

Abstract
The stem cell transcription factor Sox2 is widely recognized for its many roles during normal development and cancer. Over the last several years, it has become increasingly evident that Sox2 dosage plays critical roles in both normal and malignant cells. The work described in this review indicates that the dosage of Sox2 influences cell fate decisions made during normal mammalian development, as well as cell fate decisions in cancer, including those that influence the tumor cell of origin and progression of the cancer. Equally important, Sox2 dosage is a key determinant in the proliferation both normal cells and tumor cells, where proliferation is restricted in Sox2high cells. Collectively, the studies reviewed here indicate that tumor cells utilize the fundamental effects of Sox2 dosage to suit their own needs. Finally, we speculate that elevated expression of Sox2 helps establish and maintain tumor dormancy in Sox2-positive cancers.
Keywords: Sox2, development, cancer, dormancy, quiescence
Introduction
Interest in the stem cell transcription factor Sox2 has grown significantly over the past 10 years. Besides its diverse roles during mammalian development, Sox2 plays critical roles in both normal adult cells and tumor cells (Sarkar and Hochedlinger, 2013; Weina and Utikal, 2014). The essential roles of Sox2 first came to light with the discovery that Sox2 null embryos perish at the peri-implantation stage of development (Avillion et al., 2003). Shortly thereafter, Sox2 was found to be required for the self-renewal and pluripotency of embryonic stem (ES) cells (Chew et al., 2005). Interest in Sox2 soared after the paradigm-shifting discovery of Yamanaka and co-workers that Sox2 along with Oct4, Klf4, and c-Myc can reprogram somatic cells into a pluripotent stem cell state (induced pluripotent stem cells) (Takahashi and Yamanaka, 2006). With the recognition that Sox2 is a major player in stem cell biology and the growing interest in cancer stem cells, numerous reports appeared over the past 10 years describing the connection between Sox2 and more than twenty different types of human cancer (Wuebben and Rizzino, 2017).
Much has been learned about Sox2 since its discovery over 20 years ago. Sox2 belongs to the SRY-related gene family, which comprises ~20 members. Each member of this family contains a well-conserved high mobility group domain that mediates DNA binding. Sox2 has several unusual features. The Sox2 gene locus does not contain an intron and its single exon is located within a gene desert and present in an intron of a long non-coding RNA, Sox2-OT (Sox2-overlapping transcript). Highly relevant to the subject of this review, Sox2 expression is controlled at multiple levels, which enables its dosage to be tightly controlled. Sox2 transcription is regulated by differential utilization of over 20 enhancers, including a super-enhancer located ~100 kilobases downstream of the single Sox2 exon. Sox2 expression is also controlled at the transcriptional and post-transcriptional levels by a large array of non-coding RNAs, including Sox2-OT, and over 15 microRNAs. Additionally, Sox2 stability, subcellular localization, and function are controlled by six different types of post-translational modifications dispersed across at least a dozen of its 319 amino acids (human SOX2 is encoded by 317 amino acids). Readers interested in details regarding the diverse regulatory mechanisms that control Sox2 expression and function are directed to an earlier review (Wuebben and Rizzino, 2017). In this review we distinguish between reports investigating mouse and human Sox2, by referring to Sox2 for mouse studies and SOX2 for human studies. Sox2 is used when discussing its general properties.
Although much has been learned about Sox2 over the past 20 years, many basic questions remain unanswered. To provide a deeper understanding of Sox2, this review focuses on a fundamental property of Sox2; namely, its function in both normal and tumor cells is highly dosage dependent. In the first two sections below, we review studies demonstrating that the levels of Sox2 affect key cell fate decisions during development, and the decision of fetal cells to proliferate or remain quiescent. In the latter two sections, we review studies demonstrating parallel effects of Sox2 in tumor cells where its dosage influences both tumor cell fate decisions, and the balance between tumor cell quiescence and proliferation. In the Conclusion section, we discuss several important questions awaiting answers as we move forward in our understanding of how Sox2 controls key properties of both normal and tumor cells, including the possibility that elevated expression of Sox2 is a significant factor in establishing and maintaining tumor dormancy in Sox2-positive cancers.
Sox2 Dosage Influences Cell Fate Decisions During Development:
Work with mouse embryonic stem (ES) cells provided the first indication that the levels of stem cell transcription factors, such as Sox2, need to be very carefully regulated. The self-renewal and pluripotency of these cells are strictly dependent on several transcription factors, including Sox2 and Oct4. In 2000, Niwa et al. reported that either small decreases or small increases in Oct4 would induce the differentiation of ES cells (Niwa, Miyazaki, and Smith, 2000). Particularly surprising was the finding that increasing Oct4 ~50% above basal levels in ES cells induced their differentiation into cells that expressed markers of mesoderm and primitive extraembryonic endoderm; whereas, reducing the levels of Oct4 led ES cells to differentiate into cells that express markers of trophectoderm. Subsequently, our laboratory demonstrated that increasing the levels of Sox2 in ES cells also changed their fate (Kopp et al., 2008). Increasing Sox2 (~2-fold or less) with the aid of an inducible promoter in ES cells induced their differentiation into multiple cell types that expressed markers of neuroendoderm, mesoderm and trophectoderm. Several years later, the levels of Oct4 and Sox2 levels were found to influence the efficiency of reprogramming somatic cells into induced pluripotent stem cells (Papapetrou et al., 2009; Yamaguchi et al., 2011; Carey et al., 2011). In the case of Sox2, elevating its expression reduced reprogramming efficiency while increasing the frequency at which partially reprogrammed cells were generated (Yamaguchi et al., 2011; Carey et al., 2011).
