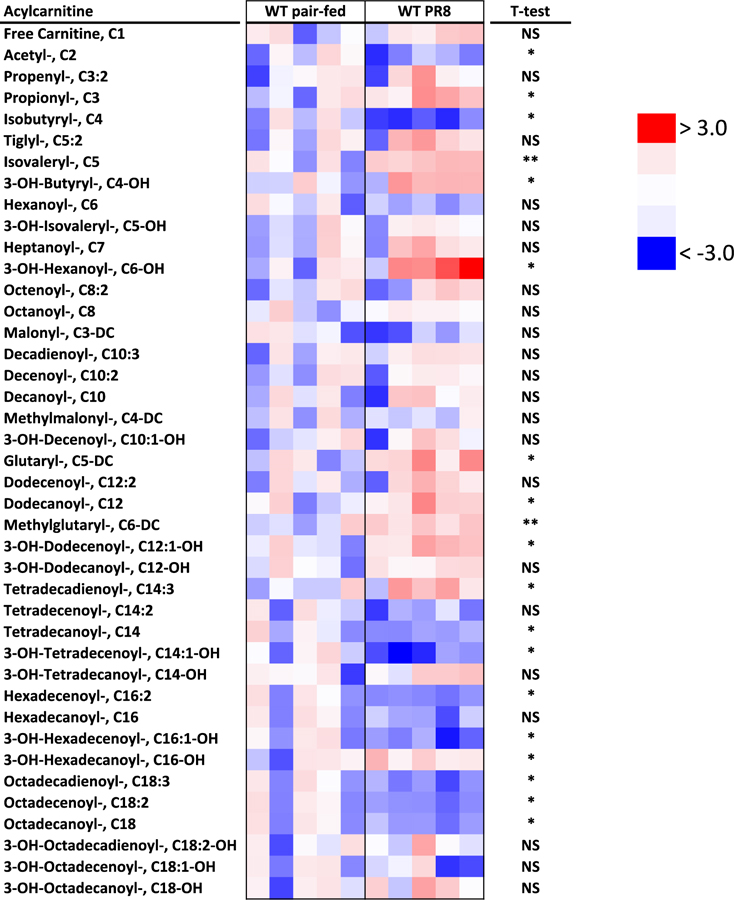
During PR8 infection, the observed depression in blood glucose implied impaired metabolic reserve. Metabolic reserve is maintained through glucose replenishing and sparing processes, gluconeogenesis and fatty acid oxidation, respectively. In order to characterize alterations in biochemical pathways that may impact metabolic reserve in infected mice, we conducted untargeted metabolomic studies in livers isolated from infected and pair-fed WT mice (Figure 2). Using a dual platform of gas chromatography mass spectrometry and liquid chromatography tandem mass spectrometry, 334 metabolites were measured comprising lipids, amino acids, dipeptides, cofactors, vitamins, and nucleotides (Supplemental Table 1). Approximately 81 metabolites were identified as significantly changed (P < 0.05) in infected mice. To characterize the metabolic response, the data were further categorized into 8 superpathways and 64 subpathways. At the superpathway level (Figure 2A), significant distinctions could not be made during infection, although in general, lipids exhibited decreased Log2 mean fold changes and a wider distribution of values compared to other categories. At the subpathway level (Figure 2B), finer distinctions between infected and pair-fed animals could be made. Lipids (pink boxplots) were noted as some of the most significantly impacted metabolites in this study. Bile acids were depressed, indicating decreased synthesis and liver dysfunction. Essential fatty acyl and long chain CoA species were also reduced, along with several metabolites involved in CoA synthesis (Supplemental Table 2). Focusing on long chain fatty acids (Figure 2C), arachidonate was elevated, consistent with inflammation, while C16-C22 long chain fatty acids were reduced. This depression in acyl CoA species also translated into a depression in long chain acylcarnitines, particularly C14, C16 and C18 chains (P < 0.05, Table 1). The relative reductions of fatty acid acyl CoA and carnitine esters in infected mice alluded to hepatic adaptations in mitochondrial β-oxidation of long chain fatty acids during infection.
Figure 2. Metabolic adaptations in the liver during PR8 influenza infection.
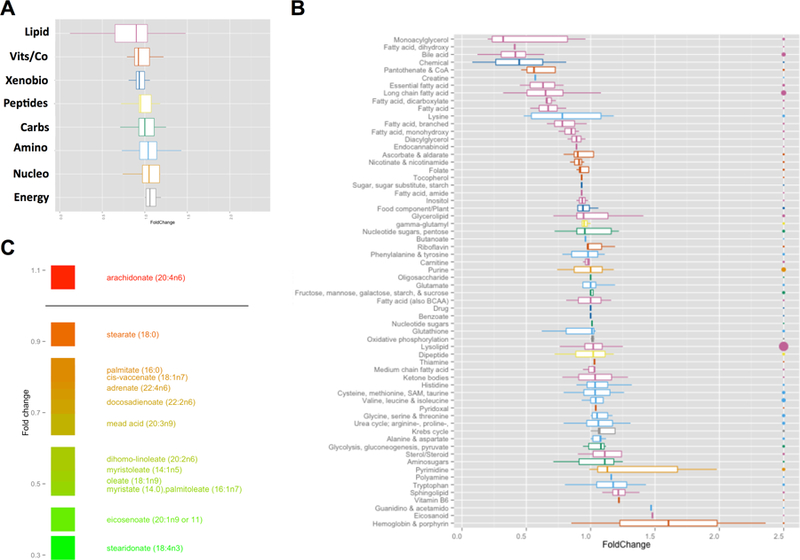
Livers were harvested from infected and pair-fed mice. Liver homogenates were normalized for protein content and analyzed using GC-MS and LC-MS/MS platforms. (A) Boxplots of Log2 fold changes for metabolic superpathways. Pink – lipids, Orange – vitamins and cofactors, Blue - xenobiotics, Yellow – peptides, Green – carbohydrates, L. blue – amino acids, L. orange – nucleotides, Grey – energy metabolites. (B) Boxplots of Log2 fold changes for metabolic subpathways. Color coding from (A) superpathways applies. Size of the bubble on the right axis represents number of metabolites for each subpathway. (C) Log2 fold changes for long chain fatty acids.
