

Abstract
A single pool of multipotent retinal progenitor cells give rise to the diverse cell types within the mammalian retina. Such cellular diversity is due to precise control of various cellular processes like cell specification, proliferation, differentiation, and maturation. Circadian clock genes can control the expression of key regulators of cell cycle progression and therefore can synchronize the cell cycle state of a heterogeneous population of cells. Here we show that the protein encoded by the circadian clock gene brain and muscle arnt-like protein-1 (Bmal1) is expressed in the embryonic retina and is required to regulate the timing of cell cycle exit. Accordingly, loss of Bmal1 during retinal neurogenesis results in increased S-phase entry and delayed cell cycle exit. Disruption in cell cycle kinetics affects the timely generation of the appropriate neuronal population thus leading to an overall decrease in the number of retinal ganglion cells, amacrine cells, and an increase in the number of the late-born type II cone bipolar cells as well as the Müller glia. Additionally, the mislocalized Müller cells are observed in the photoreceptor layer in the Bmal1 conditional mutants. These changes affect the functional integrity of the visual circuitry as we report a significant delay in visual evoked potential implicit time in the retina-specific Bmal1 null animals. Our results demonstrate that Bmal1 is required to maintain the balance between the neural and glial cells in the embryonic retina by coordinating the timing of cell cycle entry and exit. Thus, Bmal1 plays an essential role during retinal neurogenesis affecting both development and function of the mature retina.—Sawant, O. B., Jidigam, V. K., Fuller, R. D., Zucaro, O. F., Kpegba, C., Yu, M., Peachey, N. S., Rao, S. The circadian clock gene Bmal1 is required to control the timing of retinal neurogenesis and lamination of Müller glia in the mouse retina.
Keywords: circadian biology, Bmal1 retinal neurogenesis, VEP, cell cycle
The neural retina has long been used as a model system to understand how cellular diversity within the CNS is achieved. It is an excellent system to use because of its well-defined structure and stereotypical cellular composition. Though there are only 6 main types of retinal neurons, each of these have several different subtypes with distinct morphologies, intrinsic properties, and connectivity patterns. All of these diverse neurons are generated from a common pool of multipotent retinal progenitor cells (RPCs) (1–3). Deciphering the mechanisms that result in the generation of such cellular diversity will provide better insights into the creation of complex neural networks. In the mammalian retina, there is a temporal order to the generation of the different retinal cell types with the cones, horizontal cells, and retinal ganglion cells (RGCs) being early-born cells followed by amacrines, rods, and bipolar cells. Müller glia is the only nonneuronal cell type and are the last cells to be born. This birth order is evolutionarily conserved and may reflect the order in which these cell types have evolved (3).
How a multipotent RPC adopts a certain fate remains an open question. Both intrinsic and extrinsic factors are involved in the generation of the various retinal cell types. Many of the intrinsic factors function autonomously. These include transcription factors that can initiate complex regulatory programs within the RPCs and appear to play a more deterministic role in directing the RPCs toward specific fates. For example, the basic helix loop helix transcription factor Math5 is necessary but not sufficient for RGC fate, as Math5 expression can be detected in the RPCs that differentiate into nearly all the cell types within the retina (4, 5). Similarly, the expression of another transcription factor orthodenticle homeobox 2 (Otx2) restricts the RPCs to either adopt the photoreceptor or bipolar cell lineage (6). Many of these identified transcription factors are involved in late stages of RPC fate specification when the cells are already fated to become a certain neuronal type (3, 7). What determines how the RPCs are restricted to one fate is an ongoing area of research.
Extrinsic soluble factors, like hedgehog, fibroblast growth factor, and many others, mainly influence the proliferation of the progenitor pools and their ability to exit the cell cycle (8). The timing of cell cycle exit affects the total numbers of a certain cell type that are born. If the cells exit the cell cycle too early, it results in an increase in the numbers of early-born cells at the cost of late-born cells. Thus, cell cycle components are able to drive the cellular processes during retinal neurogenesis and are key regulators of cell fate specification (9, 10). Factors that promote cell cycle progression like cyclins and cyclin-dependent kinases are highly expressed in the RPCs, but their expression is down-regulated in the differentiated retinal cells, consistent with a function in maintaining cell proliferation (11–13). Conversely, cyclin-dependent kinase inhibitors are involved in promoting cell cycle exit, and their loss can result in increased proliferation (14, 15). Despite the importance of the cell cycle in controlling the cell fate, very little is known about how this is achieved and the molecular factors that regulate the progression of the cell cycle.
The circadian clock can regulate the gating of the cell cycle, and several of the cell cycle genes are under the control of the clock. The core clock proteins, brain and muscle arnt-like protein 1 (BMAL1) and circadian locomotor output cycles kaput (CLOCK) are transcription factors that dimerize and regulate the transcription of targets genes, which include Period and Cryptochrome, the negative regulators of BMAL1/CLOCK. Thus, as levels of the negative regulators increase, they repress their own transcription. This cycle of transcriptional and post-translational activation and repression continues and can be reset by external cues like light, hormones, and food (16, 17). In the liver, the expression of the cell cycle checkpoint kinase WEE1 and the cyclin-dependent kinase inhibitor P21 are regulated by the core circadian clock gene Bmal1 (18, 19). In the hair follicles, loss of Bmal1 leads to an up-regulation of P21 and a block in the G1 phase of the cell cycle, consistent with a role for the clock control genes in regulating cell cycle progression (20).
Protein products of the circadian genes are expressed in several cell types within the eye and can regulate a wide variety of cellular processes, both during development and in the adult (21–25). However, the functional role for the circadian clock genes during retinal neurogenesis is unknown. Here we show that Bmal1 is required during retinal neurogenesis and regulates cell fate specification. Loss of Bmal1 results in more cells failing to exit the cell cycle, thus adopting a different fate at the cost of the early-born cells. We see a marked reduction in the numbers of Brn3b+ RGCs and Calretinin+, choline acetyltransferase (ChAT)+ amacrine cells followed by an increase in the number of late-born Recoverin+ type II cone bipolar and sex-determining region Y-box 9 (Sox9)+ Müller glia cells. In addition, the Müller glia cells are mislocalized, leading to lamination defects in the retina. These neurogenesis defects also lead to functional deficits in axonal conductance. Thus, our data demonstrate an important role for circadian gene Bmal1 in regulating retinal neurogenesis and retinal function.
MATERIALS AND METHODS
Mouse strains
Bmal1 LoxP (018985) and Chx10Cre (005105) mice were purchased from The Jackson Laboratory (Bar Harbor, ME, USA). Chx10Cre mice were crossed to Bmal1Fl/Fl mice (homozygous for LoxP sites) to generate Bmal1Fl/+; Chx10Cre mice. Bmal1Fl/+; Chx10Cre mice were crossed to Bmal1Fl/Fl mice to generate Bmal1Fl/Fl; Chx10Cre (Bmal1 CKO) and Bmal1Fl/Fl or Bmal1Fl/+ (controls) mice. Animal use protocol was approved by the Institutional Animal Care and Use Committees at the Cleveland Clinic, and all procedures adhered to the Association for Research in Vision and Ophthalmology guidelines for the Use of Animals in Vision Research.
Real-time quantitative PCR
Total RNA was extracted from neural retina using an RNeasy Mini Kit (Qiagen, Hilden, Germany) according to the manufacturer’s instructions. The Verso cDNA Kit (Thermo Fisher Scientific, Waltham, MA, USA) was used to generate cDNA for qPCR from 250 ng of total RNA. Real-time PCR was performed on the 7900HT fast real-time PCR system (Thermo Fisher Scientific) using a Radiant SYBR Green Hi-ROX qPCR Kits (Alkali Scientific, Pompano Beach, FL, USA). Relative fold changes in Bmal1 expression were determined using the comparative Ct method (2ΔΔCt method) using β-actin as a reference gene. Primers for Bmal1 were F-5′-GGCCCACAGTCAGATTGAAA-3′ and R-5′-GCTGAACAGCCATCCTTAGC-3′ and for β-actin were F-5′-TTCTTTGCAGCTCCTTCGTT-3′ and R-5′-ATGGAGGGGAATACAGCCC-3′.