Proper regulation of Sox2 not only alters the fate of ES cells, it is also required for major cell fate decisions during mammalian development. One of the first indications that Sox2 levels may be critical during development was the finding that ~10% of humans with severe eye defects (anophthalmia/absent eye and severe microphathalmia/small eye) have SOX2 mutations (Fantes et al., 2003; Fitzpatrick et al., 2005; Hagstrom et al., 2005; Ragge et al., 2005; Zenteno et al., 2005). SOX2 mutations have also been linked to defects in human brain development, in particular hippocampal and parahippocampal malformations (Sisodiya et al., 2006). Shortly after defects in human eye and brain development were reported, Taranova et al. demonstrated a cause-and-effect relationship between aberrantly low Sox2 expression and defects in eye development (Taranova et al., 2006). These workers observed that conditional ablation of Sox2 in distal retinal progenitor cells of the embryo (E13.5) resulted in severe defects in the developing eye, reminiscent of microphthalmia. Similarly, using two different genetically engineered Sox2 hypomorphic mutant mouse models, they demonstrated that reduction of Sox2 below 40% of normal in the developing eye led to microphthalmia as a result of defects in progenitor cell differentiation.
Correct Sox2 levels are also critical for other developing organs, including the stomach, esophagus and lungs. Sox2 expression was first reported in the developing gut and lung epithelium in the chick (Ishii et al., 1998). Later, Que et al. demonstrated that abnormally low Sox2 expression alters cell fate decisions in these tissues. In this study, Sox2 was shown to be expressed at high levels in regions of the foregut endoderm that give rise to the anterior stomach and the esophagus (Que et al., 2007). Although mice with one Sox2 allele disrupted appear to develop normally, mice that expressed hypomorphic Sox2 alleles, where Sox2 expression dropped below 50% of normal, exhibited esophageal atresia and tracheoesophageal fistula where the trachea and esophagus do not separate (Que et al., 2007). In mice that express even lower levels of Sox2 (~20% compared to control mice) the esophagus was abnormal and tracheoesophageal fistula did not occur. There were also defects in the developing stomach when Sox2 levels were abnormally low. In the developing stomach, Sox2 is widely expressed, but is restricted to the anterior stomach before birth. Consistent with this pattern of expression, Que et al. observed that all hypomorphic Sox2 embryos exhibit forestomach abnormalities characteristic of the posterior stomach (Que et al., 2007).
Aberrant Sox2 expression also leads to defects in lung development (Gontan et al., 2008). In the developing lung, Sox2 is expressed at high levels in the non-branching regions of primary lung bud, but is not detectable in the branching regions. To study the roles of Sox2 during lung development, Gontan et al. developed a mouse model where Sox2 could be elevated in the airway epithelium with the aid of a doxycycline (Dox) inducible transgene (Gontan et al., 2008). In this model, Sox2 is overexpressed when Dox binds to the reverse tet-trans-activator, which is driven by a promoter (SPC) that is expressed in the epithelial cells of the developing lung. Addition of Dox during development resulted in the death of mice after birth, because they are unable to oxygenate their blood. This study demonstrated that elevating Sox2 prevented branching in the developing airway driving cells into a committed progenitor state, which blocked their responses to signals that induce branching. Additionally, this study reported that overexpression of Sox2 increased the number of neuroendocrine basal cells as well as cells that resemble a gastric/intestinal pattern of expression. Collectively, the studies described in this section demonstrate that abnormal Sox2 expression, either too little or too much, adversely affects cell fate decisions during development (Table 1).
Table 1.
Effects of Sox2 Dosage on Cell Fate Decisions
| Tissue | Sox2 Levels | Outcome | Reference |
|---|---|---|---|
| Embryonic Stem Cells | Decreased | Differentiation | Chew et al., 2005 |
| Embryonic Stem Cells | Increased | Differentiation | Kopp et al., 2008 |
| Somatic Cells | Increased | Reprogramming to induced pluripotent stem cells | Takahashi and Yamanaka, 2006; Papapetrou et al., 2009; Yamaguchi et al., 2011; Carey et al., 2011 |
| Eye | Decreased | Microphthalmia | Fantes et al., 2003; Fitzpatrick et al., 2005; Hagstrom et al., 2005; Ragge et al., 2005; Zenteno et al., 2005 |
| Esophagus | Decreased | Esophageal atresia, tracheoesophageal fistula | Que et al., 2007 |
| Forestomach | Decreased | Develop characteristics of posterior stomach | Que et al., 2007 |
| Airway Epithelium | Increased | Commitment to progenitor state, decreased branching, increased neuroendocrine basal cells | Gontan et al., 2008 |
| Lung Tumorigenesis | Increased | Squamous lung cancers originating from bronchiolar cells | Xu et al., 2013 |
| Lung Tumorigenesis | Decreased | Adenocarcinomas originating from alveolar cells | Xu et al., 2013 |
| Prostate Cancer | Increased | Castration resistance and increased expression of neuroendocrine markers | Mu et al., 2017 |
Sox2 Dosage Influences Cell Proliferation During Development
In addition to critical roles in cell fate decisions, Sox2 levels also influence cell proliferation in many tissues during development, including cells of the developing central nervous system, eye, stomach and lung. Studies from the Pevny laboratory demonstrated that altering Sox2 levels influences cell proliferation in the developing central nervous system and in postnatal eye (Hutton and Pevny, 2011; Suzenko et al., 2013). The relationship between high Sox2 levels and cell quiescence was developed even further by the Muhr laboratory. In 2014, Hagey and Muhr reported that slow cycling radial glia cells in the embryonic cortex, which exhibit stem-like properties (Hutton and Pevny, 2011), express higher levels of Sox2 than their more proliferative progeny (intermediate progenitors cells), which are committed to differentiate (Hagey and Muhr, 2014). Similarly, they observed that Sox2high cells in subventricular zone of the brain are less likely to incorporate BrdU. Importantly, they demonstrated a causative connection between higher levels of Sox2 and cell quiescence by overexpressing and knocking down Sox2 in the ventricular zone of the developing embryo. Relative to their controls, overexpression of Sox2 substantially reduced incorporation of BrdU; whereas, knocking down of Sox2 by shRNA increased the percentage of cells that incorporated BrdU.
