

Abstract
Nerve growth factor (NGF) plays a central role in multiple chronic pain conditions. As such, anti-NGF monoclonal antibodies (mAbs) that function by antagonizing NGF downstream signaling are leading drug candidates for non-opioid pain relief. To evaluate anti-canine NGF (cNGF) mAbs we sought a yeast surface display platform of cNGF. Both mature cNGF and pro-cNGF displayed on the yeast surface but bound conformationally sensitive mAbs at most 2.5-fold in mean fluorescence intensity above background, suggesting that cNGF was mostly misfolded. To improve the amount of folded, displayed cNGF, we used comprehensive mutagenesis, FACS, and deep sequencing to identify point mutants in the pro-region of canine NGF that properly enhance the folded protein displayed on the yeast surface. Out of 1,737 tested single point mutants in the pro region, 49 increased the amount of NGF recognized by conformationally sensitive mAbs. These gain-of-function mutations cluster around residues A-61-P-26. Gain-of-function mutants were additive, and a construct containing three mutations increased amount of folded cNGF to 23-fold above background. Using this new cNGF construct, fine conformational epitopes for tanezumab and three anti-cNGF mAbs were evaluated. The epitope revealed by the yeast experiments largely overlapped with the tanezumab epitope previously determined by X-ray crystallography. The other mAbs showed site-specific differences with tanezumab. As the number of binding epitopes of functionally neutralizing anti-NGF mAbs on NGF are limited, subtle differences in the individual interacting residues on NGF that bind each mAb contribute to the understanding of each antibody and variations in its neutralizing activity. These results demonstrate the potential of deep sequencing-guided protein engineering to improve the production of folded surface-displayed protein, and the resulting cNGF construct provides a platform to map conformational epitopes for other anti-neurotrophin mAbs.
Keywords: conformational epitope mapping, deep mutational scanning, nerve growth factor pro region, tanezumab, yeast display
1 |. INTRODUCTION
Nerve growth factor (NGF) was the first discovered member of the neurotrophin family, which also includes brain-derived neurotrophic factor (BDNF), neurotrophin-3, and neurotrophin-4. This family of proteins regulates the development, function, and survival of neurons in the peripheral and central nervous system (Huang & Reichardt, 2001; Lessmann, Gottmann, & Malcangio, 2003). Neurotrophins activate downstream signaling pathways by binding the pan-neuro-trophin receptor, p75NTR, and to the family of tropomyosin receptor kinases (TrkA, TrkB, and TrkC) with various affinities. NGF is synthesized as a pre-pro protein. The N-terminal pre sequence is released during translocation to the endoplasmic reticulum while the pro-peptide is often but not always cleaved by proprotein convertases prior to secretion. The NGF mature domain is approximately 120 amino acids and arranged as noncovalent homodimers where each monomer conformation possesses a cysteine knot created by three disulfide bonds (Park & Poo, 2013; Rattenholl, Lilie, et al., 2001).
Multiple studies have demonstrated the high levels of NGF during peripheral nerve injury, inflammation, and chronic pain (Slosky, Largent-Milnes, & Vanderah, 2015; Watson, Allen, & Dawbarn, 2008). As a consequence, researchers have developed anti-NGF monoclonal anti-bodies (mAbs) as potential medicines to modulate chronic pain and many other conditions. These mAbs function by interfering with binding to p75NTR and/or TrkA. Humanized Tanezumab (La Porte et al., 2014) is furthest along in a phase III clinical trial. Comprehensive reviews of clinical studies with tanezumab and others mAbs are found elsewhere (Chang, Hsu, Hottinger, & Cohen, 2016; Kumar & Mahal, 2012). As these signaling pathways are highly conserved in higher mammals, here we have investigated a panel of mAbs against canine NGF (cNGF). Canine NGF was used for analysis as this is of interest to Zoetis Animal Health and varies from human NGF by only three amino acids.
An important step in evaluating the neutralizing capacity of antibodies is to determine the epitopes that they target. To that end, yeast surface display (Chao et al., 2006) is a validated platform to determine fine conformational epitopes for complicated proteins (Adams, Mora, Walczak, & Kinney, 2016; Doolan & Colby, 2015; Kowalsky, Faber, et al., 2015). Typically, a set of mutants of the target protein is displayed on the surface of yeast, assessed for antibody binding, with loss of binding mutants mapping to the epitope. A major limitation of the method is the requirement that the displayed target protein be in a conformation recognizable by the antibody. This is an issue with cNGF and consistent with a previous study showing that the related neurotrophin BDNF displayed on the surface of yeast in a mostly inactive conformation (Burns et al., 2014).
A previous directed evolution study showed that mutations to the pro region could enhance the folding of the related neurotrophin human BDNF in Saccharomyces cerevisiae (Burns etal., 2016). Evidence suggests that neurotrophin pro regions act as chaperones to assist folding of a mature neurotrophin as they pass through the secretory pathway (Hauburger, Kliemannel, Madsen, Rudolph, & Schwarz, 2007; Kliemannel, Golbik, Rudolph, Schwarz, & Lilie, 2007; Nomoto, Takaiwa, Mouri, & Furukawa, 2007; Rattenholl, Ruoppolo, et al., 2001). The pro peptide is monomeric and highly flexible as shown by the lack of electron density in a solved structure of a proNGF complex (Feng et al., 2010) and biophysical analysis in vitro (Kliemannel et al., 2004). Two domains are sufficient to process and express active mouse NGF (Suter, Heymach, Shooter, & Shooter, 1991) (Box 3 and Box 5, shown for canine and human NGF—see Figure 1a). Three dibasic sites are proteolytically cleaved during processing of mature NGF through the secretory pathway (Nomoto et al., 2007; Pagadala, Dvorak, & Neet, 2006).
FIGURE 1.