Western blot
Isolated retinas were lysed in lysis buffer for protein extraction as previously described (26). Blots were probed with antibodies against BMAL1 (ab3350; Abcam, Cambridge, MA, USA) and β-actin (4970L; Cell Signaling Technology, Danvers, MA, USA). Immunoblots were visualized using IRDye 800CW donkey anti-rabbit secondary antibody (925-32213; Li-Cor Biosciences, Lincoln, NE, USA). Membranes were scanned with an Odyssey infrared scanner (Li-Cor Biosciences).
Immunohistochemistry and imaging
Dissected retinas were fixed in 4% paraformaldehyde for 30–60 min at room temperature (RT), washed in PBS several times, permeabilized for 30 min using 1% Triton X-100, and blocked for 1 h at RT with PBS containing 3% bovine serum albumin and 0.03% Triton X-100. Retinas for flatmount preparation were incubated with primary antibodies (1:100) overnight at 4°C. For cryosections, enucleated eyes were fixed in 4% paraformaldehyde for 60–90 min at RT (for postnatal eyes) or 30–45 min at 4°C (for embryonic eyes), washed in PBS several times, and cryoprotected using an increasing concentration of sucrose (10%:20%:30%) and kept overnight in 30% sucrose at 4°C. Eyes were mounted in mounting medium. Cryosections (8–12 µm) were washed in PBS several times, permeabilized for 1–5 min using 1% Triton X-100 or Tween-20 and blocked for 1 h at RT with PBS containing 3% bovine serum albumin and 0.03% Triton X-100 or Tween-20. Antigen retrieval [for Brn3b, Ki-67, proliferating cell nuclear antigen (PCNA) antibodies] was done by boiling slides in 10 mM sodium citrate pH 6 for 20 min. Cryosections were incubated with primary antibodies overnight at 4°C. Antibodies used in this study as follows: Goat anti-Brn3b (sc-31987; Santa Cruz Biotechnology, Dallas, TX, USA), rabbit anti-calretinin (ab702; Abcam), goat anti-ChAT(AB144P; MilliporeSigma, Burlington, MA, USA), rabbit anti-active caspase-3 (559565; BD Biosciences, San Jose, CA, USA), rabbit anti-cone arrestin (AB15282; MilliporeSigma), mouse anti-glutamine synthetase (MAB302; MilliporeSigma), mouse anti-Ki67 (180192Z; Thermo Fisher Scientific), mouse anti-PCNA (ab29; Abcam), rabbit anti-recoverin (AB5585; MilliporeSigma), rabbit anti-Sox-9 (AB5535; MilliporeSigma), rabbit anti-tyrosine hydroxylase (TH; AB152; MilliporeSigma); and secondary antibodies were Alexa Fluor donkey anti-goat 488/594, donkey anti-mouse 488/594, and Alexa Fluor donkey anti-rabbit 488/594 (Thermo Fisher Scientific). Imaging was done using a Leica laser scanning confocal microscope (TCSSP2; Leica Microsystems, Buffalo Grove, IL, USA). Unless specified, cell counts are per ×40 field for Fig. 1E, I, per ×63 field for Fig. 1H, L, M and Supplemental Fig. S3C, per retinal cross section for Figs. 1D, 2F, 4E, F, and 5C, F, G and Supplemental Fig. S4E or per the whole mounted retina for Fig. 2C.
Figure 1.

Effective deletion of Bmal1 and decrease in early-born retinal neurons. A) Immunoblot of retinal lysates probed with BMAL1 antibody, demonstrating loss of BMAL1 protein in the Bmal1Fl/Fl;Chx10Cre (Bmal1 CKO) retinas at E14 and E16. White arrowhead indicates BMAL1 protein band (∼69 KD). Note that at E12, BMAL1 could not be detected in the control retina. Same blots were probed with a β-actin antibody, as a loading control. B) Quantification of relative BMAL1 expression compared with the β-actin expression. C–J) Representative images of Calretinin+ amacrine (C, E) and Brn3b+ RGCs (G, I) in the control (left panels) and Bmal1 CKO (right panels) retinas. Reduction in Calretinin+ amacrine cells was observed as early as E15 (C, D). The graph represents densities of Calretinin+ cells at E15 (D) and P9 (F) and Brn3b+ cells at E15 (H) and P9 (J). K) Representative images of cone photoreceptors labeled with Arrestin (red) and S-opsin (green) in the control (left panels) and Bmal1 CKO (right panels) retinas. L, M) Cone numbers were not significantly different between the mutants and the controls in the dorsal (L) or ventral (M) regions. F, H) Control group includes Bmal1Fl/Fl and Bmal1Fl/+ mice. Scale bar, 30 µm (C), 50 µm (E, G, I), 20 µm (K). Error bars ± sem, n = 4–9. *P < 0.05, **P < 0.01.
Figure 2.

Loss of Bmal1 results in a decrease in cholinergic amacrine cells but not dopaminergic amacrine cells. A, B) Representative images of TH+ dopaminergic amacrine cells. C) Densities of TH+ cells were unaltered between control and Bmal1 CKO retinas. D, E) Representative images of ChAT+ cholinergic amacrine cells. D′, D″) Higher magnification images of the boxed area in D. E′, E″) Higher magnification images of the boxed area in E. F) Numbers of ChAT+ cells were significantly reduced in the Bmal1 CKO retinas compared with the control, and this reduction was more prominent in the ganglion cell layer. Scale bar, 500 µm (D, E), 50 µm (D′–E″). Error bars ± sem, n = 5–6. *P < 0.05.
Figure 4.