More recently, Hagey et al. extended the relationship between elevated Sox2 and cell quiescence by studying several endoderm-derived organs, specifically the developing stomach, lung, esophagus, and spinal cord (Hagey et al., 2018). Although Sox2-positive cells in these tissues proliferate, Hagey et al. determined that murine cells in these tissues, which express higher levels of Sox2, are less proliferative than other cells in the tissue that express lower levels of Sox2. The inverse relationship between Sox2 expression and BrdU incorporation is particularly striking in the developing stomach and spinal cord (see Figure 1B and Figure 4A, respectively, in their study). Moreover, they reported that expression of a dominant-negative form of Sox2 in the developing mouse stomach endoderm and in the developing spinal cord of the chick increased in BrdU incorporation. Conversely, overexpression of Sox2 decreased BrdU incorporation in the developing mouse stomach endoderm and in the developing spinal cord of the chick. Thus, Sox2 levels help determine the proliferative status of cells in at least four developing tissues. Interestingly, the study by Hagey et al. also noted that in both neural and endoderm cells, genes bound by Sox2 were enriched for gene ontology terms related to stem cell proliferation. This raises the possibility that high Sox2 levels limit cell proliferation by similar mechanisms in multiple developing cell types.
Figure 1.
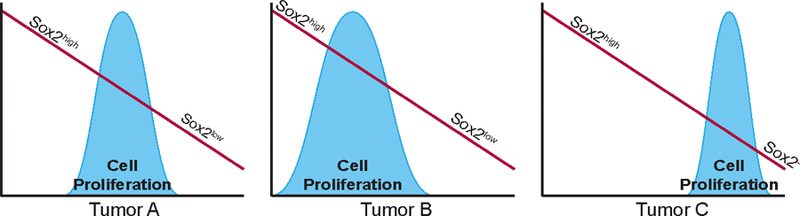
Sox2 Levels and Tumor Growth. Sox2 levels in tumor cells are optimized to maximize tumor growth. Optimal Sox2 expression is dependent on the genetic context of the tumor.
Although the mechanisms by which Sox2 inhibits proliferation are poorly understood, some progress on this front has been made. Hagey and Muhr demonstrated that Sox2 binds to the Ccnd1 promoter (cyclin D1 promoter) in mouse cortical neural progenitor cells (Hagey and Muhr, 2014). In addition, they showed that expression of a Ccnd1 promoter/reporter gene construct decreased ~3-fold when Sox2 was overexpressed in chick neural progenitor cells. Further analysis determined that elevating Sox2 allows it bind to bind to low affinity SOX2 sites (degenerate Sox2 motifs) upstream of the Ccnd1 promoter. This is also the case for low affinity sites in other genes downregulated by Sox2. Further, Hagey and Muhr reported that several of the genes downregulated by Sox2, including Ccnd1, contain gene sequences consisting of a Sox2 motif and a Tcf/Lef motif separated by 4- to 10 base pairs (Hagey and Muhr, 2014). They demonstrated the significance of these adjacent motifs with the finding that the physical association of Lef1 with the corepressor Tle1 (Gro/Tle corepressor family) in HEK293 cells increased when Sox2 was ectopically expressed. Additionally, the expression of Ccnd1 promoter-luciferase gene construct in chick neural progenitor cells was decreased more strongly when Sox2 and Tle1 were expressed together with the promoter/reporter gene construct than when Sox2 or Tle1 were expressed individually.
Other studies by Lui et al. provided additional insights into the mechanisms by which high levels of Sox2 limit cell proliferation (Liu et al., 2013). Sox2 is expressed at very high levels in quiescent support cells of the mammalian auditory sensory epithelium, which also express the cyclin-dependent kinase inhibitor p27kip1. Using an inducible CreER/lox system, Sox2 was ablated in the support cells at three different postnatal stages. Ablation of Sox2 in a subset of support cells (inner pillar cells) led to a reduction of p27kip1 and increased cell proliferation. Using a different inducible CreER/lox system, p27kip1 was ablated in support cells. Similar to ablation of Sox2, ablation of p27kip1 promoted proliferation in a sub-population of support cells, but Sox2 did not decrease. Collectively, these studies support an epistatic relationship between Sox2, p27kip1, and quiescence: p27kip1 is upregulated by Sox2 and elevation of p27kip1 inhibits proliferation of support cells. Consistent with these findings, Lui. et al. used chromatin immunoprecipitation (ChIP) studies to show that Sox2 binds to the p27kip1 gene locus. These workers also demonstrated that Sox2 upregulated a p27kip1 promoter-luciferase gene construct in Hela cells and mouse embryonic fibroblasts.
Sox2 Dosage and Tumor Cell Fate Decisions
Sox2 dosage not only plays major roles in cell fate decisions during development, it also influences tumor cell fate decisions. This connection between Sox2 levels and tumor cell fate is supported strongly by the findings of Xu et al. who investigated mutant K-Ras-induced lung cancer (Xu et al., 2014). These workers demonstrated that Sox2 expression determines the cell of origin and the histopathology of lung tumors formed when mutant K-Ras (K-Ras G12D) is expressed conditionally in lung cells. In this study, mutant K-Ras was induced by treatment with tamoxifen in CC10-CreER/K-Ras mice. When mutant K-Ras was induced in CC10+ alveolar cells, it preferentially led to the formation of adenomas and adenocarcinomas in a subset of alveoli cells. In contrast, even though mutant K-Ras was expressed in bronchiolar cells, these cells did not undergo transformation and papillary adenocarcinomas did not form.
In the course of examining the mechanisms by which mutant K-Ras induced lung tumors, Xu et al. determined that mutant K-Ras upregulated Notch signaling in alveolar cells, which, in turn, repressed Sox2 (Xu et al., 2014). Surprisingly, by blocking the activation of Notch signaling, with a dominant negative form of the mastermind-like protein (DNMaml1), in the context of induced mutant K-Ras (CC10-CreER/K-Ras/DNMaml1 mice), adenomas and clusters of hyperplastic cells formed, but adenocarcinomas did not. Moreover, in contrast to CC10-CreER/K-RasG12D mice, the lesions that formed in the CC10-CreER/K-Ras/DNMaml1 mice expressed Sox2 and markers of squamous carcinoma of the lung, which is consistent with the known expression of SOX2 in human squamous cell lung cancers (Xu et al., 2014). To directly test the effects of Sox2 on the formation of alveolar adenocarcinomas, Xu et al. conditionally overexpressed Sox2 along with mutant K-Ras. In these mice, inducible overexpression of Sox2 suppressed formation of alveolar adenocarcinomas. Together, these studies indicate that expression of mutant K-Ras, through the activation of Notch signaling and the suppression of Sox2 expression, leads to alveolar adenocarcinomas. Equally interesting, adenocarcinomas with squamous cell features form in alveoli when CC10-CreER/LSL-Sox2 mice are treated with tamoxifen and SOX2 is overexpressed on its own without mutant K-Ras.