Table 1: Acylcarnitine profile of livers during infection with PR8 influenza.
Acylcarnitine profiles were performed in liver homogenates normalized for protein content. Metabolite values were converted to Z-scores and represented a heatmap based on their relative concentrations. *P < 0.05.
 |
3.3. PR8 infection is associated with a depression in components of long chain β- oxidation
Since infected animals had a metabolomic signature consistent with alterations in long chain fatty acids, we next examined the components of hepatic β-oxidation. Pair-fed and infected mice were euthanized at 5 days, and livers were harvested and snap frozen until use. During infection, multiple steps in the β-oxidation of long chain fatty acids were disrupted at the mRNA level (P < 0.05, Figure 3A) including the carnitine cycle (carnitine palmitoyl transferase 2 (Cpt2), carnitine acylcarnitine translocase (Cact)), fatty acid dehydrogenases (very long chain acyl coA dehydrogenase (Acadvl), long chain acyl coA dehydrogenase (Acadl), long chain hydroxyacyl coA dehydrogenase (Hadha) and the final common pathway responsible for feeding electrons to the respiratory chain, the electron transport flavoprotein (Etfa)). This depression in fatty acid oxidation transcripts coincided with decreased Pparα mRNA (P < 0.01), a positive regulator of β-oxidation mRNA expression (Figure 3B). Increased Fasn mRNA (P < 0.05) suggested that the liver had switched from β-oxidation to fatty acid synthesis (Figure 3C). The perturbations in β-oxidation could also be seen were also seen on the protein level (Figure 3D). VLCAD, LCAD and HADHA were reduced by ≥50% (P < 0.05). Overall, the mRNA and protein results, along with the reduction in blood glucose, suggest a mild depression of long chain β-oxidation during influenza infection in mice.
Figure 3. Fatty acid oxidation mRNA and protein is decreased during PR8 influenza infection.
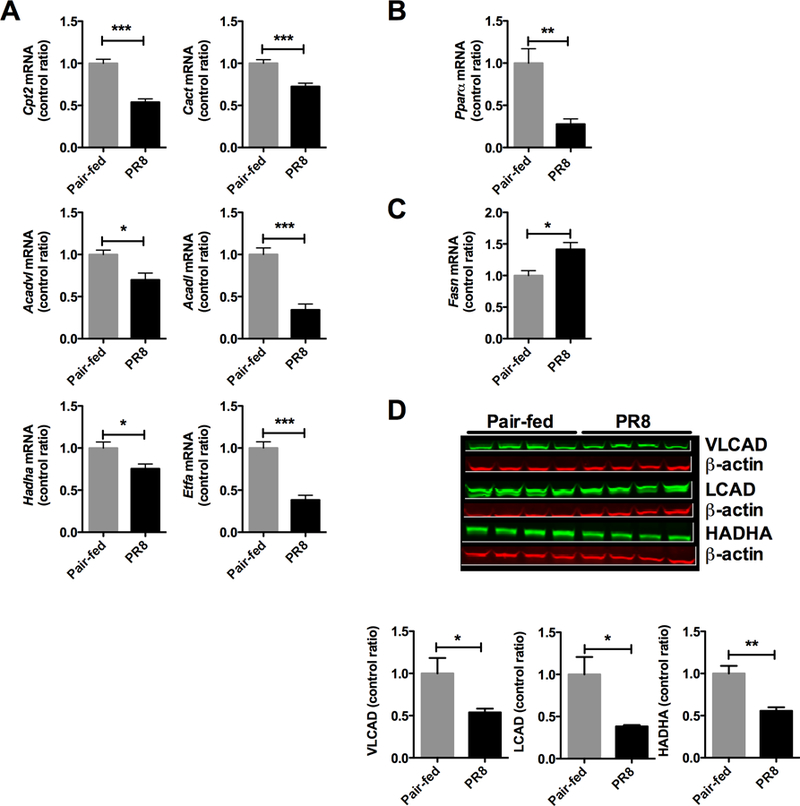
mRNA was extracted from infected and pair-fed livers and qRT-PCR was performed. (A) mRNA expression for components of the fatty acid oxidation pathway, Cpt2 – carnitine palmitoyl transferase 2, Cact – carnitine acylcarnitine translocase, Acadvl – very long chain acyl CoA dehydrogenase, Acadl – long chain acyl CoA dehydrogenase, Hadha – hydroxy acyl CoA dehydrogenase, Etfa – electron transport flavoprotein. (B) Pparα mRNA expression in livers, Pparα - peroxisome proliferator activated receptor alpha. (C) Fasn mRNA expression in livers, Fasn – fatty acid synthase. (D) Fatty acid oxidation proteins by immunoblot. * P < 0.05, ** P < 0.01, *** P< 0.001.
3.4. The hepatic transcriptional signature shows an antiviral response to influenza
In order to identify master regulators responsible for the metabolic changes seen, we performed transcriptional profiling of infected and pair-fed livers. Starting with stringent criteria (Figure 4A, P < 10−7), infection yielded 35 differentially upregulated genes (Supplemental Table 3). Not surprisingly, some of the highest values seen were related to the hepatic acute phase response: serum amyloid A1 (Saa1) and lipocalin 2 (Lcn2), which were confirmed by qRT-PCR (Figure 4B). In addition to the acute phase response, increased interferon response factor 7 (Irf7) was also observed suggestive of anti-viral innate immunity (Figure 5A-B). Gene ontology analysis (Figure 4C, Log2 > 1.5X, P < 0.01) confirmed that the innate immune response was activated in the liver. Several innate immune pathways were involved in the anti-viral response and included pattern recognition receptor signaling (toll like receptors, (TLRs)), viral nucleic acid sensing (RIG-I, MDA5) and interferon signaling (type I interferons). These results provide evidence for hepatic innate immune activation during PR8 influenza infection.
Figure 4. Transcriptional signature is consistent with an innate immune response in the liver.