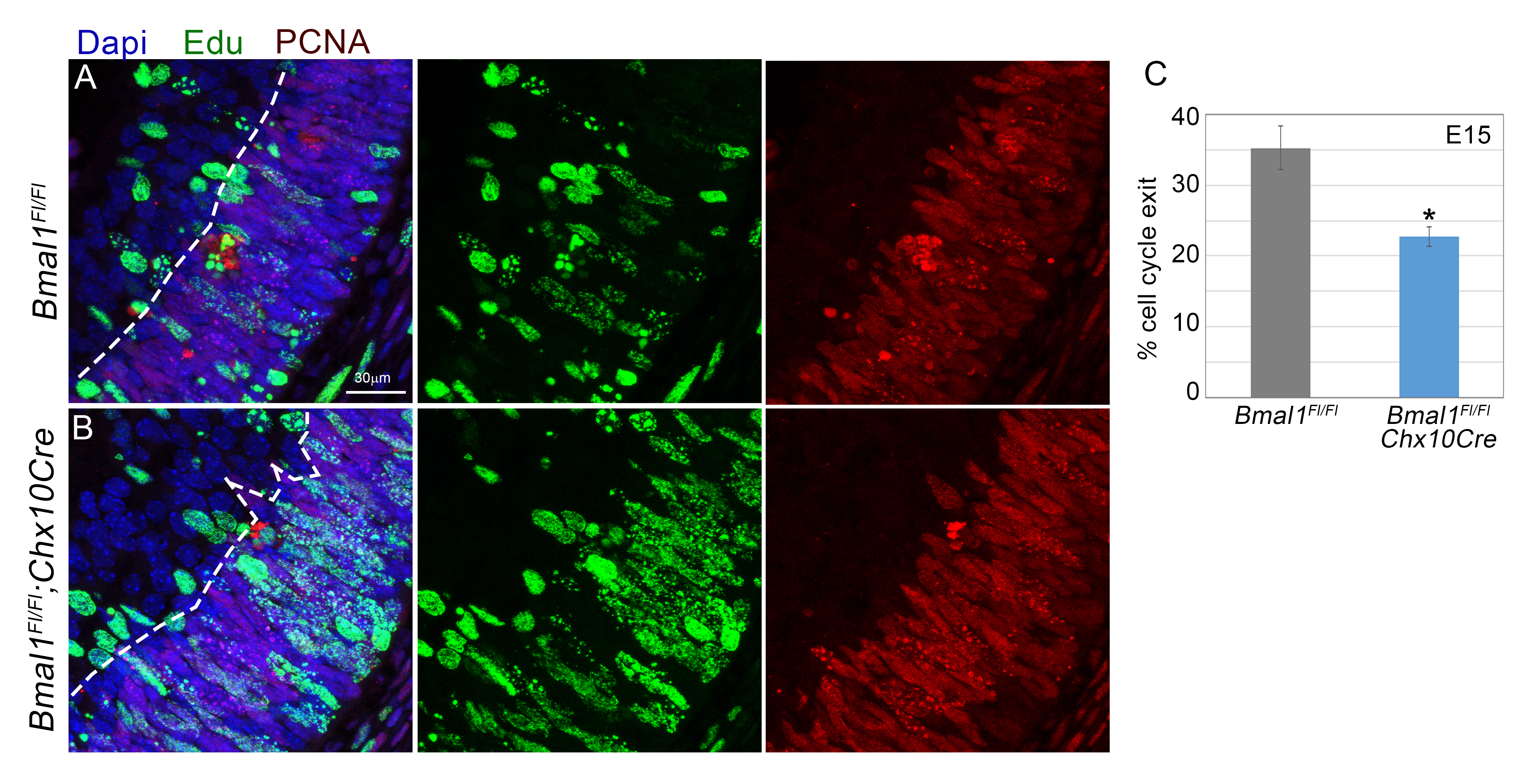
Decrease in cell cycle exit that is due to loss of Bmal1. A–D) E15 retinal cross sections labeled with EdU (red; A′–D′), Ki67 (green; A″–D″), and DAPI (blue; A–D, EdU, Ki67, DAPI merged). Yellow, green, and white arrow heads within the INBL indicate EdU+, Ki67+, and dual+ (EdU+ Ki67+) cells, respectively. INBL is mainly nonproliferative (Ki67 negative) in the central region (A) and sparsely proliferative in the peripheral region (C) in the control retinas. Bmal1 deletion (B, D) resulted in an increase in EdU+ cells (red) in the nonproliferative zone (arrowheads), indicating a failure/delay in cell cycle exit. E) Quantification of all EdU+ cells in the nonproliferative zone at E15. F) Significant decrease in the cell cycle exit [(EdU+ Ki67−)/all EdU+] was observed in Bmal1 CKO compared with the control. Embryos were collected 22 h after EdU injection. Scale bar, 30 μm. Error bars ± sem, n = 3–5. *P < 0.05, **P < 0.01.
Figure 5.

Disorganization of the Müller glia cells and an increase in type II cone bipolar cells that is due to loss of Bmal1. A, B) Representative images of Recoverin+ type II cone bipolar cells. A′, B′ represent the boxed areas in A, B. Scale bar, 100 μm. C) Numbers of Recoverin+ type II cone bipolar cells were significantly increased in the Bmal1 CKO retinas compared with the control. D, E) Retinal sections from the control (D, D′) and Bmal1 CKO (E, E′) visualized with DAPI (blue), Sox9 (green), and glutamine synthetase (red) to mark the Müller glia cells. Sox9+ Müller glia nuclei are restricted within the INL in the control retina (D, D′). In Bmal1 CKO retinas Sox9+ Müller glia nuclei are mislocalized (arrowheads) in the ONL and outer plexiform layer (E, E′). Scale bar, 100 μm (D, E), 50 μm (D′, E′). F) Quantification of Müller glia cells per section in the ONL. G) Total number of Müller glia cells per section within the INL are not significantly different between the control and Bmal1 CKO groups. Error bars ± sem, n = 3–4. *P < 0.05, **P < 0.01.
Analysis of cell cycle entrance and exit
To label proliferating cells, 5-ethynyl-2′-deoxyuridine (EdU; 100 µg/gm of body weight) was injected intraperitoneally. At embryonic day (E)15, embryonic eyes were harvested ∼5 h after EdU injections. At postnatal day (P)5, postnatal eyes were harvested 1 h after EdU injections. For cell cycle exit analysis, EdU injections were done on E13 or E14, and embryonic eyes were harvested after 48 or 22 h, respectively, on E15. Eyes were collected and processed for cryosections as described in the previous section. EdU staining was performed as per the manufacturer’s instructions using the Click-iT imaging kit (Thermo Fisher Scientific). For cell cycle exit analysis, sections were double labeled with EdU (S-phase marker) and cell proliferation marker (Ki67 or PCNA). Percent cell cycle exit was calculated as a ratio of EdU+ cells that are negative for cell proliferation marker to all EdU+ cells.
Visual evoked potential
Visual evoked potentials (VEPs) were performed based on the previously established protocol (27, 28). After overnight dark adaptation, mice were anesthetized with ketamine (80 mg/kg) and xylazine (16 mg/kg), and their pupils were dilated with 3 types of eyedrops (1% cyclopentolate HCl; 2.5% phenylephrine HCl; and 1% mydriacyl). In the experiment, the mice were placed on a heating pad in order to maintain body temperature when the mice were under anesthesia. Three Grass platinum needle electrodes were used subcutaneously: the active electrode was positioned along the midline of the visual cortex; the reference electrode was put into the cheek, and the ground electrode was placed into the tail. VEP signals were differentially amplified (Gain × 10,000, band-pass 1–100 Hz), digitized (2000 Hz), averaged, and stored using UTAS-E3000 system (LKC Technologies, Gaithersburg, MD, USA). Achromatic strobe flash stimuli were presented in a Ganzfeld under dark-adapted conditions and light adaptation. For dark-adapted VEPs, the interstimulus interval ranged from 1.1 to 6 s, increasing with stimulus intensity from −0.2 to 2.1 log cd s/m2; 60–20 successive responses were averaged to obtain a VEP waveform. For light-adapted VEPs, a constant interstimulus interval of 1.1 s was used and 80–70 successive responses were averaged for a series of stimulus intensities (0.0–1.9 log cd s/m2) presented superimposed upon the adapting field. The mouse VEP is dominated by a negative component, which is referred to as N1 (29). The implicit time of the N1 component was measured at the negative peak. The positive component right before the N1 is referred to as P1, and the positive component just after the N1 is referred to as P2. The P1-N1 amplitude was measured from the P1 peak to the N1 trough, and the P2-N1 amplitude was measured from the P2 peak to the N1 trough.
Statistical analysis
Statistical significance was determined using SigmaPlot 10.0 software (Systat Software, San Jose, CA, USA). Results comparing 2 groups were analyzed using Student’s t test. The VEP data were analyzed using 2-way repeated measures ANOVA followed by pairwise multiple comparison using Tukey’s test.