The study by Xu et al. also determined why expression of mutant K-Ras in bronchiolar cells did not lead to transformation (Xu et al., 2014). Sox2 expression is not detectable in the distal alveolar region of the lung, but it is expressed in normal airway epithelium, specifically the proximal trachea, bronchi and bronchioles (Tompkins et al., 2009). This raised the possibility that expression of Sox2 in bronchiole cells blocked the ability of mutant K-Ras to transform bronchiole cells and induce papillary tumors. This possibility was tested by examining tumors formed in CC10-CreER/K-Ras/Sox2flox/+ mice where tamioxifen induces mutant K-Ras and Sox2 expression is reduced by half. In contrast to CC10-CreER/K-Ras mice, CC10-CreER/K-Ras/Sox2flox/+ mice developed papillary adenocarcinomas (Xu et al., 2014). Collectively, the studies of Xu et al. made two important points. First, Sox2 levels alter the tumor cell of origin. Specifically, reduction of Sox2 in the context of mutant K-Ras leads to the formation of papillary adenocarcinoma, which are not seen when mutant K-Ras is expressed on its own. Second, overexpression of Sox2 in the context of mutant K-Ras alters the histopathology of alveoli lesions. Sox2 overexpression along with mutant K-Ras generated lesions with squamous characteristics.
SOX2 has also been implicated in a major tumor cell fate decision during the progression of prostate cancer, namely the transition of castration resistant prostate cancer to the highly deadly form of prostate cancer with neuroendocrine histopathology. Several studies have linked SOX2 expression to this highly aggressive form of prostate cancer (Yu et al., 2014; Russo et al., 2016; Mu et al., 2017; Ku et al., 2017). In 2017, studies by Mu et al. and Ku et al. provided a causative link between SOX2 expression and the progression to prostate cancer with neuroendocrine histopathology (Mu et al., 2017; Ku et al., 2017). These studies focused on the combined loss of TP53 and RB1 during progression of prostate cancer, which increases from ~5% in primary prostate cancer, to ~40% in castration resistant prostate cancer, to ~75% in castration resistant prostate cancer with neuroendocrine-like histopathology (Mu et al., 2017). The study by Mu et al. provided the clearest causative link between SOX2 and neuroendocrine prostate cancer (Mu et al., 2017). In this study, TP53 and RB1 were knocked down together in the androgen-dependent prostate cancer cell line LNCaP. The combined knockdown of TP53 and RB1 had three prominent effects: 1) upregulation of neuroendocrine markers, such as synaptophysin; 2) upregulation of SOX2; and 3) resistance of LNCaP tumor growth to the antiandrogen enzalutamide. Remarkably, when SOX2 upregulation was blocked by the expression of SOX2 shRNA in the context of knockdown of TP53 and RB1, the upregulation of neuroendocrine markers was reversed and tumor growth of the genetically modified LNCaP cells was again inhibited by enzalutamide (Mu et al., 2017). This study not only provided a link between SOX2 and prostate tumor cell fate, it is also linked SOX2 to prostate cancer resistance to antiandrogens.
Sox2 Dosage and Tumor Cell Quiescence
Over the past four years, several studies have directly linked tumor cell quiescence to high levels of Sox2. One of the first in vivo studies to link elevated Sox2 levels and tumor cell quiescence was reported by Vanner et al. (Vanner et al., 2014) This study used a mouse model of sonic hedgehog medulloblastoma where one allele of the Ptch1 gene was deleted. Subjecting these mice to 3 Gy radiation led to ~80% of the mice forming medulloblastomas that possessed a small population of label-retaining cells (<5%) that express Sox2. This rare, infrequently-cycling Sox2-positive population was shown to be responsible for propagating the tumor and they were the first cells to proliferate and repopulate the tumor after the tumor bearing mice had been treated with either Ara C (cytarabine) or vismodegib (a hedgehog inhibitor), which targeted the proliferating cells of the tumor. As such, the Sox2-positive population represents the cancer stem cell population of the tumor that serves as a reserve cancer stem cell population and which contributes to drug resistance.
The study by Vanner et al. linked Sox2 to the slow cycling cancer stem cell population of the tumor, but did not demonstrate a direct cause-and-effect relationship between Sox2 and tumor cell quiescence (Vanner et al., 2014). Malladi et al. provided a more direct link between SOX2 and tumor cell quiescence (Malladi et al., 2016). These investigators examined the properties of cells that they referred to as latency competent cancer (LCC) cells. These cells were isolated from early stage human lung and breast tumor cell lines that had been engrafted by intracardial injection. The engrafted cells dispersed to different organs of the host where they remained primarily as single cells or small cell clusters for several months. Although these disseminated cells exhibit little or no growth, they retained tumor-initiating potential. When isolated from different tissues and transplanted orthotopically, they formed actively growing tumors, similar to the parental cell line from which they were isolated. In fact, in a limiting tumor cell dilution assay, these LCC cells exhibited a 4- to 15-fold increase in the frequency of tumor-initiating cells.
Characterization of the lung LCC cells by gene set enrichment analysis demonstrated that they exhibited a stem-like alveolar type I and bipotent progenitor cell signature when compared to their parental tumor cell line. They also expressed higher levels of SOX2 than their parental population. Germane to this review, knockdown of SOX2 in lung LCC cells by shRNA led to a decrease in DKK1 (a Wnt signaling antagonist and direct transcriptional target of Sox2) and an increase in cell proliferation under low-mitogen conditions, where these cells exhibit limited growth (Malladi et al., 2016).