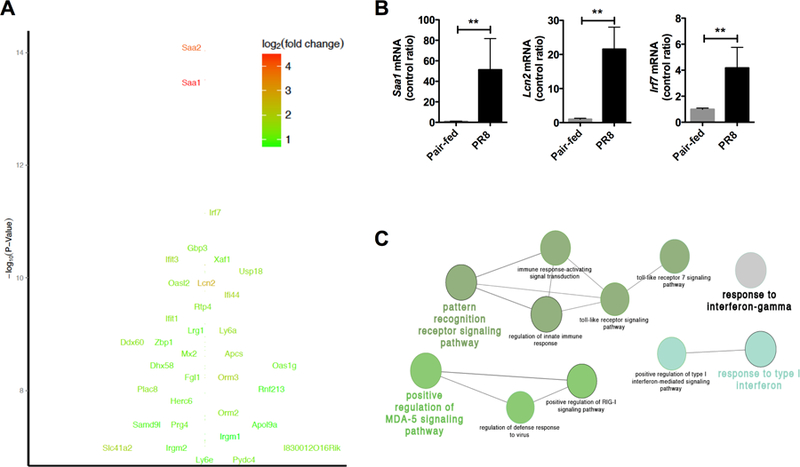
mRNA was extracted from infected and pair-fed livers and mRNA expression analysis and qRT-PCR was performed. (A) Log10 P-value of differentially expressed genes. (B) qRT-PCR confirmation of top three mRNA targets. (C) Transcriptional profile and gene ontology analysis of hepatic response. Circle size indicates number of genes with >1.5x increase and P < 0.01.
Figure 5. Innate immune activation in the liver during PR8 influenza infection.
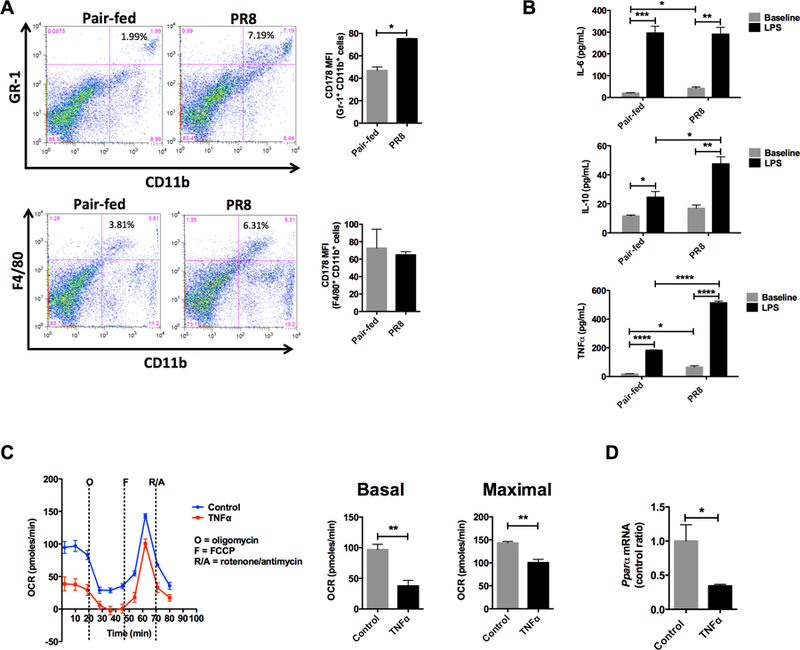
Collagenase digested livers were examined for immune cell populations. (A) Increased inflammatory myeloid cells (Gr-1+ CD11b+) and Kupffer cells (F4/80+ CD11b+) by flow cytometry. Representative flow cytometry (left figure). Fasl (CD178+) MFI on inflammatory macrophages and Kupffer cells (right figure). Experiments were repeated three or more times. (B) Cytokine production by adherent Kupffer cells, Il-6 – interleukin 6, Il-10 – interleukin 10, TNFα -tumor necrosis factor alpha. (C) Basal and maximal palmitate oxidation in Hep3B cells exposed to 24 hours of TNFα. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. All experiments were performed 3 or more times.
3.5. Innate immune activation in the liver with PR8 infection
As a result of the significant transcriptional signature suggesting hepatic sensing of influenza, we hypothesized that there were significant changes in hepatic immune cell populations. The liver is a highly immunologic organ with immune cells constituting 15% or more of the total cell count [21, 22]. To examine the changes in specific cell populations in the liver, we analyzed collagenase digested liver samples using flow cytometry. Cells of the adaptive immune system, T-cells and B-cells, showed little change with influenza infection (data not shown). In contrast, there were significant changes in innate immune cells. Inflammatory myeloid cells (Gr-1+ CD11b+) increased 3X or more, and tissue resident macrophages (F4/80+ CD11b+), known as Kupffer cells, increased approximately 2X with infection (Figure 5A). Also of note, the invading inflammatory myeloid cells (Gr-1+ CD11b+) were primed to induce tissue damage with increased FasL on their surface (Figure 5A, upper bar graph).
Kupffer cells play a central role in host defense as well as liver disease [23]. With increased numbers of Kupffer cells in the liver, we profiled their cytokine response in vitro. Supernatant from Kupffer cells from infected animals showed secretion of hepatotoxic inflammatory cytokines (IL-6 and TNFα) at baseline (Figure 5B). Evaluation of the full activation potential of Kupffer cells using LPS resulted in enhanced secretion of IL-6 and TNFα, as well as the anti-inflammatory cytokine IL-10. In order to assess the effects of the inflammatory cytokines produced by Kupffer cells on mitochondrial long chain β-oxidation, a liver cell line (Hep3B) was incubated with the prototypical inflammatory cytokine, TNFα. Following incubation with TNFα for 24 hours (Figure 5C), the basal (P < 0.01) and maximal (P < 0.01) oxidation of palmitate was depressed demonstrating impaired flux through the long chain β-oxidation pathway. Similar to our findings in the mouse livers during infection, Pparα mRNA was also decreased in Hep3B (Figure 5D).
Despite this robust hepatic immune response, the liver was free of replicating virus (data not shown) and histology was essentially normal. Hematoxylin and eosin stains (Supplemental Figure 1A) failed to show any gross abnormalities such as profound infiltrates or necrosis. Although small amounts of scattered microvesicular steatosis (Supplemental Figure 1B) were barely visible, this finding was not consistent. Mitochondrial number and morphology (Supplemental Figure 1C) were also noted to be normal by electron microscopy. However, with this normal histology, significant tyrosine nitration (3-nitrotyrosine) was located around the hepatic vessels as well as the parenchyma by immunohistochemistry (Supplemental Figure 2) suggestive of inducible and endothelial nitric oxide synthase activation (NOS) via inflammatory cytokines [24, 25].