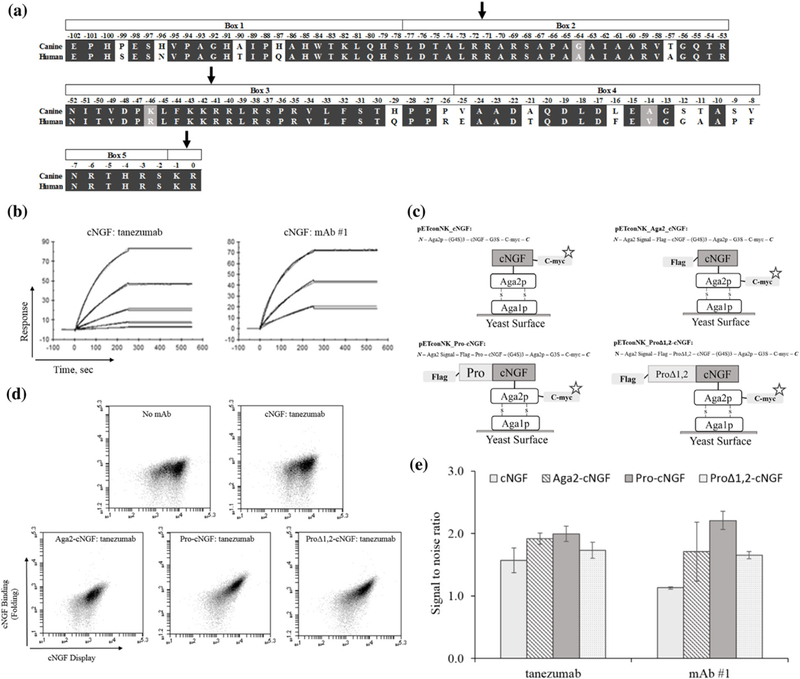
cNGF yeast display constructs are mostly misfolded as probed by conformationally sensitive mAbs. (a) Sequence alignment of the canine and human pro regions of NGF. Domain boundaries and dibasic protease cleavage sites are shown. (b) Surface plasmon resonance sensorgrams of cNGF:conformational mAb binding. cNGF was immobilized on a CM5 surface by amine coupling and either tanezumab or mAb#1 was injected and flowed over surface at various concentrations starting at 100 nM and titrating down with threefold dilutions, flowed over the chips. (c) Four different cNGF constructs tested in the present work. (d and e) Flow cytograms (d) and bar charts (e) showing increase in fluorescence in cNGF binding channel probed by tanezumab and mAb #1 (error bars, standard error of the mean, n ≥ 3). The signal:noise ratios were obtained by calculating the ratio of the MFI of the sample to the MFI in the absence of biotinylated mAb
In the current study we developed a yeast display platform for the production of folded cNGF. We used yeast surface display, saturation mutagenesis, fluorescence activated cell sorting (FACS), and deep sequencing to identify mutations in the pro-region that enhanced display of folded cNGF. Mutational libraries created using this engineering pipeline revealed new insight into the role of the neurotrophin pro region. Combinations of mutations yielded constructs with a 23-fold increase in the signal to noise ratio of display of folded cNGF over background as measured by mean fluorescence intensity (MFI). This engineered pro-cNGF allowed us to generate conformational epitope maps of multiple anti-cNGF antibodies. All anti-cNGF mAbs had an overlapping footprint with tanezumab but each had several site-specific differences. This research improves our understanding of sequence-function relationships in pro sequences for neurotrophins and highlights the power of deep sequencing to augment classical directed evolution experimental pipelines (Wren-beck, Faber, & Whitehead, 2017).
2. |. MATERIALS AND METHODS
2.1 |. Plasmid constructs
pETconNK_cNGF, pETconNK_Aga2_cNGF, pETconNK_procNGF, and pETconNK_proΔ1,2-cNGF were prepared by cloning custom codon-optimized genes (GenScript, Piscataway, NJ) into pETconNK (Wrenbecketal., 2016) (Addgene plasmid #81169) using standard restriction cloning. Sequences were verified by Sanger sequencing (Genewiz, South Plainfield, NJ), with full sequences listed in Supporting Note 1.
2.2 |. Preparation of anti-NGF mAbs
Tanezumab, a humanized anti-NGF mAb, was expressed recombinantly in CHO cells based upon the published sequences (La Porte et al., 2014) on hIgG2/kappa constant regions. This antibody was purified using Protein A resin, dialyzed into PBS, and sterile filtered. Three different caninized antibodies (mAb #1, mAb #2, and mAb #3) were supplied by Zoetis Inc. These antibodies were also expressed in CHO cells, affinity purified using Protein A resin, and dialyzed into either PBS or 20 mM Na Acetate, 150 nM NaCl, pH 7.4 buffer. Concentrations were assessed via A280 absorbance using the Edelhoch method and ranged from 1.5 mg/ml to 6.45 mg/ml final concentration. At least 1.5 mg/ml mAbs in PBS were biotinylated at a molar ratio of 1:20 mAb: biotin using the EZ-link NHS-biotin kit following the manufacturer’s instruction (Life Technologies, Carlsbad, CA). Biotinylated mAbs were then desalted into PBS using Zeba Spin desalting columns (Thermo Fisher, Waltham, MA) following the manufacturer’s instruction and stored at 4°C.
2.3 |. TF-1 cell proliferation assay
Functional potency of antibodies against cNGF was evaluated in a cell proliferation assay utilizing the TF-1 cell line which expresses human TrkA (Kitamura et al., 1989). TF-1 cells (American Type Culture Collection [ATCC], Rockville, MD) were maintained in ATCC modified RPMI 1640 medium (Life Technologies) supplemented with 10% FBS (Life Technologies) and 2ng/ml recombinant human GM-CSF (R&D Systems Inc., Minneapolis, MN) and incubated at37°C with 5%CO2. On the day of experiment, TF-1 cells were washed twice with DPBS (Life Technologies) before resuspending in proliferation assay medium: ATCC modified RPMI 1640 supplemented with 1% FBS and 10μg/ml gentamicin. The TF-1 proliferation assay was performed in 96 well microplates (Corning Inc., Corning, NY) by incubating 15,000 cells per well with anti-NGF antibodies at concentrations indicated and 2 ng/ml recombinant cNGF. cNGF was generated at Zoetis in stable CHOK1 cells. After a 72 hr culture period, a CellTiter-GLO luminescent assay kit (Promega, Madison, WI) was employed to evaluate the effects of anti-NGF antibodies on canine ß-NGF induced cellular proliferation. After addition of CellTiter-GLO reagent according to manufacturer’s instructions, cell lysates were transferred to a white 96 well Optiplate (Perkin Elmer, Waltham, MA) before reading luminescence on a Spectromax M5e microplate reader (Molecular Devices, San Jose, CA). Maximal response in the assay is defined proliferation in the presence of cNGF only (no antibody). Minimal response is defined as measured proliferation without cNGF. Calculated inhibition (NGF neutralization) values for anti-cNGF antibodies are expressed as a percentage of minimal and maximal responses. The resulting percent inhibition/neutralization data was plotted with GraphPad Prism 7 (GraphPad software, San Diego, CA) for IC50 determination using a 4-parameter curve fit.