A direct link between elevated levels of SOX2 and inhibition of tumor cell growth has also been described in studies reported by our laboratory. In our studies, we overexpressed SOX2 with the aid of a Dox inducible promoter in nine SOX2-positive human tumor cell lines representing glioblastoma and medulloblastoma, as well as cancers of the breast, prostate and pancreas (Cox et al., 2012; Wuebben et al., 2016). For each tumor cell line, elevating SOX2 led to a dose-dependent decrease in cell proliferation in culture. Notably, this is not just a short-term inhibition of cell proliferation. Cells maintained for over two months under conditions of elevated SOX2 continued to be growth inhibited (Cox et al., 2012). Importantly, the growth inhibitory effects of elevated SOX2 were observed in tumor cell lines that express high, moderate or low levels of SOX2. Collectively, our studies indicate that SOX2 levels in each tumor cell line are set to the level that is optimal for their growth (Figure 1).
Elevated SOX2 not only inhibits growth in culture, it also inhibits tumor growth in vivo. In the case of pancreatic tumor cell lines, we have demonstrated that once tumors have formed, elevating SOX2 in vivo with the aid of an inducible promoter halted tumor growth (Wuebben et al., 2016). Relative to their control counterparts, SOX2 elevated tumors exhibited a drastic reduction in Ki-67 staining and a small decrease in cleaved caspase 3 staining. Thus, when SOX2 was elevated in vivo, tumor growth was arrested without an apparent increase in cell death. Equally important, we demonstrated that knocking down SOX2 in pancreatic tumor cells in vivo with an inducible shRNA directed against SOX2 also reduced tumor growth (Wuebben et al., 2016). Collectively, the knockdown and overexpression studies indicate that SOX2 levels are optimized to maximize tumor growth. Too much or too little Sox2 decreases tumor growth.
The finding that elevating SOX2 can inhibit tumor growth raises an important question, namely, if elevated SOX2 inhibits tumor cell growth does this mean that SOX2 levels cannot rise during tumor progression without blocking further tumor growth? In fact, the evidence in several cancers indicates that SOX2 levels do rise during cancer progression. In the case of prostate cancer, discussed earlier in this review, SOX2 expression rises as a patient’s tumor progresses from an androgen-dependent stage to an androgen-independent tumor (castration resistant prostate cancer) and rises dramatically as the prostate progresses to a deadly form characterized by cells with neuroendocrine histopathology. For this cancer, the work of Mu et al. and Ku et al. demonstrated that the combined loss of TP53 and RB1 provides at least part of the answer why SOX2 levels can rise during prostate cancer progression (Mu et al., 2017; Ku et al., 2017). Knocking down TP53 and RB1 results in cells that can actively proliferate even though Sox2 levels rose significantly. Thus, Sox2 levels can rise in cancer when there are other changes in the cancer that are able to counterbalance the growth inhibitory properties of Sox2.
The finding that elevating SOX2 with the aid of an inducible promoter reduces tumor cell growth contrasts with findings reported by others where SOX2 was stably overexpressed in tumor cells and growth was increased (Jia et al., 2011; Li et al., 2013; Nakatsugawa et al., 2011; Herreros-Villanueva et al., 2013). This apparent contradiction has been addressed previously (Wuebben and Rizzino, 2017) and is only briefly addressed here. When tumor cells were engineered for inducible expression of SOX2, the engineered population being generated was allowed to expand in the absence of elevated SOX2 during the drug selection period (Cox et al., 2012; Wuebben et al., 2016). Only after the cells had been engineered was SOX2 elevated and cell proliferation assessed. In contrast, when tumor cells are engineered to stably overexpress SOX2, they are exposed continually to elevated SOX2 during the drug selection period. Any cells that are growth inhibited by stable overexpression of SOX2 will be lost during the drug selection process.
Conclusions:
The goal of this review was to focus attention on the critical roles that Sox2 dosage plays in both normal and malignant cells. Work described in this review indicates that the dosage of Sox2 plays prominent roles in cell fate decisions made during development, as well as cell fate decisions in cancer. The latter include cell fate decisions that influence the tumor cell of origin as well as progression of the cancer. We also described work showing how elevated levels of Sox2 limit the growth of both normal cells and tumor cells. These findings indicate that tumor cells have usurped a fundamental property of Sox2 to suit their own needs.
Complete ablation of Sox2 and conditional knockout of Sox2 demonstrated the need for Sox2 during both embryogenesis as well as later stages of mammalian development. Knockdown studies in a wide range of tumor cell types also established the need for SOX2 in tumor growth. Similarly, studies reporting enhanced growth of tumor cells when SOX2 was stably overexpressed have also been used to argue for the importance of SOX2 in different types of tumor cells. While we agree that SOX2 plays essential roles in over 20 different types of human cancers, we urge caution in concluding that elevating SOX2 on its own, without making other changes in the tumor cells, leads to a general increase in tumor cell proliferation. As discussed in the previous section, elevating SOX2 with the aid of an inducible promoter in nine different tumor cell lines leads to growth inhibition in each case. This includes several tumor cell lines where stably overexpressing SOX2 was reported to increase growth. Additionally, we have verified this in at least 10 other tumor cell lines (Metz, Wilder and Rizzino, unpublished data). Thus, we argue that for the vast majority of cells in a tumor cell population, elevating SOX2 leads to growth inhibition not growth stimulation. This not is only borne-out by our studies, but also supported by the tumor studies reported by Vanner et al. and Malladi et al. (Vanner et al., 2014; Malladi et al., 2016). Collectively, multiple lines of evidence support the conclusion that once Sox2 is expressed at levels above that needed to promote tumor growth, further increases in Sox2 promote growth inhibition.
Our understanding of Sox2 has grown significantly over the past twenty years, yet there are still fundamental questions surrounding how Sox2 alters the behavior of both normal and tumor cells. One of the most pressing questions is how high levels of Sox2 lead to growth inhibition. Answering this fundamental question will not only provide significant insights into the function of Sox2 during development, it will also provide insights into the molecular mechanisms by which Sox2 regulates tumor cell growth. Understanding how Sox2 regulates tumor growth could prove helpful in improving the treatment of patients with Sox2-positive cancers that respond poorly to current therapies, which include advanced glioblastoma, prostate cancer, triple negative breast cancer, and recurrent sonic hedgehog driven medulloblastoma.