RESULTS
Loss of Bmal1 results in reduced numbers of early-born retinal neurons
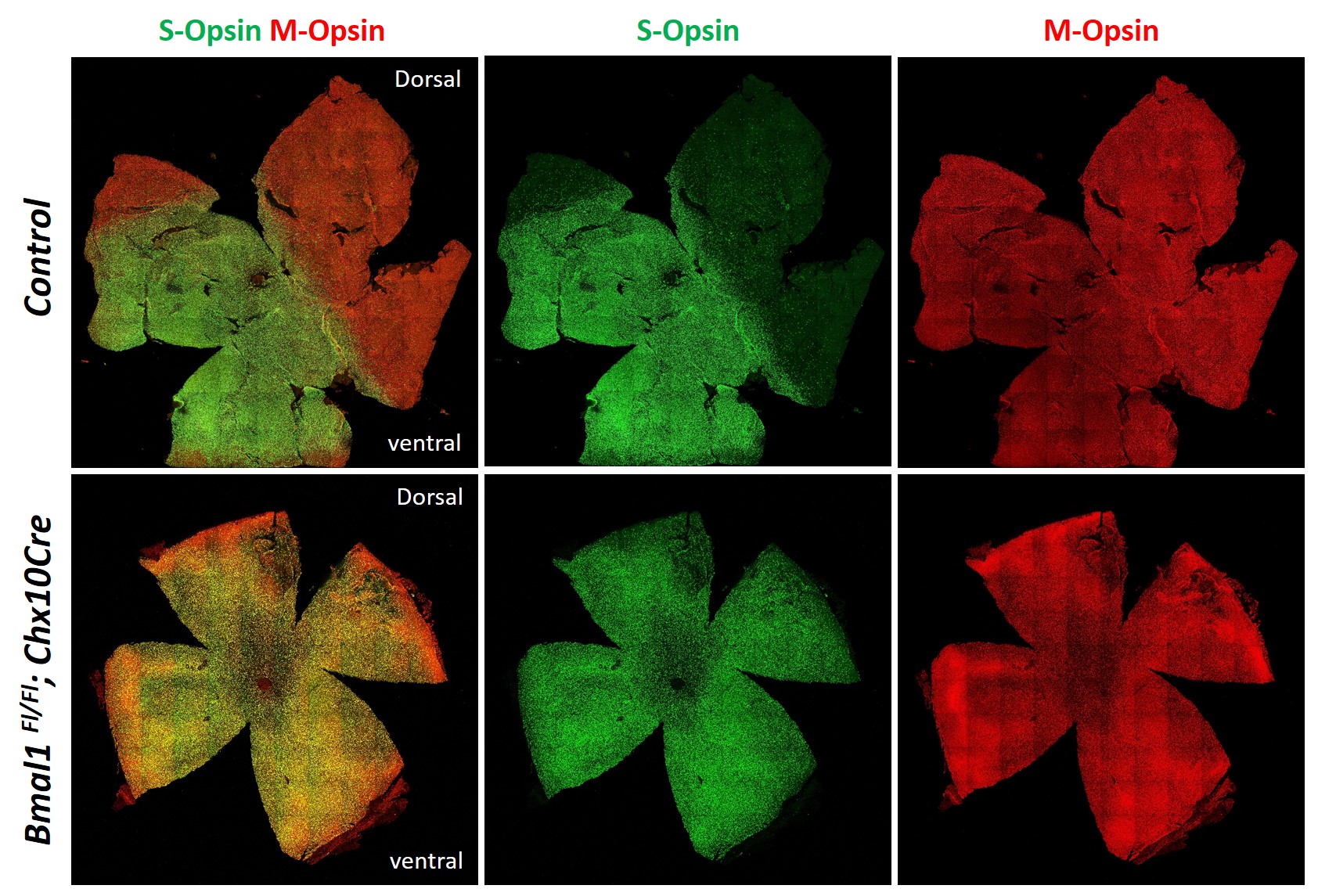
To assess the role of Bmal1 during retinal neurogenesis, we used the Chx10Cre transgene to delete Bmal1 from all the retinal progenitors. In these transgenic mice, Cre expression is detected in the RPCs as early as E12.5 (8). Cre-mediated deletion was confirmed using quantitative PCR to detect the Bmal1 transcript (Supplemental Fig. S1), and immunoblots were probed with an anti-BMAL1 antibody (Fig. 1A, B) to quantitate protein levels at E14 and E16. The BMAL1 protein is either not expressed in the retina at E12 or was below the level of detection but could be detected from E14 onwards. In the presence of the Cre transgene, BMAL1 was reduced but not completely absent, reflecting the chimeric nature of Cre-mediated deletion at E16. During this stage of retinal development, the majority of the cells are competent to give rise to RGCs, cones, horizontal cells, and amacrine cells (30). To determine if Bmal1 function is required for the generation of these early-born cells, we counted the number of Brn3b+ RGCs, Calretenin+ amacrine cells, and cones. Loss of Bmal1 resulted in a reduction of Calretinin+ amacrine cells at E15 (Fig. 1C, D) and P9 (Fig. 1E, F). Though the Br3b+ RGCs were reduced in numbers at E15 (Fig. 1G, H) it was not significant. However, in the postnatal retina the numbers of Brn3b+ RGCs were significantly reduced (Fig. 1I, J). These differential effects on the amacrine and RGC populations could be due to the mosaic expression of the Cre transgene that results in partial deletion of the protein at E15 and thus some of the Brn3b+ cells may still express Bmal1. Alternatively, there is also an overall increase in programmed cell death in the Bmal1 conditional knockout (CKO) at P5 (Supplemental Fig. S4) that could partially account for the lower numbers of RGCs. Stronger decrease in Calretinin+ cells compared with the Brn3b+ cells could also be due to the differential expression pattern of Bmal1 at the single cell level. Recent study has shown that Bmal1 is expressed in RPCs but that perhaps intermediate- to late-stage RPCs that generate amacrine cells could have relatively higher Bmal1 expression due to the heterogeneity in the populations of RPCs and the overall levels of Bmal1 within these populations (31). To further assess if Bmal1 is required for the generation of other early-born cells, we also quantitated the cone photoreceptors. Surprisingly, we did not detect any change in the numbers of the cone cells despite these cells being early-born (Fig. 1K–M). However, similar to our previous findings (23), Bmal1 is required for the S-opsin patterning as we see a disruption of the S-opsin gradient in the Bmal1 CKO retinas (Fig. 1K and Supplemental Fig. S2).
Gamma-aminobutyric acid-ergic (GABAergic) and glycinergic are the 2 main subsets of amacrine cells, and the majority of the GABAergic amacrine cells are specified around E14-E16 (30). The likelihood that this subset could be most affected by the loss of Bmal1 was much higher, given that Bmal1 deletion resulted in loss of amacrine cells at E15. To gain better insights into the type of the GABAergic amacrine cells that require Bmal1, we counted the TH+ and ChAT+ cells in the retina. Bmal1 deletion did not affect dopaminergic TH+ amacrine cells (Fig. 2A–C); however, there was a significant reduction in the number of ChAT+ amacrine cells (Fig. 2D–F). Taken together, these results suggest that Bmal1 is required for the generation and maintenance of Brn3b+ RGCs and Calretinin+, ChAT+ amacrine cells.
Loss of Bmal1 results in increased number of S-phase cells within the retina
Brn3b+ RGCs and Calretinin+, ChAT+ amacrine cells are the earliest-born cells within the mouse retina. Based on the reduction in their numbers, we hypothesized that Bmal1 may be required for the proliferation of either the RPCs or the lineage-committed RPCs, and thus loss of Bmal1 would result in lower numbers of proliferating cells in the retina. To test this hypothesis, we used thymidine analog EdU to mark cells in the S-phase of the cell cycle (Fig. 3A). In the Bmal1 CKO retinas at E15 (Fig. 3B, C), there is an ∼15% increase (P < 0.014) in the number of EdU+ cells compared with the control retinas. These results suggest that in the absence of Bmal1, cells might be inappropriately re-entering the S-phase instead of exiting the cell cycle. To determine the cell cycle state of the S-phase cells, we performed a pulse chase EdU experiment. EdU pulse will label only the dividing cells, and the chase allows the identification of the progeny derived from the dividing cells. We injected pregnant dams with EdU at the gestational age of E14 and harvested the embryos 22 h postinjection. In the control retinas, most of the EdU+ cells could be detected in the proliferative outer neuroblastic layer (ONBL), which is also Ki67+ but not in the nonproliferative inner neuroblastic layer (INBL) (Fig. 4A). Interestingly, in the Bmal1 CKO both in the central region of the retina (Fig. 4A, B) as well as in the periphery (Fig.4C, D) there was a significantly (P = 0.005) higher number of cells in the non proliferative zone (Fig. 4E). To determine if these cells continue to re-enter the cell cycle, we quantitated the percent of cells that exited the cell cycle (details provided in the Materials and Methods section). Because of the large number of Ki67 positive cells in the ONBL, it was challenging to identify individual nonproliferative nuclei within the ONBL and hence we restricted our cell cycle exit analysis to the INBL. In the Bmal1 CKO retinas, we observed an increased number of EdU+ (Fig.4A–D), Ki67+ (Fig. 4A–D) cells that fail to exit the cell cycle (Fig. 4F). To further demonstrate that the loss of Bmal1 prevents the subset of RPCs from exiting the cell cycle, we used a different strategy to label the proliferating cells. We injected EdU in the pregnant dams at E13 and harvested the embryonic retinas 48 h later. A 48-h chase experiment will result in eventual dilution of the EdU signal as more cells divide, become postmitotic, and migrate out of the ONBL. We hypothesized that persistent EdU labeling will be observed in the Bmal1 CKO retinas because of their slow cell cycle exit. This protocol resulted in a sparse EdU labeling of the proliferating cells and allowed us to quantify relative proportion of the cells that exited the cell cycle (EdU+ PCNA−) (Supplemental Fig. S3). Consistent with what we observed in the INBL, in the mutants, there are a greater number of cells that continue to proliferate and fail to exit the cell cycle at the appropriate time, resulting in delayed cell cycle exit. One caveat of a longer chase experiment is that slow-cycling cells will still retain the EdU signal over a longer time period and will be positive for PCNA. This would not necessarily mean that these cells are re-entering the cell cycle but are just slowly cycling cells. However, in the Bmal1 CKO, because short pulses for EdU (5 h) showed greater numbers of cells that are in the S-phase, our pulse chase experiment results suggest that these cells failed to exit the cell cycle appropriately. Furthermore, we detected a similar increase in EdU+ cells at P5 (Fig. 3D–F), which suggests that Bmal1 function is required throughout retinal neurogenesis. Despite this increase in proliferation, we and others (22) have not seen any changes in the retinal thickness in the Bmal1 mutants (unpublished data). One possible explanation is that many of the inappropriately cycling cells may also be dying, as we do see an increased number of active caspase-3+ nuclei in the mutants compared with the controls (Supplemental Fig. S4).