2.4 |. Surface plasmon resonance
Surface Plasmon Resonance was performed on a Biacore T200 (GE Healthcare, Pittsburgh, PA) to measure binding affinities of each antibody to nerve growth factor (NGF). A 2.5 μg/ml cNGF in 10 mM Sodium Acetate pH 4 (GE Healthcare, BR-1003–49), 5 μg/ml human NGF (R&D Systems Cat #256-GF/CF) in 10 mM Sodium Acetate pH 4 and 1 μg/ml human proNGF (Alomone labs Cat. # N-280) in 10 mM Sodium Acetate pH 5 (GE Healthcare, BR-1003–51) was immobilized by amine coupling using EDC/ NHS for a final density ~250 RU (resonance unit) on CM5 sensor flow cells 2–4, respectively. Flow cell 1 is used as an internal reference to correct running buffer effects. Antibody binding was measured at 15°C with a contact time of 250 s and flow rate of 30 μl/min. The dissociation period was 300 s. Regeneration was performed with regeneration buffers (10 mM Glycine pH1.5 and 10 mM NaOH) and flow rate at 20 μl/min for 60 s each. Running/dilution buffer (1X HBS-EP, GE Healthcare, BR-1006–69, 10X including 100 mM HEPES, 150 mM NaCl, 30 mM EDTA and 0.5% v/v surfactant P20, pH7.4, 1:10 in filtered MQ H2O) was used as negative control at the same assay format. Data were analyzed with Biacore T200 Evaluation software by using the method of double referencing. The resulting curve was fitted with the 1:1 binding model.
2.5 |. Yeast surface display expression and binding
Cellular fluorescence was measured using a BD Accuri C6 flow cytometer. Yeast cells displaying cNGF variants were detected using anti-cymc-FITC (Miltenyi Biotec, San Diego, CA) and an anti-FLAG tag alexa fluor 647-conjugated antibody (R&D System, Minneapolis, MN). Binding to biotinylated mAbs was detected using streptavidin-R-phycoerythrin conjugate (Thermo Fisher). Apparent dissociation constants were determined according to Chao et al. (2006) by titrating mAb at labeling concentrations from 0.064 nM to 262 nM. Titrations were performed in triplicate on at least two separate days.
2.6 |. Preparation of mutagenesis libraries
Comprehensive single site saturation mutagenesis (SSM) libraries were constructed using nicking mutagenesis exactly as described (Wren-beck et al., 2016). All mutagenic oligos were designed using Quik Change primer design Program (Agilent) and were ordered from IDT (Coralville, IA). For pro-cNGF, two separate libraries were prepared: library 1 covered residues Glu-102-Asn-52 and library 2 covered residues Ile-51-Arg0. For proΔ1,2-cNGF, the library covered residues Gln-55-Arg0. For conformational epitope mapping pro.v4-cNGF was split into two libraries, with library 1 covering residues Ser1-Asp60 and library 2 covering residues Pro61-Ala120. Library plasmid DNA was transformed into chemically competent S. cerevisiae EBY100, grown, and stored in yeast storage buffer at −80°C exactly according to published protocols (Medina-Cucurella & Whitehead, 2018).
2.7 |. Screening of Pro-cNGF and ProA1,2-cNGF libraries
1 × 107 cells were grown from freezer stocks in 1 ml of SDCAA for 6 hr at 30°C and re-inoculated at OD600 = 1.0 in 1 ml of SGCAA at 18°C for 16 hrs. 2 × 107 yeast libraries were labeled with either biotinylated tanezumab or mAb #1 at 5 nM for 30mins at room temperature in PBS-BSA. After centrifugation and washing, cells were secondarily labeled with 60 μl of anti-cymc-FITC and 50 μl of streptavidin-R-phycoerythrin conjugate in 1.89 ml of PBS-BSA for 10 mins at 4°C. Sorting was done on a BD Influx Cell Sorter at the Michigan State University Flow facility. For each sort 200,000 cells were collected (approx. 100-fold the theoretical diversity at the amino acid level) using a diagonal gate set to collect the top 2–3% of the displaying population (full statistics in Supplementary Table S1). Collected cells from each population were recovered at 30°C for 30 hrs in 10 ml of SDCAA and 100 μl of penicillin-streptomycin, washed, and then stored in 1 ml of yeast storage buffer at a concentration of 4 × 107 cells per ml at −80°C.
2.8 |. Determination of conformational epitopes
NGF conformational epitopes for all four mAbs were determined using yeast surface display, comprehensive mutagenesis, FACS, and deep sequencing exactly as previously described (Medina-Cucurella & Whitehead, 2018). Supplemental information contains the average dissociation constant values (Table S2), the percentage collected from library screening (Table S3), primers used for deep sequencing (Table S4), and statistics results (Table S5).
2.9 |. Deep sequencing preparation
Libraries were prepared for deep sequencing according to Kowalsky, Klesmith, et al. (2015) using Method B with both PCR reactions set to 14 cycles. The libraries were pooled and sequenced on an Illumina MiSeq using 2 × 250 bp pair-end reads at the Michigan State University Genomic Sequencing Core facility or the University of Illinois at Chicago DNA Service facility. Primer sequences used for each library are listed in Supplementary Table S4.
2.10 |. Data analysis
A modified version of Enrich 0.2 software as described in Kowalsky, Klesmith, et al. (2015) was used to compute enrichment ratios from the raw sequencing files. Custom python scripts available at Github (user: JKlesmith) were used to normalize the enrichment ratios (ER;) defined as:
| (1) |
where fi,sel is the frequency of variant i in the selected population, and fi, ref is the frequency of variant i in the reference population. Libraries statistics results are listed in Supplementary Table S5. For the pro region sorting experiments, we define an enrichment score (ESi) for each mutant i as the enrichment ratio of the selected mutant minus the wild-type enrichment ratio:
| (2) |
For conformational epitope mapping experiments we define a relative binding term for each mutant as the log transform of the mean fluorescence for variant i, normalized to the relative mean fluorescence of the wild-type construct, :
| (3) |
This equation can be written in terms of experimental observables according to:
| (4) |
where σ´ is the log normal fluorescence standard deviation of the clonal population, and Φ is the percentage of cells collected by the sorting gate on the flow cytometer (Supplementary Table S3) (Kowalsky, Klesmith, et al., 2015).
2.11 |. Data availability
Full datasets including normalized fitness metrics, pre- and post-selection read counts, and raw log base two enrichment scores for each variant can be found in Supplementary Data S1–S8. Raw sequencing reads for this work have been deposited in the SRA(SAMN07693504-SAMN07693526).
3 |. RESULTS
3.1 |. Initial cNGF constructs are improperly folded on the yeast surface
We sought a yeast surface display platform of cNGF to evaluate binding of candidate anti-cNGF mAbs. Mature cNGF is 97.5% pairwise identical to human NGF (hNGF), with an additional 17 substitutions (out of 103 total residues) on the pro-sequence (Figure 1a). We assessed proper folding of cNGF on the yeast surface using two conformationally sensitive mAbs. Soluble tanezumab—an anti-hNGF mAb—and an anti-cNGF mAb (mAb#1) recognized soluble recombinant cNGF as shown by surface plasmon resonance (Figure 1b, Supplementary Table S2). However, neither tanezumab nor mAb#1 recognized denatured cNGF as demonstrated by lack of signal by Western blotting (data not shown). Recombinant cNGF increased proliferation of TrkA-expressing TF-1 cells (Kitamura et al., 1989), and this proliferation could be blocked by the anti-cNGF mAbs (Supplementary Figure S1). Thus, both tanezumab and mAb#1 recognize a conformational epitope on cNGF and all mAbs function as cNGF antagonists. This biochemical data are corroborated with a previously published co-structure of tanezumab with hNGF (La Porte et al., 2014) that reveals binding mainly at the homodimer interface between subunits in a conformation that requires properly folded hNGF. In the remainder of this work we use the term “folded” to describe cNGF that is recognized by these conformationally sensitive mAbs.