As discussed earlier in this review, some progress has been made in understanding how elevating Sox2 in normal cells influences cell cycle machinery, including the inhibition of cyclin D1 expression and the upregulation of the cyclin-dependent kinase inhibitor p27kip1 (Hagey and Muhr, 2014; Liu et al., 2013). While this is a start, more in-depth studies are needed to determine how Sox2 levels influence cell proliferation. Moreover, it will be important to determine whether elevating Sox2 in normal cells and tumor cells limits cell proliferation by a well-conserved mechanism(s) or whether the molecular mechanisms used to inhibit growth when Sox2 is elevated vary considerably and are context-dependent. Given that elevating SOX2 in many different tumor cell lines representing at least five different cancers leads to growth inhibition, we suggest that a well-conserved mechanism is primarily responsible for inhibiting cell growth when Sox2 levels are elevated.
Finally, it is important to consider how sustained elevation of Sox2 could impact the behavior of both normal and tumor cells over the longer-term. Based on the work discussed in this review, we speculate that the effects of high Sox2 levels may not simply promote short-term growth inhibition of normal and tumor cells. The findings of Malladi et al. discussed earlier demonstrate that SOX2-positive lung tumor cells can remain largely dormant for months, and there is every reason to believe that this is not the upper limit (Malladi et al., 2016). Moreover, this is unlikely to be an isolated case. Thus, we proffer the hypothesis that high SOX2 levels play a significant factor in establishing and maintaining prolonged growth arrest of the cancer stem cell population of SOX2-positive cancers. Intriguingly, this may also be true for other Sox family members. The studies conducted by Malladi et al. determined that dormant breast cancer tumor cells were enriched for the expression of the SRY-related gene SOX9 and that depletion of this gene decreased the number of dormant tumor cells present in the brain of athymic mice (Malladi et al., 2016). Additionally, other groups have shown that elevating Sox9 in four melanoma tumor cell lines and in a rat non-transformed chondrocyte cell line inhibits proliferation (Passeron et al., 2009; Panda et al., 2001). Further support for the roles of Sox2 and Sox9 in tumor dormancy come from the work of Aguirre-Ghiso and co-workers. These researchers reported that NR2F1, a dormancy associated transcription factor elevated in several cancers (Sosa et al. 2014; Borgen et al. 2018), drives the expression of SOX9 and SOX2 in dormant tumor cells (Sosa et al., 2014). Collectively, these findings suggest that regulation of cell proliferation during both development and cancer progression may extend to other Sox genes. If this is the case, understanding mechanistically how Sox2 and other Sox9 promote tumor cell quiescence could provide strategies for addressing a major unmet need in the treatment of cancer, namely eradicating dormant tumor cells. Dormant tumor cells pose a major challenge clinically. These cells are responsible for tumor recurrence in patients whose primary tumor was removed years earlier (Demicheli et al., 1996; Morgan et al., 2009; Ghajar, 2015; Linde, Fluegen, and Aguirre-Ghiso, 2016). Additionally, dormant tumor cells are largely resistant to current therapies, such as DNA damaging agents and radiation, that eradicate actively proliferating tumor cells. In the future, learning more about the function of SOX2 in both normal and tumor cells could help pave the way for meeting the significant challenge posed by dormant tumor cells.
Acknowledgments
Phillip Wilder is thanked for reading this review and Heather Rizzino is thanked for editorial assistance.
Funding: Work in this laboratory is supported in part by a grant from the National Institute of General Medical Sciences (GM106397) and funds from the state of Nebraska through the Pediatric Cancer Research Group and the Fred & Pamela Buffett Cancer Center. Core facilities of the Fred & Pamela Buffett Cancer Center are supported by a Cancer Center Support grant from the National Cancer Institute, P30 CA036727.
Footnotes
Disclosure of Potential Conflicts of Interest: No potential conflicts.
References
- Avilion AA, Nicolis SK, Pevny LH, Perez L, Vivian N, Lovell-Badge R (2003). Multipotent cell lineages in early mouse development depend on SOX2 function. Genes and Development 17(1), 126–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borgen E, Rypdal MC, Sosa MS, Renolen A, Schlichting E, Lønning PE, Synnestvedt M, Aguirre-Ghiso JA, Naume B (2018). NR2F1 stratifies dormant disseminated tumor cells in breast cancer patients. Breast Cancer Research 20(1), 120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carey BW, Markoulaki S, Hanna JK, Faddah DA, Buganim Y, Kim J, Ganz K, Steine EJ, Cassady JP, Creyghton MP, Welstead GG, Gao Q, Jaenisch R (2011). Reprogramming factor stoichiometry influences the epigenetic state and biological properties of induced pluripotent stem cells. Cell Stem Cell 9(6), 588–598. [DOI] [PubMed] [Google Scholar]
- Chew JL, Loh YH, Zhang W, Chen X, Tam WL, Yeap LS, Li P, Ang YS, Lim B, Robson P, Ng HH (2005). Reciprocal transcriptional regulation of Pou5f1 and Sox2 via the Oct4/Sox2 complex in embryonic stem cells. Molecular and Cellular Biology 25(14), 6031–6046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox JL, Wilder PJ, Desler M, Rizzino A (2012). Elevating SOX2 levels deleteriously affects the growth of medulloblastoma and glioblastoma cells. PLoS One 7(8), e44087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demicheli R, Abbattista A, Miceli R, Valagussa P, Bonadonna G (1996). Time distribution of the recurrence risk for breast cancer patients undergoing mastectomy: further support about the concept of tumor dormancy. Breast Cancer Research and Treatment 41(2), 177–185. [DOI] [PubMed] [Google Scholar]