Figure 3.

Increased cell proliferation that is due to loss of Bmal1. A–C) Embryonic retina from the control (A, a′, a″) and Bmal1 CKO mice (B, b′, b″), labeled with the S-phase marker EdU (Green). C) Number of EdU positive cells is higher in the Bmal1 CKO retina compared to the controls. D–F) The increase in cell proliferation continues into the post-natal retina. There is a higher number of EdU positive cells (Red) at P5 in the mutants (E, F) compared to the control (D, F). a′, a″, b′, b″, d′, d″, e′, e″) represent the boxed areas in A, B, D, E. C, F) Quantification of EdU+ cells at E15 (C) and P5 (F). Error bars are ±SEM, n = 6–8. Control group in C and F includes Bmal1Fl/Fl and Bmal1Fl/+ mice. *P < 0.05, **P < 0.01.
Bmal1 is essential for the correct localization of the Müller glia cells
BMAL1 is required during retinal neurogenesis for the cells to exit the cell cycle appropriately. Thus, in the absence of BMAL1, both early- and late-born cells would be affected. To assess whether any of the late-born cell numbers are altered, we counted the Recoverin+ type II cone bipolar cells and Sox9+ Müller glia cells. In the Bmal1 CKO retinas, there is a modest but significant increase in the total number of Recoverin+ cells compared with the control (Fig. 5A–C). Despite the increase in the number of Recoverin+ cells, their localization did not appear to be different between the mutant and the control. Müller glia cells are the only nonneuronal cells in the retina originating from RPCs and are the last fate to be specified. In the control retinas, Sox9+ Müller glia cells can be detected as a single row of cells within the inner nuclear layer (INL) (Fig. 5D). Interestingly, in the Bmal1 CKO retina, the Müller glia cells within the INL are highly disorganized, with several of the cells mislocalized to the outer nuclear layer (ONL) and outer plexiform layer (Fig. 5E). We observed a significant increase (P < 0.001) in Sox9+ Müller glia in the ONL of Bmal1 CKO retinas compared with the littermate control retinas at P21 (Fig. 5F). Despite their disorganization within the INL, densities of the Müller cells within the INL remained unaltered between control and Bmal1 CKO groups (Fig. 5G). Furthermore, the mislocalized Sox9+ Müller cells continue to survive till P90 (unpublished data), suggesting that the lamination defects are persistent.
Bmal1 is required for the correct organization of the retinal circuitry
To evaluate whether the observed histochemical changes in RGC and amacrine neurons can affect visual transmission, we measured the VEP in response to the flash stimulus under dark-adapted and light-adapted conditions. VEP measurements were done at P21 and 4 mo of age. The mouse VEP waveforms display a characteristic negative peak (N1), which is preceded by P1 (first positive peak) and followed by P2 (second positive peak) (Fig. 6A) (29). The implicit time to attain N1 decreases with increased flash intensities. At P21, there is no difference between the Bmal1 control and CKO for the P1-N1 (Fig.6B, C) or the N1-P2 (Fig. 6C, E) amplitudes under dark adapted conditions. Moreover, at all flash intensities, though there was no difference in N1 latency between the control and Bmal1 CKO group (Fig. 6D), the implicit time to the second positive peak (P2) was almost significantly increased in the Bmal1 CKO group (P = 0.057) (Fig. 6E). Under light adapted conditions, the Bmal1 CKO mice display shorter N1 implicit time (faster response) at low flash intensity (P < 0.001 and 0.002 for 0.0 and 0.4 log cd s/m2, respectively) (Fig. 6F, I), which suggests that Bmal1 CKO animals respond faster to the lower flash intensities compared with the littermate controls under light-adapted conditions. Although not statistically significant, under light-adapted conditions, the implicit time to the second positive peak (P2) was longer in the Bmal1 CKO group compared with the control group (Fig. 6J). In the 4-month old animals, under the dark adapted conditions the VEP response between the control and Bmal1 CKO was indistinguishable (Fig. 7A–E). Under light adapted conditions, again the amplitudes were not different (Fig. 7F–H) but the N1 implicit time was significantly higher (slower response) for high-intensity flash stimulus under light-adapted conditions (P = 0.012 and 0.007 for 1.4 and 1.9 log cd s/m2, respectively) (Fig. 7I). Thus, the loss of Bmal1 results in specific changes in neural circuitry, and the age-related differences may reflect degenerative changes within the retina.
Figure 6.

Visual transduction defects that are due to loss of Bmal1. A, F) Representative VEP traces obtained from P21 control (gray traces) and Bmal1 CKO mice (orange traces) under dark-adapted (A) and light-adapted (F) conditions. N1 represents negative peak, which is preceded by P1 (first positive peak) and followed by P2 (second positive peak). B–E, G–J) Light-adapted N1 latencies were faster (lower implicit time) at P21 (I) in Bmal1 CKO, whereas the time taken to reach the P2 was slower (higher implicit time) under both light-adapted (J) and dark-adapted (E) conditions. Dark-adapted N1 latencies (D) and VEP amplitudes for dark-adapted (B, C) or light-adapted (G, H) conditions were not significantly altered between the groups. Control group includes Bmal1Fl/Fl and Bmal1Fl/+ mice. Error bars ± sem, n = 5–6. #P = 0.057 between the groups (E), *P < 0.05 for pairwise comparison between the groups for specific flash intensity (I).
Figure 7.

A, F) Representative VEP traces obtained from 4-mo-old control (gray traces) and Bmal1 CKO mice (orange traces) under dark-adapted (A) and light-adapted (F) conditions. N1 represents negative peak, which is preceded by P1 (first positive peak) and followed by P2 (second positive peak). B–E, G–J) Light-adapted N1 latencies were slower (higher implicit time) at 4 mo of age (I) in Bmal1 CKO mice compared with controls. Dark-adapted N1 (D) and P2 (E) latencies, light-adapted P2 latencies (J) and VEP amplitudes for dark-adapted (B, C) or light-adapted (G, H) conditions were not significantly altered between the groups. F, H) Control group includes only Bmal1Fl/Fl mice. Error bars ± sem, n = 6. *P < 0.05 for pairwise comparison between the groups for specific flash intensity (I).