We tested folding of four different cNGF yeast display constructs (Figure 1c). First, mature cNGF was fused with a N-terminal Aga2p domain, a N-terminal (G4S)3 linker and a C-terminal c-myc epitope tag (cNGF; pETconNK_cNGF). Second, the mature cNGF was displayed with an N-terminal Aga2p pre sequence, an N-terminal FLAG epitope tag, a C-terminal (G4S)3 linker, a C-terminal Aga2p, and a terminal c- myc epitope tag (Aga2-cNGF; pETconNK_Aga2_cNGF). Our third construct was identical to Aga2-cNGF but included the full-length pro region between the C-terminus of the Aga2 pre sequence and the N-terminus of the mature cNGF (pro-cNGF; pETconNK_Pro-cNGF). Finally, a classical study defined sections of the pro sequence into five “boxes” on the basis of sequence conservation (Suter et al., 1991) (Figure 1a). Truncation experiments showed that only Box 3 and Box 5 of the pro sequence were necessary and sufficient to produce active mouse NGF. Thus, our fourth construct proΔ1,2-cNGF was identical to pro-cNGF except Box 1 and Box 2 were deleted from the pro sequence. Although all variants displayed on the yeast surface, yeast cells labeled with saturating amounts of mAbs yielded signals of only 1.1 (cNGF) to 2.2 (pro-cNGF) in the signal:noise ratio (Figures 1d and 1e). This signal:noise ratio was calculated by measuring the ratio of sample MFI over the MFI in the absence of biotinylated antibody (“background”) for the subset of cmyc+ and fsc/ssc+ cells (to ensure measurement of individual yeast cells). Since similar experiments from our research group show 50 to over 100-fold above background for diverse protein-protein interactions (Kowalsky, Faber, et al., 2015; Kowalsky & Whitehead, 2016), we conclude that cNGF surface displays in a mostly misfolded form.
3.2 |. Comprehensive analysis of pro mutations that improve cNGF folding
A previous directed evolution study showed that mutations within the pro sequence could enhance proper folding of the mature neuro-trophin BDNF on the yeast surface (Burns et al., 2016). Based on this precedent, we sought mutants that improve the expression of folded cNGF. A flowchart of the experimental pipeline utilizing comprehensive mutagenesis, FACS, and deep sequencing is shown in Figure 2a. Comprehensive single-site saturation mutagenesis (SSM) libraries for pro-cNGF and proΔ1,2-cNGF were prepared by nicking mutagenesis (Wrenbecketal., 2016) and transformed into S. cerevisiae EBY100. The SSM libraries covered an average of 84.3% of all possible single missense and nonsense mutations (1737 mutations for pro-cNGF, 943 mutations for proΔ1,2-cNGF; Supplementary Table S5). Libraries were labeled with either biotinylated tanezumab or mAb #1 at 5 nM and sorted by FACS. Tanezumab was chosen as structural information of the mAb-NGF complex is known, while mAb #1 was chosen as a representative anti-cNGF as it had the highest initial signal:noise ratio. We collected the top 3% by cell fluorescence in the mAb channel, along with a reference population of yeast cells that passed through the cell sorter. After each sort, plasmid DNA was isolated, prepared, and deep sequenced. We evaluated each mutant by a relative enrichment score (ES) defined as the enrichment ratio of the mutant in the sorted population minus the enrichment ratio of the wild-type sequence. In this scoring system, a mutant with positive ES improves cNGF folding relative to the wild-type sequence.
FIGURE 2.
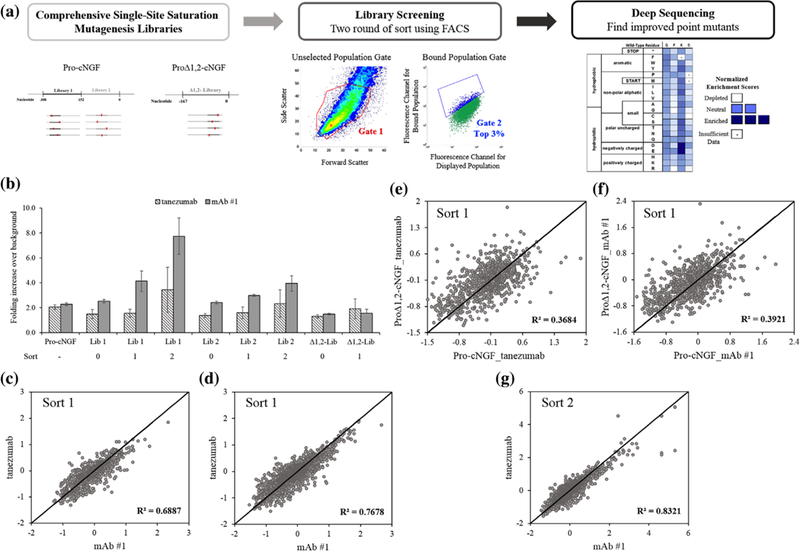
Identifying sequence-function relationships for pro region engineering using deep sequencing. (a) Comprehensive site-saturation mutagenesis libraries were constructed for the pro region and sorted twice by FACS to collect the top 3% of cells using a diagonal gate set for fluorescent channels corresponding to mAb binding and surface display. Collected populations were deep sequenced, compared with the reference population and converted to an enrichment score. (b) Increase in fluorescence channel associated with mAb binding for libraries before and after sorts (error bars, standard error of the mean, n ≥ 3). (c–g) Correlation in enrichment scores for each mutation for different mAbs and initial constructs
After the first sort, bulk populations from all three libraries showed increased fluorescence associated with cNGF folding (Figure 2b). We made a number of observations based on these near-comprehensive datasets after the first sort. First, most mutations, including premature stop codons, centered near an ES of 0 (for pro-cNGF: missense −0.12 ±0.52, nonsense −0.53 ±0.36; proΔ1,2-cNGF: missense −0.14 ±0.45, nonsense −0.03 ±0.44; mean values at 1s.d.). This low signal to noise for loss of function mutations is not surprising after the first sort because the starting constructs have a fluorescence intensity in the binding channel barely above background. Second, for each construct the correlation between each mAb was high: R2 = 0.69 for proΔ1,2-cNGF and R2 = 0.77 for pro-cNGF (Figures 2c and 2d). Because the observed reproducibility is similar to that seen in replicates using this deep sequencing methodology (Klesmith, Bacik, Wrenbeck, Michalczyk, & Whitehead, 2017), we cannot differentiate between experimental noise inherent in the deep sequencing pipeline and true biological differences of folding probed by individual mAbs. Third, correlation between pro-cNGF and proΔ1,2-cNGF was statistically significant (p-value 3.1 × 10−05 tailed paired t-test) but comparatively and unexpectedly lower (Figures 2e and 2f). Thus, mutations that confer differences in cNGF folding are, to a certain extent, context sensitive and suggest that higher order models would be needed to capture the chaperone function of the pro sequence. Fourth, there are many mutations that improve cNGF folding. Using an ES cut-off of three standard deviations above 0 we identified 49 (2.8%) pro-cNGF mutations that improve folding. Finally, because pro-cNGF had more beneficial mutations and a higher bulk population fluorescence than proΔ1,2-cNGF (Figure 2b), we moved ahead with pro-cNGF alone for the next round of sorting.