- Fantes J, Ragge NK, Lynch SA, McGill NI, Collin JR, Howard-Peebles PN, Hayward C, Vivian AJ, Williamson K, van Heyningen V, Fitzpatrick DR (2003). Mutations in SOX2 cause anophthalmia. Nature Genetics 33(4), 461–463. [DOI] [PubMed] [Google Scholar]
- Fitzpatrick DR, van Heyningen V (2005). Developmental eye disorders. Current Opinion in Genetics & Development 15(3), 348–353. [DOI] [PubMed] [Google Scholar]
- Ghajar CM (2015). Metastasis prevention by targeting the dormant niche. Nature Reviews Cancer 15(4), 238–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gontan C, de Munck A, Vermeij M, Grosveld F, Tibboel D, Rottier R (2008). Sox2 is important for two crucial processes in lung development: branching morphogenesis and epithelial cell differentiation. Developmental Biology 317(1), 296–309. [DOI] [PubMed] [Google Scholar]
- Hagey DW, Klum S, Kurtsdotter I, Zaouter C, Topcic D, Andersson O, Bergsland M, Muhr J (2018). SOX2 regulates common and specific stem cell features in the CNS and endoderm derived organs. PLoS Genetics 14(2), e1007224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagey DW, Muhr J (2014). Sox2 Acts in a Dose-Dependent Fashion to Regulate Proliferation of Cortical Progenitors. Cell Reports 9(5), 1908–1920. [DOI] [PubMed] [Google Scholar]
- Hagstrom SA, Pauer GJ, Reid J, Simpson E, Crowe S, Maumenee IH, Traboulsi EI (2005). SOX2 mutation causes anophthalmia, hearing loss, and brain anomalies. American Journal of Medical Genetics Part A 138A(2), 95–98. [DOI] [PubMed] [Google Scholar]
- Herreros-Villanueva M, Zhang JS, Koenig A, Abel EV, Smyrk TC, Bamlet WR, de Narvajas AA, Gomez TS, Simeone DM, Bujanda L, Billadeau DD (2013). SOX2 promotes dedifferentiation and imparts stem cell-like features to pancreatic cancer cells. Oncogenesis 2, e61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutton SR, Pevny LH (2011). SOX2 expression levels distinguish between neural progenitor populations of the developing dorsal telencephalon. Developmental Biology 352(1), 40–47. [DOI] [PubMed] [Google Scholar]
- Ishii Y, Rex M, Scotting PJ, Yasugi S (1998). Region-specific expression of chicken Sox2 in the developing gut and lung epithelium: regulation by epithelial-mesenchymal interactions. Developmental Dynamics 213(4), 464–475. [DOI] [PubMed] [Google Scholar]
- Jia X, Li X, Xu Y, Zhang S, Mou W, Liu Y, Liu Y, Lv D, Liu CH, Tan X, Xiang R, Li N (2011). SOX2 promotes tumorigenesis and increases the anti-apoptotic property of human prostate cancer cell. Journal of Molecular Cell Biology 3(4), 230–238. [DOI] [PubMed] [Google Scholar]
- Kopp JL, Ormsbee BD, Desler M, Rizzino A (2008). Small increases in the level of Sox2 trigger the differentiation of mouse embryonic stem cells. Stem Cells 26(4), 903–911. [DOI] [PubMed] [Google Scholar]
- Ku SY, Rosario S, Wang Y, Mu P, Seshadri M, Goodrich ZW, Goodrich MM, Labbe DP, Gomez EC, Wang J, Long HW, Xu B, Brown M, Loda M, Sawyers CL, Ellis L, Goodrich DW (2017). Rb1 and Trp53 cooperate to suppress prostate cancer lineage plasticity, metastasis, and antiandrogen resistance. Science 355(6320), 78–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Xu Y, Chen Y, Chen S, Jia X, Sun T, Liu Y, Li X, Xiang R, Li N (2013). SOX2 promotes tumor metastasis by stimulating epithelial-to-mesenchymal transition via regulation of WNT/β-catenin signal network. Cancer Letters 336(2), 379–89. [DOI] [PubMed] [Google Scholar]
- Linde N, Fluegen G, Aguirre-Ghiso JA (2016). The Relationship Between Dormant Cancer Cells and Their Microenvironment. Advances in Cancer Research 132, 45–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z, Walters B, Owen T, Brimble MA, Steigelman KA, Zhang L, Lagarde MMM, Valentine MB, Yu Y, Cox BC, Zuo J (2013). Regulation of p27Kip1 by Sox2 maintains quiescence of inner pillar cells in the murine auditory sensory epithelium. The Journal of neuroscience 32(31),10530–10540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malladi S, Macalinao DG, Jin X, He L, Basnet H, Zou Y, de Stanchina E, Massague J (2016). Metastatic latency and immune evasion through autocrine inhibition of Wnt. Cell 165(1), 45–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan TM, Lange PH, Porter MP, Lin DW, Ellis WJ, Gallaher IS, Vessella RL (2009). Disseminated tumor cells in prostate cancer patients after radical prostatectomy and without evidence of disease predicts biochemical recurrence. Clinical Cancer Research 15(2), 677–683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mu P, Zhang Z, Benelli M, Karthaus WR, Hoover E, Chen CC, Wongvipat J, Ku SY, Gao D, Cao Z, Shah N, Adams EJ, Abida W, Watson PA, Prandi D, Huang CH, de Stanchina E, Lowe SW, Ellis L, Beltran H, Rubin MA, Goodrich DW, Demichelis F, Sawyers CL (2017). SOX2 promotes lineage plasticity and antiandrogen resistance in TP53- and RB1-deficient prostate cancer. Science 355(6320), 84–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakatsugawa M, Takahashi A, Hirohashi Y, Torigoe T, Inoda S, Murase M, Asanuma H, Tamura Y, Morita R, Michifuri Y, Kondo T, Hasegawa T, Takahashi H, Sato N (2011). SOX2 is overexpressed in stem-like cells of human lung adenocarcinoma and augments the tumorigenicity. Laboratory Investigation 91(12),1796–80. [DOI] [PubMed] [Google Scholar]
- Niwa H, Miyazaki J, Smith AG (2000). Quantitative expression of Oct-¾ defines differentiation, dedifferentiation or self-renewal of ES cells. Nature Genetics 24(4), 372–376. [DOI] [PubMed] [Google Scholar]