3.6. Depletion of Kupffer cells modulates hepatic β-oxidation mRNA
Kupffer cells were increased in the liver during PR8 influenza infection and secreted inflammatory cytokines that inhibit fatty acid oxidation in vitro. In order to characterize the role of Kupffer cells in the modulation of hepatic long chain β-oxidation, we employed an immunomodulation strategy. Clodronate liposomes were given at Day 3 of infection, which corresponds to the appearance of illness, to deplete Kupffer cells [26]. Administration of clodronate liposomes resulted in successful depletion of Kupffer cells (Figure 6A) as determined by flow cytometry. Following this treatment, there was a recovery of Cpt2, Acadvl, Acadl, Hadha, and Etfa mRNAs during infection (P < 0.05, Figure 6B, upper and lower panels). VLCAD protein also increased, as measured by immunoblot (P < 0.05), while LCAD and HADHA remained unchanged (P > 0.05, Supplemental Figure 3). Consistent with the return of expression of multiple mRNAs for β-oxidation, Pparα mRNA (P < 0.05) also increased (Figure 6C). Despite this improvement in β-oxidation mRNA and VLCAD protein, blood glucose dropped even further (P < 0.001, Figure 6D). This further drop in blood glucose provides evidence for increased whole body glycolysis that cannot be compensated for due to an incomplete recovery of long chain β-oxidation.
Figure 6. Liposomal clodronate treatment restores expression of fatty acid oxidation mRNA.
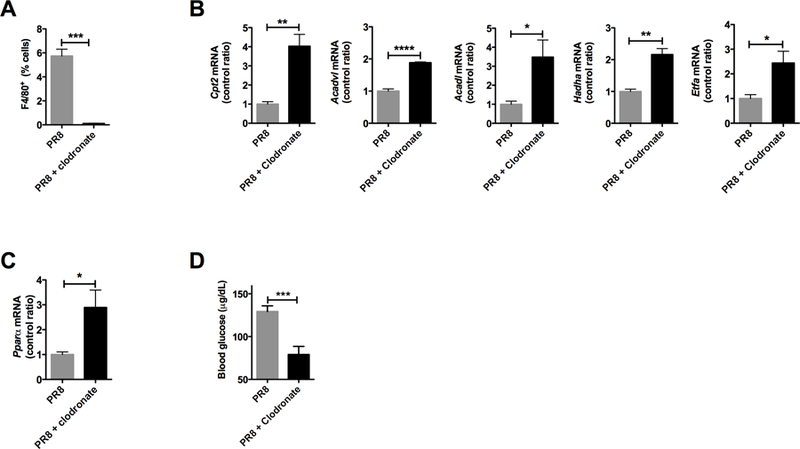
Animals infected with influenza A were injected with a single dose of liposomal clodronate to deplete Kupffer cells. (A) Flow cytometry confirms depletion of Kupffer cells. (B) mRNA expression for components of the fatty acid oxidation pathway. (C) Pparα mRNA expression. (D) Blood glucose during treatment with liposomal clodronate. *P < 0.05, **P < 0.01, *** P < 0.001, ****P < 0.0001. All experiments were performed 3 or more times.
4. DISCUSSION
Patients with IEM are at risk for developing metabolic decompensation during infectious illnesses. Many of the biochemical findings during metabolic decompensation can be traced directly to impaired intermediary metabolism in the liver [27]. As previously published by our group, WT mice exhibit inhibition of mitochondrial enzymes of the urea cycle with perturbations in urea cycle intermediates during influenza infection [13]. Our current study expands on this hepatic biochemical phenotype to include β-oxidation of long chain fatty acids. More importantly, we also identify a critical pathophysiologic process that may be a common thread in hepatic metabolic decompensation in IEM, that is, the activation of the innate immune system in the liver.
Fatty acid oxidation (FAO), also known as β-oxidation, occurs in the mitochondria of most tissues and involves the concerted effort of over 20 different enzymes [28]. This essential metabolic pathway provides the majority of energy required for daily functioning of the liver [29]. The first step in β-oxidation involves the transport of long chain fatty acids into the mitochondrial matrix via the carnitine shuttle, whereas medium and short chain fatty acids readily cross the mitochondrial membrane. Once in the mitochondrial matrix, acyl CoA dehydrogenases, displaying chain-length specificity, catalyze the breakdown of fatty acids [28]. By way of each turn of the β-oxidation cycle, acetyl CoA and NADH and FADH2 are produced. Acetyl CoA enters the tricarboxylic acid cycle, and NADH and FADH2 are fed to the electron transport chain driving ATP generation. During illness, glucose and fatty acids have an intimate relationship. β-oxidation of fatty acids plays a critical role in providing glucose-sparing metabolic reserve. The Randle cycle or glucose-fatty acid cycle, demonstrates this important concept: due to a deficiency in energy generation from fatty acids and ketones, glucose will be consumed [30].
In the studies presented herein, 3 days of clinical infection with PR8 influenza leads to a physiologic adaptation that includes a depression of β-oxidation intermediates: coenzyme A, long chain acyl-CoA and carnitine species, and acetyl-carnitine (Figure 2, Table 1). Due to the nature of the experimental design, the direction of these metabolites (i.e. production versus consumption) cannot be imparted from the data. However, given that multiple components of the long chain β-oxidation pathway were also decreased at the level of mRNA and protein (Figure 3), we can infer its compromise. This statement is further supported by the drop in blood glucose (Figure 1F), suggesting that the glucose-sparing effect of long chain β-oxidation is insufficient during influenza infection.