Whereas after the first sort libraries showed only a modest increase in cNGF folding, libraries after sort 2 obtained 3.4 and 7.7 increase in the signal:noise ratio for tanezumab and mAb#1, respectively (Figure 2b). The deep sequencing results were very similar between mAbs, with a R2 = 0.83 for the entire dataset between conformational antibodies and all 27 tanezumab mutations with an ES above 2 matched in the mAb#1 dataset (Figure 2g). A full-length heatmap of the pro-region showing site-specific preferences probed by tanezumab binding is shown in Figure 3 (full datasets for all constructs and sorts are listed in Supplementary Figures S2-S6). Consistent with Suter et al. (1991), Box 3 is more conserved than the rest of the sequence (mean ES missense mutations −0.23 vs. 0.01; p-value 2.9 × 10−11 tailed paired t-test). In fact, for most positions outside of Box 3 a large majority of missense mutations are tolerated, highlighting the inessentiality of specific sequences for the majority of the pro region.
FIGURE 3.

Per-position heatmap of enrichment scores for pro-cNGF mutants after two sorts with tanezumab
A number of intriguing sequence-function relationships were revealed by inspection of mutations enhancing cNGF folding. Of the 27 mutations with an ES score above 2, all were located either in Box 3 or close to the domain boundary in Box 2. Positive substitutions at Box 2 include substitutions at Ala-61, Thr-57, and Arg-53. Most substitutions at Ala-61 have positive ES, especially aliphatics and aromatics. We note that the nonsense codon has a slightly positive ES at this position: since a premature stop codon is unlikely to result in displayed cNGF, calculations for some slightly positive ES reported here may be within noise of the measurement. All substitutions at Thr-57 had positive ES, including major gains for mutations to Tyr, Met, Leu, Val, and the polar Gln. While Box 3 is more conserved than other pro regions, certain positions showed beneficial mutations. Most notably, most substitutions in the strongly conserved protease susceptible “KKRRLK” sequence had positive ES, including very high ES for charge reversal mutations at Lys-43Asp and Arg-40Asp.
3.3 |. Combining single mutants improves the amount of displayed, folded cNGF
To improve display of folded cNGF, we first made three isogenic constructs of the best mutations (ES values higher than 4.6) identified from the deep sequencing experiment (Box 2: A-61W, T-57Q; Box 3: K-43D) and tested their ability to recognize biotinylated tanezumab. In the yeast surface display context, all three chosen mutants showed improved MFI relative to pro-cNGF (Supplementary Figure S7). Next, we reasoned that combining mutations would result in higher amounts of folded cNGF. Pro.v1-cNGF-Pro.v3-cNGF were double mutants, while Pro.v4-cNGF contained all three mutations (Table 1). Each was tested for the expression of the folded protein by labeling with tanezumab, mAb #1, or two additional anti-cNGF mAbs (mAb #2, mAb #3). While all constructs demonstrated at least 12-fold increase in signal:noise ratio, Pro.v3-cNGF and Pro.v4-cNGF showed between a 19 and 23 increase in signal:noise ratio depending on the probe mAb (Figures 4a and 4b). Improved pro constructs gained at most a 1.9 fold-increase in surface expression over pro-cNGF (Figure 4c), showing that most of the effect of mutations centered on improving the folding of displayed cNGF.
TABLE 1.
Signal:noise ratio for Pro-cNGF constructs defined as the MFI of yeast cells labeled with specified mAb over MFI of unlabeled cells
| Library name | Mutations | Tanezumab | mAb #1 | mAb #2 | mAb #3 |
|---|---|---|---|---|---|
| Pro.v1-cNGF | T-57Q-K-43D | 6.5 ±1.5 | 7.6 ± 0.7 | 2.8 ±1.6 | 5.3 ±1.2 |
| Pro.v2-cNGF | A-61W-T-57Q | 10.1 ±2.5 | 11.7 ±1.3 | 4.3 ± 2.6 | 5.8 ± 1.0 |
| Pro.v3-cNGF | A-61W-K-43D | 14.3 ±3.7 | 19.0 ±2.8 | 6.3 ± 4.4 | 7.3+ 1.8 |
| Pro.v4-cNGF | A-61W-T-57Q-K-43D | 14.3 ± 3.4 | 20.5 ± 4.2 | 23.2 ± 2.6 | 8.7 ± 1.7 |
Errors bars represent the standard error of the mean (n ≥ 3)
FIGURE 4.