DISCUSSION
Cell cycle and neurogenesis
Our data suggest that Bmal1 regulates the timing of the cell cycle exit. Accordingly, loss of Bmal1 causes either the RPCs or the lineage-restricted progenitor cells to re-enter the cell cycle, resulting in the higher number of cells in the S-phase. These data further support the idea that the production of the different retinal neurons is under the control of an intrinsic timer that is linked to the length of the cell cycle. Indeed, it has been demonstrated that as the timing of the cell cycle changes so does the multipotency of the RPCs (9). The earliest-born cells are generated from fast-cycling RPCs, whereas the late-born cells are generated from slow-cycling RPCs (9). Thus, it would be an additional mechanism to consider that the length of the cell cycle will significantly alter the potency of the RPCs and their ability to generate certain types of neurons. Consistent with this explanation are the phenotypes reported in several cell cycle mutants that alter the dynamics of cell cycle progression. For example, loss of cyclin D1, a protein that controls cell cycle transition from G1 to the S-phase, results in a loss of RGCs and an increase in the fate of late-born cells (12). Similarly, loss of cyclin D2 alters the cell cycle duration of the cells within the ciliary marginal zone, resulting in a reduction in the ipsilateral and contralateral RGC population (32). Alternatively, loss of mediators that affect cell cycle exit, like p57(kip2), leads to distinct changes in cell proliferation and cell specification, suggesting that these cell cycle mediators affect fate specification (14). The role of Bmal1 in cell cycle kinetics of retinal progenitors is undetermined. Based on previous findings in other tissues (33–35), we speculate that Bmal1 could be regulating the gating of the S-phase entry. Gating would ensure that cells proliferate during certain times of the day or during the night. Circadian regulation of gating is believed to be an evolutionarily conserved mechanism that has been proposed to represent an adaptation of ancestral unicellular organisms to reduce UV-induced DNA damage. This could be an alternative explanation for the observed increase in the S-phase marker in the Bmal1 mutants and would suggest an evolutionarily beneficial role for the circadian clock in ensuring that cellular damage is prevented by gating cell proliferation in the retina. Several of the cell cycle regulators, including c-myc, cyclin D1, and Wee1, are regulated in a circadian manner (36, 37). Thus, it remains possible that Bmal1 could be regulating a number of these factors during retinal neurogenesis, which reflects a functional role of the circadian clock in the retina. A similar role for Bmal1 in regulating the timing of adult neurogenesis has been reported (33). In this analysis, the authors have demonstrated that the cell cycle entry and exit of the quiescent neural progenitor cells are under the control of the circadian clock. To our knowledge, there is no known analysis demonstrating a functional circadian clock in the embryonic retina, except in the chicken (38). Though Bmal1 is a component of the core circadian clock, whether its role in retinal neurogenesis is due to a disruption in diurnal rhythms is yet to be determined.
Bmal1 is required for the proliferation and survival of a subpopulation of the lineage-restricted RPCs
Despite the loss of Bmal1 from the subset of progenitor cells that are competent to give rise to both early-born and late-born cells, our analysis suggests that only a small subset of certain neuronal types is affected by the loss of Bmal1. For example, within the early-born fates, only the numbers of Brn3b+ RGCs and Calretinin+/ChAT+ amacrines are altered, but the densities of cone photoreceptors and TH+ dopaminergic amacrines were unchanged. Although we have not quantitated the rods, others have reported (22, 39) that in the Bmal1 CKO animals, rod function is unaffected but the cones are functionally compromised. Consistent with these reports and our previous studies, though we did not see any changes in the total number of cones, their spectral identity is controlled by Bmal1. Accordingly, in the Bmal1 CKO animals, at P21 (Fig. 1I) and at P180 (Supplemental Fig. S2), there is an increased S-opsin expression in the dorsal retina compared with that in the controls. It is important to mention that, though Bmal1 CKO animals have been previously analyzed by Storch et al. (22), these authors did not report any changes in the retinal organization and retinal thickness. The focus of their study was to determine the role of Bmal1 in the regulation of retinal physiology and the overall effects of Bmal1 deletion on the oscillation of circadian-dependent transcripts within the retina. Using electroretinogram (ERG) analysis, they demonstrated that in the mice lacking retinal Bmal1, rod photoreceptor function was unaffected but cone function as well as inner retinal processing was abnormal. This would be in agreement with our observed changes in the S-opsin patterning in the Bmal1 CKO retina, which would result in functional deficits in cone function. Moreover, the increased numbers of Recoverin+ bipolar cells in the Bmal1CKO animals could account for the abnormal inner retinal processing. Cone function is also compromised in other circadian clock mutants like Cry1/Cry2 double mutant animals (40). Contrary to these phenotypes, the Per1/Per2 double mutants have defects in S-opsin distribution, but the cone ERG responses are unaffected (41). Our analysis suggests that the functional changes in the Bmal1 CKO animals are due to early disruption in retinal development, and it remains to be seen whether a similar phenotype exists in other core circadian clock mutants.
One of the intriguing results from our data is the differential effect of Bmal1 on neuronal subtypes. Restricted expression of Bmal1 within a subpopulation of the committed RPCs could account for these differential effects. This could explain why the TH+ amacrine cells were unaffected but the ChAT+ population was reduced, if Bmal1 was expressed only in a ChAT+ lineage-restricted amacrine cells. We cannot rule out the possibility that the glycinergic subset of amacrine cells also requires Bmal1 for their specification. Nonetheless, our data support the idea that despite the multipotent nature of the RPCs, they may be representing multiple subpopulations that are lineage restricted. Indeed, with single cell profiling of RPCs, it is obvious that within the RPCs there is a large heterogeneity in the transcriptional profiles of any single cell. Thus, it is conceivable that there are several committed subpopulations of RPCs that are responsible for generating the different subtypes of the retinal neurons. Loss of Bmal1 had the strongest effect on amacrine numbers followed by RGCs, type II cone bipolar, and Müller glia cells. The effect of Bmal1 on these neurons perhaps reflects the intrinsic functional requirement of the circadian clock within these cell types. In the adult retina, it is known that some neurons can express their own intrinsic clock and thus regulate physiologic events in the retina (21, 42, 43). It would be interesting to speculate that Bmal1 is required for the proliferation of certain neuronal subtypes that are capable of generating rhythmic activity that is due to the presence of an intrinsic clock. Future studies with specific markers will perhaps answer this question.
Effects on retinal circuitry
Our data suggest that early loss of Bmal1 from the retina affects visual transduction. Recordings from the visual cortex of Bmal1 CKO animals demonstrate that the implicit time for the evoked potentials is shorter under the light-adapted condition at P21. Another clock gene, Rev-Erbα is also abundantly expressed in the photoreceptor layer, outer plexiform layer, and the ganglion cell layer (44). Dark-adapted ERG responses recorded from the Rev-Erbα mutant mice showed shorter latencies compared with the wild types at all intensities (45). Faster conductance at low flash intensities in Bmal1 CKO mice could be due to an increased number of Müller glia. In order to complete the visual cycle, all-trans-retinal, released from bleached photopigments must be converted to all-cis-retinal. Parts of this recycling process occurs in the retinal pigment epithelium (RPE). However, recent studies have indicated that the cone photoreceptors use Müller glia to convert all-trans-retinol to 11-cis-retinol, which is eventually converted back to the 11-cis-retinal within the cone photoreceptors [reviewed in ref. (46)]. It has also been proposed that a similar recycling pathway exists between the Müller glia and the intrinsically photosensitive RGCs in the vertebrate retinas (47). In the Bmal1 CKO mice, there is an overall increase in the number of Müller cells, and though we do not know if the mislocalized Müller cells are functional, it is conceivable that faster recycling of photopigment could contribute to the visual conductance.