A significant question that arises from our studies concerns the physiologic significance of a depression in β-oxidation during infection. The mild depression in fatty acid oxidation seen during influenza infection may reflect limited hepatic metabolic flexibility due to energetic constraints [31–33]. Infection in general is an energetically costly process. Basal metabolic rate during infection increases by ≥13% for each 1° C, and can even approach 10,000 calories daily during sepsis [34, 35]. This hypermetabolic state places increased demands on metabolic resources and requires increased intake, which oftentimes cannot be met, as it occurs at a time when caloric intake is curtailed as a result of illness [35]. This systemic energy deficiency may also be compounded by limitations in liver bioenergetics. Unlike the heart, which has a > 10-fold dynamic range of ATP production, the liver has a dynamic range of ATP production that is just 2-fold [36]. Given this limited range of energetic flexibility, it is reasonable to conceive that the liver would prioritize certain cellular processes such as protein synthesis for the acute phase response versus intermediary metabolism: bile acid synthesis, β-oxidation of fatty acids (Figure 2B) and nitrogen metabolism [13]. This reprogramming of liver metabolism may be mediated through the repression of PPARα (Figure 3B), a known positive regulator of β-oxidation.
The current data on the hepatic metabolic response to infection has been reported in various sepsis models and vary according to the stage of sepsis, dosing and the pathogen involved. The early stress response to sepsis is marked by hyperglycemia resulting from the release of neuroendocrine hormones and increased hepatic gluconeogenesis [37]. In addition, fatty acid oxidation is also increased in the early stages of sepsis, also providing a glucose-sparing effect [38–40]. Our findings are in keeping with the data reported in the later stages of sepsis, which shows a depression of hepatic metabolic function. In a study of endotoxemic rats, the rate of hepatic CPT1a-dependent palmitate oxidation and ketogenesis were reduced at 24 hours [41]. This reduction in β-oxidation may be partially due to limited substrates. During pneumococcal sepsis with caloric deprivation, rats displayed reduced ketogenesis in the liver secondary to an insufficient supply of fatty acids [42]. Our data are consistent with these findings; livers of mice infected with PR8 influenza showed a reduction in fatty acyl CoA and carnitine esters (Figure 2, Supplemental Table 2, Table 1). In addition to the data similarities, our study design was also similar with respect to caloric restriction, and implies a complex interaction between infection and food restriction that is distinct from the physiology of uninfected pair-fed animals.
The systemic immune response to PR8 infection in B6 mice is well documented, particularly with regard to serum cytokine release. Beginning with the peak of lung viral titers at 48 hours, there is a significant rise in chemokines (KC, CCL2, RANTES), proinflammatory cytokines (IL-1β, IL-6, TNFα), and interferons (IFNβ), all of which continue to rise up to 120 hours post infection [15]. In addition to this systemic rise in inflammatory mediators, WT mice displayed a significant hepatic innate immune response, much to our surprise due to the location of the primary infection. Gene ontology of expression profiling of livers displayed a marked innate immune response signature with up-regulation of toll-like receptor and viral sensing pathways (Figure 4C). This was seen in conjunction with a hepatic increase in inflammatory myeloid and Kupffer cell populations (Figure 5A). Similar hepatic innate immune activation is also seen during other non-hepatic systemic infections: sinusoidal dilatation combined with an influx of inflammatory cells, primarily Kupffer cells [43].
Although the spleen is considered a critical mediator in the clearance of blood pathogens, these functions overlap with other tissues involved in immunosurveillance, particularly the liver. With its dual blood supply from the hepatic artery and portal vein, the liver receives 30% of the total blood volume per minute [44], making it an ideal organ for immunosurveillance. As the largest organ in the body, the liver contains the largest population of resident macrophages, Kupffer cells. Kupffer cells account for up to 35% of non-parenchmyal cells in the liver and 80–90% of all tissue macrophages in the body [45]. This heterogeneous population consists of large periportal and smaller mid-zonal/perivenous Kupffer cells. As a result of being exposed to incoming molecular signals due to their location in the periportal zone and sinusoids, Kupffer cells exhibit phagocytosis, lysosomal protease activity, and cytokine output [23]. Depletion of Kupffer cells by liposomal clodronate during L. monocytogenes and B. burgdorferi infections results in increased lethality, demonstrating their key role in immunity to blood born pathogens [46, 47].
Besides systemic immunity, Kupffer cells also play a role in hepatic injury. Hepatocytes adjacent to the sinusoids may undergo damage and apoptosis due to activation of Kupffer cells, a bystander effect [48]. This local tissue innate immune reaction is likely responsible for the increase in AST and ALT (Figure 1) seen during influenza infection in mice and humans [49]. Although influenza virus could not be isolated from the liver in our model, this does not exclude the possibility of periodic viremia activating Kupffer cells. This hypothesis is further supported by the innate anti-viral signature seen in the liver (Figure 4C). Our proposed pathophysiologic mechanism of bystander hepatocyte damage is also reinforced by depletion studies. The role of Kupffer cells in inflammatory hepatotoxicity has been demonstrated in several other experimental systems using liposomal clodronate: concanavalin, alcohol, and acetaminophen induced hepatic damage and non-alcoholic fatty liver disease [50]. In our model, depletion of Kupffer cells by liposomal clodronate led to an improvement in components of β-oxidation mRNA and some proteins (Figure 6B, Supplemental Figure 3) demonstrating their role in the modulation of fatty acid metabolism. However, the lack of a full recovery with liposomal clodronate treatment implies that the nutritional management of hepatic metabolic decompensation with intravenous glucose still plays a critical role. Moreover, the further depression in blood glucose due to Kupffer cell depletion suggests that the relationship between these tissue macrophages and liver metabolism is more complex and under recognized, prompting the need for additional investigation.
Although Kupffer cell hepatotoxicity may be mediated in several ways, cells isolated from infected mice produced increased IL-6, IL-10 and TNFα in our model system (Figure 5B). These cytokines likely play a part in the hepatitis seen (Figure 1E). In another experimental system of infection, TNFα, produced by Kupffer cells and inflammatory monocytes, is a critical mediator in immune mediated liver damage during Entamoeba histolytica infection [51]. Although we did not see an increase in apoptosis in the livers of infected animals, TNFα was likely an important mediator in the inhibition of β-oxidation (Figure 5C) acting via the repression of PPARα (Figure 5D and [52]).