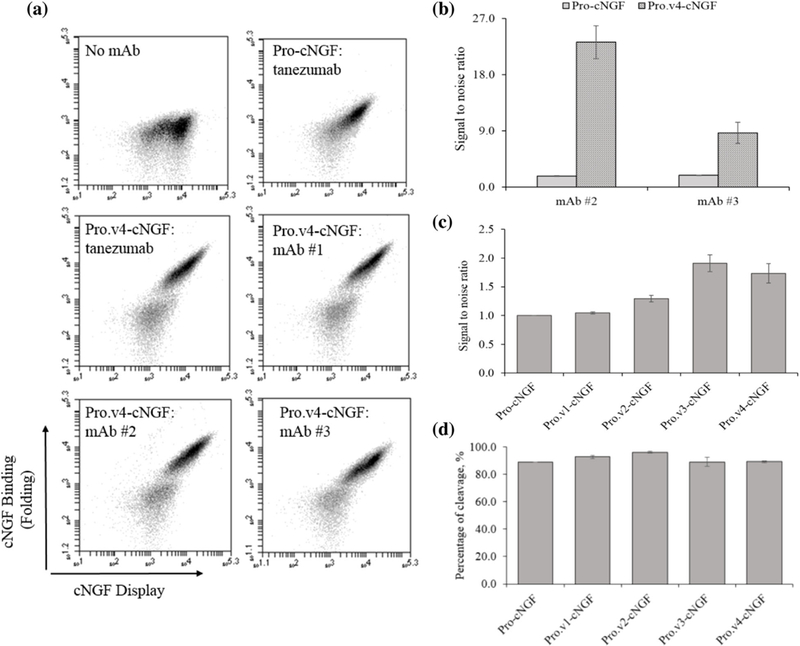
Pro-cNGF variants with multiple point mutations show improved cNGF folding. (a) Flow cytograms for pro.v4-cNGF versus pro-cNGF. (b) Increases in fluorescent channel probed by mAb#2 and mAb #3 binding for pro.v4-cNGF compared with pro-cNGF. (c) Bar charts showing the increase of all pro-cNGF variants in surface expression probed by labeling with a fluorescently conjugated anti-cmyc mAb. (d) Percentage of displayed, folded cNGF on the yeast surface (error bars, standard error of the mean, n ≥ 3)
3.4 |. Small amount of pro-cNGF displays on the yeast surface
To determine whether pro-cNGF displays on the yeast surface or whether the pro sequence is processed, wild-type cNGF, pro-cNGF and pro.v1-v4-cNGF were labeled with fluorescence conjugated antibodies against the C-terminal c-myc epitope tag and the N-terminal FLAG epitope tag. The percentage of cleavage was estimated by measuring the FLAG/c-myc ratios of pro-cNGF and pro.v1-v4-cNGF using the fluorescence mean values. As a control, these samples were compared with the ratio of cNGF (Burns et al., 2016) labeled exactly the same. In all cases, cells positive for c-myc were slightly positive for FLAG binding (Figure 4d), indicating that at least a portion of the pro sequence is proteolytically cleaved before display and suggesting that mature cNGF is displayed. All constructs, Pro.v1-v4-cNGF, showed from 88% to 96% of cleavage, meaning that a low percentage of the full-length pro-region is displayed on the yeast surface.
3.5 |. Conformational epitopes reveal similar profiles, but distinct epitopes, for tanezumab and all three mAbs
Using the pro.v4-cNGF yeast display construct we determined dissociation constants of tanezumab and mAbs #1–3 to cNGF. Yeast cells were incubated with varying amounts of mAb, washed, and labeled with secondary reagents prior to analysis by flow cytometry. Binding dissociation constants ranged from 143 ± 44 pM for mAb #3 to 800 ± 164 pM (1 s.d., n ≥ 3) for tanezumab (Figure 5, Supplementary Table S2). Interestingly, best fits of the Hill coefficient for these mAbs are all significantly below 1 (p-value <0.0106 one tail t-test; Table S2), indicating potential negative cooperativity between the dimeric displayed cNGF and dimeric mAb. Alternatively, Hill coefficients of less than 1 can arise for binding of non-equivalent binding sites. Since cNGF presumably exists in a range of folded conformations on the cell surface, both alternatives are plausible (Abeliovich, 2005).
FIGURE 5.
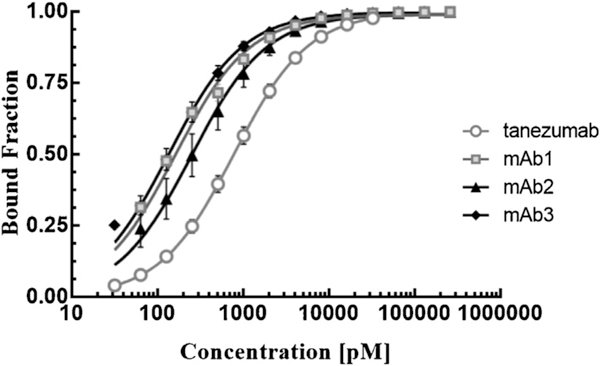
Binding titrations for surface-displayed pro.v4-cNGF for various mAbs. Each mAb was titrated at least three separate times on different days (error bars, 1.s.d. n ≥ 3)
Next, we determined the fine conformational epitopes of tanezumab and the three anti-cNGF mAbs using a previously developed method involving yeast surface display, nicking mutagenesis, and deep sequencing (Kowalsky, Faber, et al., 2015; Medina-Cucurella & Whitehead, 2018). The principle behind this method is that antigenic mutations that disrupt binding will map predominantly to the epitope positions recognized by the antibody. Compared with other epitope mapping strategies that collect the population that no longer binds antibody (Chao, Cochran, & Dane Wittrup, 2004; Mata-Fink et al., 2013), for our approach we collect the binding population. We then deep sequence the reference and binding populations. For each mutant, a relative binding term can be derived from the change in frequency of the bound population compared with a reference population. Shannon entropy (SE), a measure of sequence conservation, is then calculated. Positions with SE values less than or equal to the midpoint of the SE range are defined as belonging to the epitope (Kowalsky, Faber, et al., 2015).
In this experiment, two SSM libraries of cNGF were constructed by nicking mutagenesis, labeled with biotinylated mAb at half of the experimentally determined dissociation constant (Table S2), sorted by FACS, and deep sequenced. SSM libraries covered an average of 95.6% of all possible single nonsynonymous mutations in cNGF (1,115/1,200 for library 1 covering cNGF positions 1–60 and 1,178/1,200 for library 2 covering positions 61–120). Full statistics of library coverage is given in Tables S5, and per-position cNGF heatmaps for all mAb epitopes are given in Figure 6a and Supplementary Figures S8–S11.
FIGURE 6.