Another plausible explanation is that the reduced number of early-born GABAergic amacrine cells could be altering the local circuitry and contributing toward the VEP defects. In the mouse retina, the majority of GABAergic amacrine cells are Calretinin+ (48). Studies in humans have shown that elevation of endogenous GABA caused by the administration of GABA transporter-1 blocker tiagabine results in slower latencies and decreased VEP amplitude recorded by magnetoencephalographic signals (49). However, we still observe a slower visual conductance for dark-adapted responses at P21 and light-adapted responses at 4 mo of age. Slower visual conduction at the older age could be due to faster aging and compromised integrity of visual synaptic connectivity. We and others have reported that the loss of Bmal1 from the photoreceptors results in accelerated aging of the cones (23, 39). Several of the BMAL1 target genes, such as Robo1, Slit2, Unc5d, Pik3r1, Ablim1, and Sema3a, are important for axon guidance and maintaining synaptic connectivity (23). Members of Slit-Robo signaling pathway and Sema3a play an important function in retinal axon guidance, and their expression pattern is altered with age (50, 51). Using a Xenopus model, Campbell et al. (50) demonstrated that RGC axonal growth cones collapse in response to Sema3a is an age-dependent process, and older growth cones are more sensitive than young-stage growth cones. Based on these previous reports and our data, we propose that the circadian clock gene Bmal1 is required for maintaining the integrity of retinal circuitry in adult mice, and loss of Bmal1 results in accelerated aging.
In summary, this work provides the first evidence of a functional integration between the circadian clock gene Bmal1 and cell cycle duration during retinal neurogenesis. Further work will be needed to identify the molecular links that integrate the circadian clock to cell fate specification within the retina.
Supplementary Material
This article includes supplemental data. Please visit http://www.fasebj.org to obtain this information.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
ACKNOWLEDGMENTS
The authors are grateful to the Biological Resources Unit and Veterinary Services at Cleveland Clinic for their assistance with mouse colony management. This research was funded by grants from the U.S. National Institutes of Health/National Eye Institute EY027077-01 (to S.R.), RPB1503 (to S.R.), the Knights Templar Eye Foundation Career Starter Research Grant KTEF1806 (to O.B.S.), the U.S. Department of Veterans Affairs (to N.S.P.), a National Eye Institute P30-EY025585 Core Grant and Research to Prevent Blindness Challenge Grant. The authors declare no conflicts of interest.
Glossary
- BMAL1
brain and muscle arnt-like protein-1
- ChAT
choline acetyltransferase
- CKO
conditional knockout
- CLOCK
circadian locomotor output cycles kaput
- EdU
5-ethynyl-2′-deoxyuridine
- ERG
electroretinogram
- GABAergic
gamma-aminobutyric acid-ergic
- INBL
inner neuroblastic layer
- INL
inner nuclear layer
- ONBL
outer neuroblastic layer
- ONL
outer nuclear layer
- PCNA
proliferating cell nuclear antigen
- RGC
retinal ganglion cell
- RPC
retinal progenitor cell
- RT
room temperature
- SOX9
sex-determining region Y-box 9
- TH
tyrosine hydroxylase
- VEP
visual evoked potential
Footnotes
This article includes supplemental data. Please visit http://www.fasebj.org to obtain this information.
AUTHOR CONTRIBUTIONS
O. B. Sawant, V. K. Jidigam, R. D. Fuller, O. F. Zucaro, C. Kpegba, M. Yu, and S. Rao conducted the experiments and investigations; O. B. Sawant, V. K. Jidigam, M. Yu, N. S. Peachey, and S. Rao performed the data analysis; O. B. Sawant, M. Yu, and S. Rao wrote the manuscript; O. B. Sawant and S. Rao designed the research; and the final version of the manuscript was reviewed and approved by all the authors.
REFERENCES
- 1.Swaroop A., Kim D., Forrest D. (2010) Transcriptional regulation of photoreceptor development and homeostasis in the mammalian retina. Nat. Rev. Neurosci. 11, 563–576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Turner D. L., Cepko C. L. (1987) A common progenitor for neurons and glia persists in rat retina late in development. Nature 328, 131–136 [DOI] [PubMed] [Google Scholar]
- 3.Cepko C. (2014) Intrinsically different retinal progenitor cells produce specific types of progeny. Nat. Rev. Neurosci. 15, 615–627 [DOI] [PubMed] [Google Scholar]
- 4.Brown N. L., Patel S., Brzezinski J., Glaser T. (2001) Math5 is required for retinal ganglion cell and optic nerve formation. Development 128, 2497–2508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brown N. L., Kanekar S., Vetter M. L., Tucker P. K., Gemza D. L., Glaser T. (1998) Math5 encodes a murine basic helix-loop-helix transcription factor expressed during early stages of retinal neurogenesis. Development 125, 4821–4833 [DOI] [PubMed] [Google Scholar]
- 6.Nishida A., Furukawa A., Koike C., Tano Y., Aizawa S., Matsuo I., Furukawa T. (2003) Otx2 homeobox gene controls retinal photoreceptor cell fate and pineal gland development. Nat. Neurosci. 6, 1255–1263 [DOI] [PubMed] [Google Scholar]
- 7.Bassett E. A., Wallace V. A. (2012) Cell fate determination in the vertebrate retina. Trends Neurosci. 35, 565–573 [DOI] [PubMed] [Google Scholar]
- 8.Sakagami K., Gan L., Yang X. J. (2009) Distinct effects of Hedgehog signaling on neuronal fate specification and cell cycle progression in the embryonic mouse retina. J. Neurosci. 29, 6932–6944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Alexiades M. R., Cepko C. (1996) Quantitative analysis of proliferation and cell cycle length during development of the rat retina. Dev. Dyn. 205, 293–307 [DOI] [PubMed] [Google Scholar]
- 10.Miles A., Tropepe V. (2016) Coordinating progenitor cell cycle exit and differentiation in the developing vertebrate retina. Neurogenesis (Austin) 3, e1161697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Das G., Choi Y., Sicinski P., Levine E. M. (2009) Cyclin D1 fine-tunes the neurogenic output of embryonic retinal progenitor cells. Neural Dev. 4, 15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Das G., Clark A. M., Levine E. M. (2012) Cyclin D1 inactivation extends proliferation and alters histogenesis in the postnatal mouse retina. Dev. Dyn. 241, 941–952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Blackshaw S., Harpavat S., Trimarchi J., Cai L., Huang H., Kuo W. P., Weber G., Lee K., Fraioli R. E., Cho S. H., Yung R., Asch E., Ohno-Machado L., Wong W. H., Cepko C. L. (2004) Genomic analysis of mouse retinal development. PLoS Biol. 2, E247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dyer M. A., Cepko C. L. (2000) p57(Kip2) regulates progenitor cell proliferation and amacrine interneuron development in the mouse retina. Development 127, 3593–3605 [DOI] [PubMed] [Google Scholar]
- 15.Cunningham J. J., Levine E. M., Zindy F., Goloubeva O., Roussel M. F., Smeyne R. J. (2002) The cyclin-dependent kinase inhibitors p19(Ink4d) and p27(Kip1) are coexpressed in select retinal cells and act cooperatively to control cell cycle exit. Mol. Cell. Neurosci. 19, 359–374 [DOI] [PubMed] [Google Scholar]
- 16.Takahashi J. S. (2017) Transcriptional architecture of the mammalian circadian clock. Nat. Rev. Genet. 18, 164–179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.McMahon D. G., Iuvone P. M., Tosini G. (2014) Circadian organization of the mammalian retina: from gene regulation to physiology and diseases. Prog. Retin. Eye Res. 39, 58–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Masri S., Cervantes M., Sassone-Corsi P. (2013) The circadian clock and cell cycle: interconnected biological circuits. Curr. Opin. Cell Biol. 25, 730–734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Matsuo T., Yamaguchi S., Mitsui S., Emi A., Shimoda F., Okamura H. (2003) Control mechanism of the circadian clock for timing of cell division in vivo. Science 302, 255–259 [DOI] [PubMed] [Google Scholar]