The mechanisms by which proinflammatory cytokines inhibit long chain β-oxidation likely involve multiple signaling pathways, including excess nitric oxide and related free radicals [53]. S-nitrosylation or tyrosine nitration of metabolic enzymes may alter their metabolic activity or increase their turnover. Indeed, the livers from our infected mice showed increased periportal and parenchymal tyrosine nitration, consistent with inflammatory cytokine exposure (Supplemental Figure 2). In keeping with our findings, previous reports have also found that TNFα alone or in combination with IL-1 or IL-6, results in decreased hepatic fatty acid oxidation, ketone body production and acetyl CoA/CoA ratios in vitro and in vivo [54–56]. Interestingly, these same cytokines are responsible for the synthesis of hepatic acute phase proteins [57] suggesting that they may be an important regulator of the re-prioritization of metabolism in hepatocytes during infection. As a result, we propose that further studies targeting inflammatory cytokines like TNFα are necessary and may lead to potential therapeutic targets during hepatic metabolic decompensation.
5. CONCLUSIONS
In conclusion, our work has expanded the phenotype of hepatic metabolic adaptations to influenza infection. In WT mice, influenza infection with PR8 influenza leads to a decrease in intermediates and enzymes involved in hepatic β-oxidation, an important regulator of metabolic reserve. More importantly, our study is the first to suggest that the role of the liver as an immunologic organ is central in the pathophysiology of hepatic metabolic decompensation in IEM due to viral infection. The dual function of the liver as a major metabolic regulator and a secondary lymphoid organ responsible for immunosurveillance places this organ at risk for significant hepatotoxicity. This normal physiologic function is predicted to be deleterious in IEM. Based on this assertion, strategies aimed at modulating the hepatic innate immune response may be a viable target for intervention in the treatment of hepatic metabolic decompensation.
Supplementary Material
7. ACKNOWLEDGEMENTS
The authors would like to thank Dr. Les Biesecker for his support and the support of the Physician Scientist Development Program at NHGRI. Thanks to all the individuals who provided technical support and assistance including: Dr. Maryna Eichelberger (influenza virus), Dr. Abdel Elkahloun (Intramural Research Program Microarray Core Facility), and Dr. Shelley Hoogstraten-Miller and Ms. Irene Ginty (NHGRI Office of Laboratory Animal Medicine). Thanks to Dr. Charles Venditti for his editorial insights during preparation of this manuscript.
6. FUNDING
This work was supported by the Intramural Research Program of the National Institutes of Health.
Abbreviations:
- PR8
influenza A/PR/8
- IEM
inborn errors of metabolism
- OA
organic academia
- UCD
urea cycle disorder
- pMD
primary mitochondrial disease
- TCID
tissue culture infective dose
- MDCK
Madin-Darby canine kidney epithelial cells
- NS1
influenza non-structural protein
- CPT2
carnitine palmitoyl transferase 2
- CACT
carnitine acylcarnitine translocase
- Acadvl
very long chain acyl coA dehydrogenase
- Acadl
long chain acyl coA dehydrogenase
- Hadha
hydroxy acyl coA dehydrogenase
- Etfa
electron transport flavoprotein
- Pparα
peroxisome proliferator-activated alpha
- Fasn
fatty acid synthase
- Saa
serum amyloid A
- Lcn2
lipocalin 2
- Irf7
interferon response factor 7
- RIG-I
retinoic acid inducible gene I
- MDA-5
melanoma differentiation-associated protein 5
- TLR
toll like receptor
Footnotes
CONFLICT OF INTEREST
The authors have no conflicts of interest to declare.
9. REFERENCES
- [1].Marsden D, Larson C, Levy HL, Newborn screening for metabolic disorders, J Pediatr, 148 (2006) 577–584.e575. [DOI] [PubMed] [Google Scholar]
- [2].de Baulny HO, Benoist JF, Rigal O, Touati G, Rabier D, Saudubray JM, Methylmalonic and propionic acidaemias: management and outcome, J Inherit Metab Dis, 28 (2005) 415–423. [DOI] [PubMed] [Google Scholar]
- [3].Leonard JV, Walter JH, McKiernan PJ, The management of organic acidaemias: the role of transplantation, J Inherit Metab Dis, 24 (2001) 309–311. [DOI] [PubMed] [Google Scholar]
- [4].Singh RH, Rhead WJ, Smith W, Lee B, Sniderman King L, Summar M, Nutritional management of urea cycle disorders, Crit Care Clin, 21 (2005) S27–35. [DOI] [PubMed] [Google Scholar]
- [5].Summar M, Tuchman M, Proceedings of a consensus conference for the management of patients with urea cycle disorders, J Pediatr, 138 (2001) S6–10. [DOI] [PubMed] [Google Scholar]
- [6].Haberle J, Boddaert N, Burlina A, Chakrapani A, Dixon M, Huemer M, Karall D, Martinelli D, Crespo PS, Santer R, Servais A, Valayannopoulos V, Lindner M, Rubio V, Dionisi-Vici C, Suggested guidelines for the diagnosis and management of urea cycle disorders, Orphanet J Rare Dis, 7 (2012) 32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Singh RH, Nutritional management of patients with urea cycle disorders, J Inherit Metab Dis, 30 (2007) 880–887. [DOI] [PubMed] [Google Scholar]
- [8].Feinstein JA, O’Brien K, Acute metabolic decompensation in an adult patient with isovaleric acidemia, South Med J, 96 (2003) 500–503. [DOI] [PubMed] [Google Scholar]
- [9].Yen TY, Hwu WL, Chien YH, Wu MH, Lin MT, Tsao LY, Hsieh WS, Lee NC, Acute metabolic decompensation and sudden death in Barth syndrome: report of a family and a literature review, Eur J Pediatr, 167 (2008) 941–944. [DOI] [PubMed] [Google Scholar]