- Panda DK, Miao D, Lefebvre V, Hendy GN, Goltzman D (2001). The transcription factor SOX9 regulates cell cycle and differentiation genes in chondrocytic CFK2 cells. Journal of Biological Chemistry 276(44), 41229–41236. [DOI] [PubMed] [Google Scholar]
- Papapetrou EP, Tomishima MJ, Chambers SM, Mica Y, Reed E, Menon J, Tabar V, Mo Q, Studer L, Sadelain M (2009). Stoichiometric and temporal requirements of Oct4, Sox2, Klf4, and c-Myc expression for efficient human iPSC induction and differentiation. Proceedings of the National Academy of Science of the United States of America 106(31),12759–12764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Passeron T, Valencia JC, Namiki T, Vieira WD, Passeron H, Miyamura Y, Hearing VJ (2009). Upregulation of SOX9 inhibits the growth of human and mouse melanomas and restores their sensitivity to retinoic acid. Journal of Clinical Investigation 119(4), 954–963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Que J, Okubo T, Goldenring JR, Nam KT, Kurotani R, Morrisey EE, Taranova O, Pevny LH, Hogan BL (2007). Multiple dose-dependent roles for Sox2 in the patterning and differentiation of anterior foregut endoderm. Development 134(13), 2521–2531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ragge NK, Lorenz B, Schneider A, Bushby K, de Sanctis L, de Sanctis U, Salt A, Collin JR, Vivian AJ, Free SL, Thompson P, Williamson KA, Sisodiya SM, van Heyningen V, Fitzpatrick DR (2005). SOX2 anophthalmia syndrome. American Journal of Medical Genetics Part A 135(1), 1–7. [DOI] [PubMed] [Google Scholar]
- Sarkar A, Hochedlinger K (2013). The Sox family of transcription factors: versatile regulators of stem and progenitor cell fate. Cell Stem Cell 12(1), 15–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russo MV, Esposito S, Tupone MG, Manzoli L, Airoldi I, Pompa P, Cindolo L, Schips L, Sorrentino C, Di Carlo E (2016). SOX2 boosts major tumor progression genes in prostate cancer and is a functional biomarker of lymph node metastasis. Oncotarget 7(11), 12372–12385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sisodiya SM, Ragge NK, Cavalleri GL, Hever A, Lorenz B, Schneider A, Williamson KA, Stevens JM, Free SL, Thompson PJ, van Heyningen V, Fitzpatrick DR (2006). Role of SOX2 mutations in human hippocampal malformations and epilepsy. Epilepsia 47(3), 534–542. [DOI] [PubMed] [Google Scholar]
- Sosa MS, Falguni P, Maia AG, Estrada Y, Bosch A, Bragado P, Ekpin E, George A, Zheng Y, Lam HM, Morrissey C, Chung CY, Farias EF, Bernstein E, Aguirre-Ghiso JA (2014). NR2F1 controls tumour cell dormancy via SOX9- and RAR-driven quiescence programmes. Nature Communications [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzenko N, Crowl T, Bachleda A, Langer L, Pevny L (2013). SOX2 maintains the quiescent progenitor cell state of postnatal retinal Müller glia. Development 140(7),1445–1456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi K, Yamanaka S (2006). Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 126(4), 663–676. [DOI] [PubMed] [Google Scholar]
- Taranova OV, Magness ST, Fagan BM, Wu Y, Surzenko N, Hutton SR, Pevny LH (2006). SOX2 is a dose-dependent regulator of retinal neural progenitor competence. Genes and Development 20(9), 1187–1202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tompkins DH, Besnard V, Lange AW, Wer SE, Keiser AR, Smith AN, Lang R, Whitsett JA (2009). Sox2 is required for maintenance and differentiation of bronchiolar Clara, ciliated, and goblet cells. PLoS One 4(12), e8248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanner RJ, Remke M, Gallo M, Selvadurai HJ, Coutinho F, Lee L, Kushida M, Head R, Morrissy S, Zhu X, Aviv T, Voisin V, Clarke ID, Li Y, Mungall AJ, Moore RA, Ma Y, Jones SJ, Marra MA, Malkin D, Northcott PA, Kool M, Pfister SM, Bader G, Hochedling K, Korshunov A, Taylor MD, Dirks PB (2014). Quiescent sox2(+) cells drive hierarchical growth and relapse in sonic hedgehog subgroup medulloblastoma. Cancer Cell 26(1), 33–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weina K, Utikal J (2014). SOX2 and cancer: current research and its implications in the clinic. Clinical and Translational Medicine 3, 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wuebben EL, Rizzino A (2017). The dark side of SOX2: cancer – a comprehensive overview. Oncotarget 8(27), 44917–44943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wuebben EL, Wilder PJ, Cox JL, Grunkemeyer JA, Caffrey T, Hollingsworth MA, Rizzino A (2016). SOX2 functions as a molecular rheostat to control the growth, tumorigenicity and drug responses of pancreatic ductal adenocarcinoma cells. Oncotarget 7(23), 34890–34906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu X, Huang L, Futtner C, Schwab B, Rampersad RR, Lu Y, Sporn TA, Hogan BL, Onaitis MW (2014). The cell of origin and subtype of K-Ras-induced lung tumors are modified by Notch and Sox2. Genes and Development 28(17), 1929–1939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaguchi S, Hirano K, Nagata S, Tada T (2011). Sox2 expression effects on direct reprogramming efficiency as determined by alternative somatic cell fate. Stem Cell Research 6(2), 177–186. [DOI] [PubMed] [Google Scholar]
- Yu X, Cates JM, Morrissey C, You C, Grabowska MM, Zhang J, DeGraff DJ, Strand DW, Franco OE, Lin-Tsai O, Hayward SW, Matusik RJ (2014). SOX2 expression in the developing, adult, as well as diseased prostate. Prostate cancer and prostatic diseases 17(4), 301–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zenteno JC, Gascon-Guzman G, Tovilla-Canales JL (2005). Bilateral anophthalmia and brain malformations caused by a 20-bp deletion in the SOX2 gene. Clinical Genetics 68(6), 564–566. [DOI] [PubMed] [Google Scholar]