- 20.Lin K. K., Kumar V., Geyfman M., Chudova D., Ihler A. T., Smyth P., Paus R., Takahashi J. S., Andersen B. (2009) Circadian clock genes contribute to the regulation of hair follicle cycling. PLoS Genet. 5, e1000573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu X., Zhang Z., Ribelayga C. P. (2012) Heterogeneous expression of the core circadian clock proteins among neuronal cell types in mouse retina. PLoS One 7, e50602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Storch K. F., Paz C., Signorovitch J., Raviola E., Pawlyk B., Li T., Weitz C. J. (2007) Intrinsic circadian clock of the mammalian retina: importance for retinal processing of visual information. Cell 130, 730–741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sawant O. B., Horton A. M., Zucaro O. F., Chan R., Bonilha V. L., Samuels I. S., Rao S. (2017) The circadian clock gene Bmal1 controls thyroid hormone-mediated spectral identity and cone photoreceptor function. Cell Rep. 21, 692–706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Baba K., Ribelayga C. P., Michael Iuvone P., Tosini G. (2018) The retinal circadian clock and photoreceptor viability. Adv. Exp. Med. Biol. 1074, 345–350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Baba K., Tosini G. (2018) Aging alters circadian rhythms in the mouse eye. J. Biol. Rhythms 33, 441–445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sawant O., Horton A. M., Shukla M., Rayborn M. E., Peachey N. S., Hollyfield J. G., Rao S. (2015) Light-regulated thyroid hormone signaling is required for rod photoreceptor development in the mouse retina. Invest. Ophthalmol. Vis. Sci. 56, 8248–8257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yu M., Narayanan S. P., Wang F., Morse E., Macklin W. B., Peachey N. S. (2011) Visual abnormalities associated with enhanced optic nerve myelination. Brain Res. 1374, 36–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yu M., Sturgill-Short G., Ganapathy P., Tawfik A., Peachey N. S., Smith S. B. (2012) Age-related changes in visual function in cystathionine-beta-synthase mutant mice, a model of hyperhomocysteinemia. Exp. Eye Res. 96, 124–131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ridder W. H., III, Nusinowitz S. (2006) The visual evoked potential in the mouse--origins and response characteristics. Vision Res. 46, 902–913 [DOI] [PubMed] [Google Scholar]
- 30.Voinescu P. E., Kay J. N., Sanes J. R. (2009) Birthdays of retinal amacrine cell subtypes are systematically related to their molecular identity and soma position. J. Comp. Neurol. 517, 737–750; erratum: 518, 254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Clark B., Stein-O’Brien G., Shiau F., Cannon G., Davis E., Sherman T., Rajaii F., James-Esposito R., Gronostajski R., Fertig E., Goff L., Blackshaw S. (2018) Comprehensive analysis of retinal development at single cell resolution identifies NFI factors as essential for mitotic exit and specification of late-born cells. bioRxiv [Google Scholar]
- 32.Marcucci F., Soares C. A., Mason C. (2019) Distinct timing of neurogenesis of ipsilateral and contralateral retinal ganglion cells. J. Comp. Neurol. 527, 212–224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bouchard-Cannon P., Mendoza-Viveros L., Yuen A., Kærn M., Cheng H. Y. (2013) The circadian molecular clock regulates adult hippocampal neurogenesis by controlling the timing of cell-cycle entry and exit. Cell Rep. 5, 961–973 [DOI] [PubMed] [Google Scholar]
- 34.Roenneberg T., Foster R. G. (1997) Twilight times: light and the circadian system. Photochem. Photobiol. 66, 549–561 [DOI] [PubMed] [Google Scholar]
- 35.Nikaido S. S., Johnson C. H. (2000) Daily and circadian variation in survival from ultraviolet radiation in Chlamydomonas reinhardtii. Photochem. Photobiol. 71, 758–765 [DOI] [PubMed] [Google Scholar]
- 36.Okamura H. (2004) Clock genes in cell clocks: roles, actions, and mysteries. J. Biol. Rhythms 19, 388–399 [DOI] [PubMed] [Google Scholar]
- 37.Hunt T., Sassone-Corsi P. (2007) Riding tandem: circadian clocks and the cell cycle. Cell 129, 461–464 [DOI] [PubMed] [Google Scholar]
- 38.Lima L. H., Santos K. P., Lauro Castrucci A. M. (2011) Clock genes, melanopsins, melatonin, and dopamine key enzymes and their modulation by light and glutamate in chicken embryonic retinal cells. Chronobiol. Int. 28, 89–100 [DOI] [PubMed] [Google Scholar]
- 39.Baba K., Piano I., Lyuboslavsky P., Chrenek M. A., Sellers J. T., Zhang S., Gargini C., He L., Tosini G., Iuvone P. M. (2018) Removal of clock gene Bmal1 from the retina affects retinal development and accelerates cone photoreceptor degeneration during aging. Proc. Natl. Acad. Sci. USA 115, 13099–13104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cameron M. A., Barnard A. R., Hut R. A., Bonnefont X., van der Horst G. T., Hankins M. W., Lucas R. J. (2008) Electroretinography of wild-type and Cry mutant mice reveals circadian tuning of photopic and mesopic retinal responses. J. Biol. Rhythms 23, 489–501 [DOI] [PubMed] [Google Scholar]
- 41.Ait-Hmyed O., Felder-Schmittbuhl M. P., Garcia-Garrido M., Beck S., Seide C., Sothilingam V., Tanimoto N., Seeliger M., Bennis M., Hicks D. (2013) Mice lacking Period 1 and Period 2 circadian clock genes exhibit blue cone photoreceptor defects. Eur. J. Neurosci. 37, 1048–1060 [DOI] [PubMed] [Google Scholar]
- 42.Xu L., Ruan G., Dai H., Liu A. C., Penn J., McMahon D. G. (2016) Mammalian retinal Müller cells have circadian clock function. Mol. Vis. 22, 275–283 [PMC free article] [PubMed] [Google Scholar]
- 43.Besharse J. C., McMahon D. G. (2016) The retina and other light-sensitive ocular clocks. J. Biol. Rhythms 31, 223–243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mollema N. J., Yuan Y., Jelcick A. S., Sachs A. J., von Alpen D., Schorderet D., Escher P., Haider N. B. (2011) Nuclear receptor Rev-erb alpha (Nr1d1) functions in concert with Nr2e3 to regulate transcriptional networks in the retina. PLoS One 6, e17494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ait-Hmyed Hakkari O., Acar N., Savier E., Spinnhirny P., Bennis M., Felder-Schmittbuhl M. P., Mendoza J., Hicks D. (2016) Rev-Erbα modulates retinal visual processing and behavioral responses to light. FASEB J. 30, 3690–3701 [DOI] [PubMed] [Google Scholar]
- 46.Wang J. S., Kefalov V. J. (2011) The cone-specific visual cycle. Prog. Retin. Eye Res. 30, 115–128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Schmidt T. M., Chen S. K., Hattar S. (2011) Intrinsically photosensitive retinal ganglion cells: many subtypes, diverse functions. Trends Neurosci. 34, 572–580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Haverkamp S., Wässle H. (2000) Immunocytochemical analysis of the mouse retina. J. Comp. Neurol. 424, 1–23 [PubMed] [Google Scholar]
- 49.Muthukumaraswamy S. D., Myers J. F., Wilson S. J., Nutt D. J., Hamandi K., Lingford-Hughes A., Singh K. D. (2013) Elevating endogenous GABA levels with GAT-1 blockade modulates evoked but not induced responses in human visual cortex. Neuropsychopharmacology 38, 1105–1112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Campbell D. S., Regan A. G., Lopez J. S., Tannahill D., Harris W. A., Holt C. E. (2001) Semaphorin 3A elicits stage-dependent collapse, turning, and branching in Xenopus retinal growth cones. J. Neurosci. 21, 8538–8547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Erskine L., Williams S. E., Brose K., Kidd T., Rachel R. A., Goodman C. S., Tessier-Lavigne M., Mason C. A. (2000) Retinal ganglion cell axon guidance in the mouse optic chiasm: expression and function of robos and slits. J. Neurosci. 20, 4975–4982 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.