- [10].Yoshino M, Aoki K, Akeda H, Hashimoto K, Ikeda T, Inoue F, Ito M, Kawamura M, Kohno Y, Koga Y, Kuroda Y, Maesaka H, Murakamisoda H, Sugiyama N, Suzuki Y, Yano S, Yoshioka A, Management of acute metabolic decompensation in maple syrup urine disease: a multi-center study, Pediatr Int, 41 (1999) 132–137. [DOI] [PubMed] [Google Scholar]
- [11].McGuire PJ, Lee HS, Summar ML, Infectious precipitants of acute hyperammonemia are associated with indicators of increased morbidity in patients with urea cycle disorders, J Pediatr, 163 (2013) 1705–1710 e1701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Edmonds JL, Kirse DJ, Kearns D, Deutsch R, Spruijt L, Naviaux RK, The otolaryngological manifestations of mitochondrial disease and the risk of neurodegeneration with infection, Archives of otolaryngology--head & neck surgery, 128 (2002) 355–362. [DOI] [PubMed] [Google Scholar]
- [13].McGuire PJ, Tarasenko TN, Wang T, Levy E, Zerfas PM, Moran T, Lee HS, Bequette BJ, Diaz GA, Acute metabolic decompensation due to influenza in a mouse model of ornithine transcarbamylase deficiency, Dis Model Mech, 7 (2014) 205–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Fernandez-Sesma A, Marukian S, Ebersole BJ, Kaminski D, Park MS, Yuen T, Sealfon SC, Garcia-Sastre A, Moran TM, Influenza virus evades innate and adaptive immunity via the NS1 protein, J Virol, 80 (2006) 6295–6304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Moltedo B, Lopez CB, Pazos M, Becker MI, Hermesh T, Moran TM, Cutting edge: stealth influenza virus replication precedes the initiation of adaptive immunity, Journal of immunology, 183 (2009) 3569–3573. [DOI] [PubMed] [Google Scholar]
- [16].Oh S, McCaffery JM, Eichelberger MC, Dose-dependent changes in influenza virus-infected dendritic cells result in increased allogeneic T-cell proliferation at low, but not high, doses of virus, J Virol, 74 (2000) 5460–5469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Zeng WQ, Zhang JQ, Li Y, Yang K, Chen YP, Liu ZJ, A new method to isolate and culture rat kupffer cells, PloS one, 8 (2013) e70832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Ghazalpour A, Bennett BJ, Shih D, Che N, Orozco L, Pan C, Hagopian R, He A, Kayne P, Yang WP, Kirchgessner T, Lusis AJ, Genetic regulation of mouse liver metabolite levels, Molecular systems biology, 10 (2014) 730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Sanges R, Cordero F, Calogero RA, oneChannelGUI: a graphical interface to Bioconductor tools, designed for life scientists who are not familiar with R language, Bioinformatics, 23 (2007) 3406–3408. [DOI] [PubMed] [Google Scholar]
- [20].Bindea G, Mlecnik B, Hackl H, Charoentong P, Tosolini M, Kirilovsky A, Fridman WH, Pages F, Trajanoski Z, Galon J, ClueGO: a Cytoscape plug-in to decipher functionally grouped gene ontology and pathway annotation networks, Bioinformatics, 25 (2009) 1091–1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Crispe IN, The liver as a lymphoid organ, Annual review of immunology, 27 (2009) 147–163. [DOI] [PubMed] [Google Scholar]
- [22].Nemeth E, Baird AW, O’Farrelly C, Microanatomy of the liver immune system, Seminars in immunopathology, 31 (2009) 333–343. [DOI] [PubMed] [Google Scholar]
- [23].Bilzer M, Roggel F, Gerbes AL, Role of Kupffer cells in host defense and liver disease, Liver international : official journal of the International Association for the Study of the Liver, 26 (2006) 1175–1186. [DOI] [PubMed] [Google Scholar]
- [24].Barsacchi R, Perrotta C, Bulotta S, Moncada S, Borgese N, Clementi E, Activation of endothelial nitric-oxide synthase by tumor necrosis factor-alpha: a novel pathway involving sequential activation of neutral sphingomyelinase, phosphatidylinositol-3’ kinase, and Akt, Molecular pharmacology, 63 (2003) 886–895. [DOI] [PubMed] [Google Scholar]
- [25].Nandi J, Saud B, Zinkievich JM, Yang ZJ, Levine RA, TNF-alpha modulates iNOS expression in an experimental rat model of indomethacin-induced jejunoileitis, Mol Cell Biochem, 336 (2010) 17–24. [DOI] [PubMed] [Google Scholar]
- [26].Sturm E, Havinga R, Baller JF, Wolters H, van Rooijen N, Kamps JA, Verkade HJ, Karpen SJ, Kuipers F, Kupffer cell depletion with liposomal clodronate prevents suppression of Ntcp expression in endotoxin-treated rats, J Hepatol, 42 (2005) 102–109. [DOI] [PubMed] [Google Scholar]
- [27].Dixon MA, Leonard JV, Intercurrent illness in inborn errors of intermediary metabolism, Arch Dis Child, 67 (1992) 1387–1391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Goetzman ES, Modeling disorders of fatty acid metabolism in the mouse, Progress in molecular biology and translational science, 100 (2011) 389–417. [DOI] [PubMed] [Google Scholar]
- [29].Eaton S, Bartlett K, Pourfarzam M, Mammalian mitochondrial beta-oxidation, Biochem J, 320 ( Pt 2) (1996) 345–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Houten SM, Herrema H, te Brinke H, Denis S, Ruiter JPN, van Dijk TH, Argmann CA, Ottenhoff R, Muller M, Groen AK, Kuipers F, Reijngoud DJ, Wanders RJA, Impaired amino acid metabolism contributes to fasting-induced hypoglycemia in fatty acid oxidation defects, Hum Mol Genet, 22 (2013) 5249–5261. [DOI] [PubMed] [Google Scholar]
- [31].Kimmig R, Mauch TJ, Kerzl W, Schwabe U, Scholz R, Actions of glucagon on flux rates in perfused rat liver. 1. Kinetics of the inhibitory effect on glycolysis and the stimulatory effect on glycogenolysis, European journal of biochemistry / FEBS, 136 (1983) 609–616. [DOI] [PubMed] [Google Scholar]
- [32].Lindberg B, Darle N, The effect of glucagon and blood transfusion on hepatic circulation and oxygen consumption in hemorrhagic shock, J Surg Res, 23 (1977) 257–263. [DOI] [PubMed] [Google Scholar]