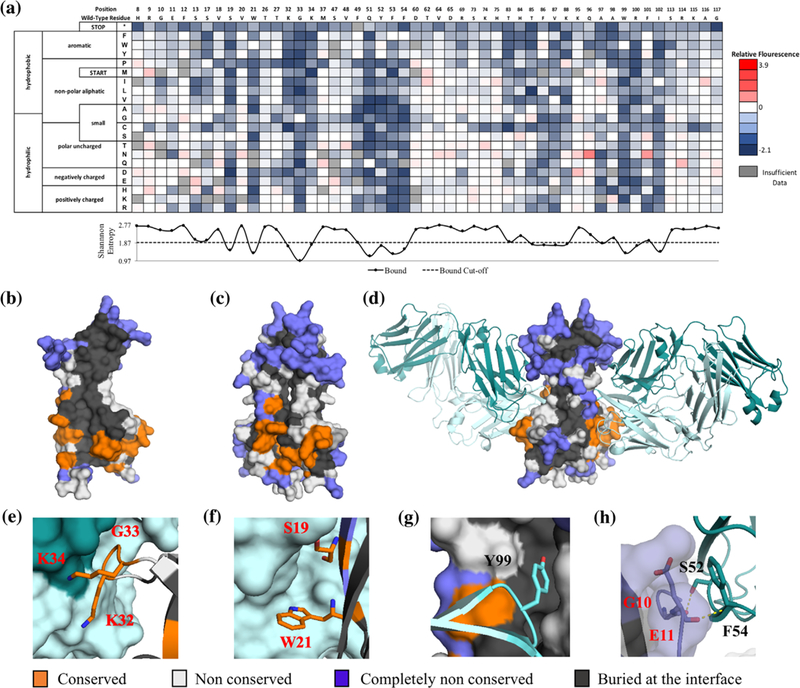
Determination of the cNGF:tanezumab conformational epitope using deep sequencing. (a) A subset of the fitness metric per position heatmap of the top 7% bound population versus the reference population for tanezumab. Shannon entropy is plotted beneath with the midpoint cut-off shown as a dashed line. (b and c) Tanezumab epitope view at the interface core of each monomer (b) and as a homodimer mature structure (c). Positions colored in orange, blue, gray, and white are epitope, completely non-conserved, buried in the core, and non-conserved positions, respectively. (d-h) Close-up views of specific tanezumab:cNGF sidechain interactions. Molecular representation of the interaction was modeled on the solved tanezumab:hNGF structure (PDB ID: 4EDW)
In our epitope mapping method, we typically start by removing positions that are structurally conserved from further analysis. We identify structurally conserved positions by collecting the population that surface displays the C-terminal c-myc epitope tag; structurally conserved positions are those that have a Shannon entropy less than or equal to the midpoint for the surface displayed population. However, our results show that mutations at most positions did not result in a change in the displayed protein (Supplementary Figure S12). These results are roughly consistent with the initial results showing misfolded cNGF still displaying on the surface. Thus, knowing the mature NGF conformation, we reasoned that structurally conserved positions are mainly at the core or the homodimer interface and can be identified as positions with less than 25% accessible surface area (ASA) evaluated from solved NGF structures. This analysis excluded 48 out of 120 residues (Figure S8). The average Shannon entropy for the tanezumab binding population for these buried positions was significantly lower than for solvent accessible positions (1.75 vs. 2.31, p-value <10−11 tailed paired t-test), indicating that conservation of the folded state of cNGF is essential to recognition by tanezumab.
As an initial control we compared our experimentally determined epitope with the previously published X-ray crystal structure of tanezumab Fab bound to hNGF (La Porte et al., 2014). 11/120 positions mapped to the tanezumab epitope (S19, W21, K32, G33, K34, F49, Y52, K88, A97, W99, and R100) while 38/120 positions were completely non-conserved (Figures S8 and 6a). As shown in Figures 6b–h, epitope positions form a contiguous patch that largely maps to the binding footprint of tanezumab previously described by La Porte et al. (2014). Of the 18 NGF positions within 4Å of tanezumab and with Cα-Cβ vectors pointing towards the antibody, seven were identified as hits in our pipeline, while six were structurally conserved. Of the remainder, only one epitope position (R114) was identified as nonconserved; this Arg makes a hydrogen bonding interactions with the main chain of tanezumab. Our deep sequencing approach identifies strong conservation of the cNGF loop centered around K32-K34 (Figure 6e) and positions central to the interface (S19, W21) (Figure 6f) that presumably make strong van der Waals contacts. The remaining epitope hits were second shell residues (e.g., W99, R100) buttressing these original contacts. However, many epitope positions at the homodimer interface where tanezumab binds to hNGF (e.g., F54, T56, T85, F86, T106, A107, and C108) are invisible to our method since they are structurally conserved (Figure 6g). Another shortcoming is that positions recognized by the antibody using main chain contacts (e.g., G10, E11, F12, and H84) are also invisible, as mutations to other amino acids will not disrupt antibody binding. Tanezumab binds to a ridge on NGF with side chains of residues R9-F12, V111, and R114 pointing directly away from the antibody (Figure 6h). All of these positions are completely non-conserved (Figure 6a).
Finally, we evaluated the binding interaction with three canine anti-cNGF mAbs. Shannon entropy results were very similar between tanezumab and each mAb, with a R2 = 0.88,0.89, and 0.77 for mAb #1, mAb #2, and mAb #3, respectively (Figure 7). All mAbs shared 5/11 conserved positions as the tanezumab epitope, but possessed different sequence-binding profiles and therefore unique epitopes (Supplementary Figure S9–S11).
FIGURE 7.
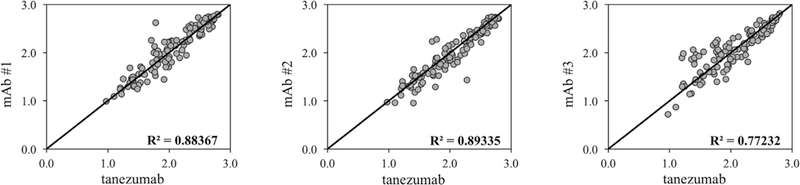
Correlation in Shannon entropy of the binding populations between tanezumab and other mAbs
Interesting, there were two mutations, V6M and M92F that show increased relative fluorescence in the binding population for all four mAbs (Supplementary Figure S8–S11). Our epitope mapping method cannot discriminate whether such mutations improve the binding affinity to a given mAb or whether the mutation increases the amount of folded cNGF. However, we speculate that the latter is the case as these two residues are surface exposed positions located distal to the tanezumab epitope, and all four mAbs bind in overlapping but atomically different epitopes.
4. |. DISCUSSION AND CONCLUSION
In this study we engineered a pro-region to enhance folding of cNGF on the yeast surface. This yeast platform enabled our team to map conformational epitopes for tanezumab and a number of anti-cNGF mAbs. During the course of this research we showcased the power of deep sequencing to augment directed evolution workflows to improve protein properties (Wrenbeck et al., 2017), gained new insight into sequence-function relationships for neurotrophin pro regions, and identified fine conformational epitopes for anti-cNGF mAbs.
4.1 |. Pro-region engineering pipeline
cNGF and pro-cNGF displayed on the yeast surface but in mostly misfolded forms. This adds to a growing body of literature showing that while the quality control machinery in the S. cerevisiae endoplasmic reticulum (Ellgaard & Helenius, 2003) can impact the overall amount of protein displayed on the surface (Klesmith et al., 2017; Whitehead, Chevalier, Song, & Dreyfus, 2012), for any given grossly misfolded protein some will still pass through quality control checkpoints in the secretory pathway and display on the yeast surface (Kowalsky, Faber, et al., 2015; Kowalsky & Whitehead, 2016; Park et al., 2006; Whitehead et al., 2012).