- [33].Soboll S, Regulation of energy metabolism in liver, Journal of bioenergetics and biomembranes, 27 (1995) 571–582. [DOI] [PubMed] [Google Scholar]
- [34].Dubois EF, The Mechanism of Heat Loss and Temperature Regualtion Stanford University Press, Stanford, CA, 1937. [Google Scholar]
- [35].Borgen L, Total parenteral nutrition in adults, The American journal of nursing, 78 (1978) 224–228. [PubMed] [Google Scholar]
- [36].Phillips D, Covian R, Aponte AM, Glancy B, Taylor JF, Chess D, Balaban RS, Regulation of oxidative phosphorylation complex activity: effects of tissue-specific metabolic stress within an allometric series and acute changes in workload, Am J Physiol Regul Integr Comp Physiol, 302 (2012) R1034–1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Van Cromphaut SJ, Hyperglycaemia as part of the stress response: the underlying mechanisms, Best practice & research. Clinical anaesthesiology, 23 (2009) 375–386. [DOI] [PubMed] [Google Scholar]
- [38].Yamamoto T, Rat liver peroxisomal and mitochondrial fatty acid oxidation in sepsis, Surgery today, 23 (1993) 137–143. [DOI] [PubMed] [Google Scholar]
- [39].Whelan SP, Carchman EH, Kautza B, Nassour I, Mollen K, Escobar D, Gomez H, Rosengart MA, Shiva S, Zuckerbraun BS, Polymicrobial sepsis is associated with decreased hepatic oxidative phosphorylation and an altered metabolic profile, Journal of Surgical Research, 186 (2014) 297–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Tappy L, Chiolero R, Substrate utilization in sepsis and multiple organ failure, Critical care medicine, 35 (2007) S531–534. [DOI] [PubMed] [Google Scholar]
- [41].Takeyama N, Itoh Y, Kitazawa Y, Tanaka T, Altered hepatic mitochondrial fatty acid oxidation and ketogenesis in endotoxic rats, Am J Physiol, 259 (1990) E498–505. [DOI] [PubMed] [Google Scholar]
- [42].Wannemacher RW Jr., Pace JG, Beall RA, Dinterman RE, Petrella VJ, Neufeld HA, Role of the liver in regulation of ketone body production during sepsis, The Journal of clinical investigation, 64 (1979) 1565–1572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Srivastava B, Gimson A, Hepatic changes in systemic infection, Best Pract Res Clin Gastroenterol, 27 (2013) 485–495. [DOI] [PubMed] [Google Scholar]
- [44].Dancygier H, The Liver as an Immune Organ, Clinical Hepatology, Vol 1, (2010) 141–152. [Google Scholar]
- [45].Jenne CN, Kubes P, Immune surveillance by the liver, Nature immunology, 14 (2013) 996–1006. [DOI] [PubMed] [Google Scholar]
- [46].Ebe Y, Hasegawa G, Takatsuka H, Umezu H, Mitsuyama M, Arakawa M, Mukaida N, Naito M, The role of Kupffer cells and regulation of neutrophil migration into the liver by macrophage inflammatory protein-2 in primary listeriosis in mice, Pathology international, 49 (1999) 519–532. [DOI] [PubMed] [Google Scholar]
- [47].Lee WY, Moriarty TJ, Wong CH, Zhou H, Strieter RM, van Rooijen N, Chaconas G, Kubes P, An intravascular immune response to Borrelia burgdorferi involves Kupffer cells and iNKT cells, Nature immunology, 11 (2010) 295–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Polakos NK, Cornejo JC, Murray DA, Wright KO, Treanor JJ, Crispe IN, Topham DJ, Pierce RH, Kupffer cell-dependent hepatitis occurs during influenza infection, Am J Pathol, 168 (2006) 1169–1178; quiz 1404–1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Adams DH, Hubscher SG, Systemic viral infections and collateral damage in the liver, Am J Pathol, 168 (2006) 1057–1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Baffy G, Kupffer cells in non-alcoholic fatty liver disease: the emerging view, J Hepatol, 51 (2009) 212–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Helk E, Bernin H, Ernst T, Ittrich H, Jacobs T, Heeren J, Tacke F, Tannich E, Lotter H, TNFalpha-mediated liver destruction by Kupffer cells and Ly6Chi monocytes during Entamoeba histolytica infection, PLoS pathogens, 9 (2013) e1003096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Kim MS, Sweeney TR, Shigenaga JK, Chui LG, Moser A, Grunfeld C, Feingold KR, Tumor necrosis factor and interleukin 1 decrease RXR alpha, PPAR alpha, PPAR gamma, LXR alpha, and the coactivators SRC-1, PGC-1 alpha, and PGC-1 beta in liver cells, Metabolism-Clinical and Experimental, 56 (2007) 267–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Kitade H, Kanemaki T, Sakitani K, Inoue K, Matsui Y, Kamiya T, Nakagawa M, Hiramatsu Y, Kamiyama Y, Ito S, Okumura T, Regulation of energy metabolism by interleukin-1beta, but not by interleukin-6, is mediated by nitric oxide in primary cultured rat hepatocytes, Biochim Biophys Acta, 1311 (1996) 20–26. [DOI] [PubMed] [Google Scholar]
- [54].Beylot M, Vidal H, Mithieux G, Odeon M, Martin C, Inhibition of hepatic ketogenesis by tumor necrosis factor-alpha in rats, Am J Physiol, 263 (1992) E897–902. [DOI] [PubMed] [Google Scholar]
- [55].Nachiappan V, Curtiss D, Corkey BE, Kilpatrick L, Cytokines inhibit fatty acid oxidation in isolated rat hepatocytes: synergy among TNF, IL-6, and IL-1, Shock, 1 (1994) 123–129. [DOI] [PubMed] [Google Scholar]
- [56].Pailla K, Lim SK, De Bandt JP, Aussel C, Giboudeau J, Troupel S, Cynober L, Blonde-Cynober F, TNF-alpha and IL-6 synergistically inhibit ketogenesis from fatty acids and alpha-ketoisocaproate in isolated rat hepatocytes, JPEN J Parenter Enteral Nutr, 22 (1998) 286–290. [DOI] [PubMed] [Google Scholar]
- [57].Bauer M, Press AT, Trauner M, The liver in sepsis: patterns of response and injury, Current opinion in critical care, 19 (2013) 123–127. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.