We used an engineering pipeline involving comprehensive sitesaturation mutagenesis, FACS, and deep sequencing to improve surface displayed, folded cNGF. This engineering pipeline enabled us to evaluate the functional effect for over 2,000 individual point mutants to the pro-cNGF and proΔ1,2-cNGF constructs. We then created multi-site constructs by combining the best mutations cherry picked from the deep sequencing datasets. There are several advantages of this workflow compared with traditional directed evolution approaches. First, precise single-site mutagenesis libraries constructed from nicking mutagenesis allow us to assign a functional effect to a single mutation; libraries constructed by error-prone PCR often have multiple mutations per gene, which then need to be deconvoluted. Second, deep sequencing allows us to perform experiments in parallel: we performed duplicate sorts using different conformational mAbs, allowing one to identify mutations beneficial to both mAbs. Third, the small library size allows us to simplify steps for yeast transformation and sample handling, shorten FACS times, and allow us to complete our screening in 2 sorts compared 4–7 sorts for larger libraries (“go small, get them all”). One slight disadvantage of screening single point mutants is the chance of missing epistatic mutations but in the present work identified beneficial pro mutants could be combined additively.
Our best construct identified by deep sequencing greatly improved the folding of displayed cNGF and was sufficient for the conformational epitope mapping performed. However, we suspect that there is room for further optimization of the yeast display of folded cNGF as (I.) the max signal:noise ratio of pro.v4-cNGF is still lower than that seen for other antibody-antigen interactions probed by our lab; (II.) two mutations on mature cNGF could presumably improve folding of the pro.v4-cNGF construct; and (III.) the Hill coefficient for all antibodies was less than one, which is consistent with cNGF existing in a range of folded and partially misfolded configurations on the yeast surface. Further optimization could be done by selecting similar beneficial point mutants identified from the deep sequencing datasets (Figure 3). We speculate that the combination of an additional subset of these point mutations would facilitate production of mature cNGF from yeast (Burns et al., 2016), although this is beyond the scope of the present work.
4.2 |. Pro region sequence-function relationships
Although the classic work from Suter et al. (1991) showed that Box 3 and 5 of the pro region were sufficient to produce active mouse NGF, mutations for the truncated variant proΔ1,2-cNGF did not improve folding as noticeably as for pro-cNGF. Given that many of the strongest beneficial mutations were in Box 2, we speculate that Box 2 encodes some chaperone activity necessary for complete cNGF folding. However, further studies should be performed to understand domain boundaries and functional differences between constructs.
Conserved and beneficial mutations cluster around Box 3 as was suggested for mouse NGF (Suter et al., 1991). Notable lack of conservation in Box 3 centered around a cleavage site for proprotein convertases at Lys-43-Arg-40. These four positions demonstrated improved profiles for almost all the mutations. Indeed, Lys-43Asp was one of the mutations introduced in our engineered pro-cNGF that in turn, disrupt recognition of this KR dipeptide by endogenous Kex2 protease in S. cerevisiae. Interestingly, both pro-cNGF and prov.4-cNGF display a mixture of full-length pro-cNGF and at least partially cleaved cNGF, suggesting that enhanced folding conferred by the Lys-43Asp mutation does not result from differential protease cleavage.
A previous study utilizing hydrogen-deuterium exchange with the pro region and mature NGF determined thatTrp-83-Ala-63 in the pro sequence (Trp-84-Gly-64 canine numbering) was involved in the intramolecular chaperone-like interactions with mature human NGF (Kliemannel et al., 2007). Inconsistent with this experiment, we observed all positions in this region to be non-conserved, with most mutations centered around an ES of 0. Our deep sequencing experiments probe folding of cNGF but do not directly assess the chaperone function of specific pro sequence variants, so further work is necessary to reconcile this apparent discrepancy.
4.3 |. Mapping epitopes targeted by anti-cNGF mAbs
We were able to determine conformational epitopes for tanezumab and three potential canine mAbs. Our experimentally determined tanezumab epitope largely overlapped with the previously published tanezumab-mNGF structure (La Porte et al., 2014), although many epitope positions (i.) at the dimerization interface, and (ii.) not participating in side chain contacts with antibody were invisible to our deep sequencing method. These shortcomings will be shared with all mutational-based epitope mapping methods. The three anti-cNGF mAbs had different sequence-binding profiles at the epitope. We conclude from these results that our yeast display platform for cNGF is able to map fine conformational epitopes for candidate anti-NGF mAbs.
Although our epitope mapping workflow cannot directly measure if cNGF is displayed as a monomer or dimer on the yeast surface, the results strongly suggests that our best constructs display as mature dimer cNGF. Mutations at the homodimer interface core away from the epitope will disrupt the folded state of cNGF necessary for mAb recognition, and indeed these are depleted in our experiments. These results are consistent with our lab’s previous determination of the conformational epitope tumor necrosis factor (TNF)-infliximab (Kowalsky, Faber, et al., 2015), where mutations at the TNF homo-trimeric interface disrupted antibody binding. However, we note that the solution-based measurements of cNGF-mAb binding are high fM-low pM, whereas measurements from the yeast surface are mid- to high-pM. While Gai and Wittrup have shown the rough equivalence in affinity between yeast- and solution measurements (Gai & Wittrup,2007), their dataset included only a single fM binder and thus we do not necessarily expect equivalence for these high affinity binders.
In conclusion, we used a deep sequencing pipeline to develop a yeast display platform for folded cNGF. This contribution highlights the power of deep sequencing to identify nearly all beneficial point mutants in a protein in a simplified workflow. Since a major limitation of yeast display is proper folding of complicated mammalian proteins, this work complements a recent directed evolution study (Burns et al., 2016) to show that effective strategies exist to overcome such limitations. Our pipeline could be a promising platform to increase the production of highly active titers of the other members of the neurotrophin family, to determine the specificity and affinity to their respective receptors, and to enable the epitope mapping for therapeutics against neuronal diseases such as the brain disorders caused by BDNF.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by a Zoetis research grant, the Johansen-Crosby endowed chair (both to T.A.W.), and a NIH T32 Biotechnology Training Grant (Award # T32-GM110523) (to A.M.C). The authors have no stated conflicts of interest.
Nomenclature
- PBS
Phosphate buffered saline
- FBS
fetal bovine serum
- DPBS
Dubelcco’s phosphate buffered saline
- PBS-SBA
phosphate buffered saline with bovine serum albumin
- SDCAA
synthetic dextrose medium supplemented with casamino acids
- SGCAA
synthetic galactose medium supplemented with casamino acids
Footnotes
SUPPORTING INFORMATION
Additional supporting information may be found online in the Supporting Information section at the end of the article.
REFERENCES
- Abeliovich H (2005). An Empirical Extremum Principle for the Hill Coefficient in Ligand-Protein Interactions Showing Negative Cooper-ativity. Biophysical Journal, 89, 76–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams RM, Mora T, Walczak AM, & Kinney JB (2016). Measuring the sequence-affinity landscape of antibodies with massively parallel titration curves. Elife, 5, 5980–5985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burns ML, Malott TM, Metcalf KJ, Puguh A,Chan JR,&Shusta EV (2016). Pro-region engineering for improved yeast display and secretion of brain derived neurotrophic factor. Biotechnology Journal, 11,425–436. [DOI] [PubMed] [Google Scholar]
- Burns ML, Malott TM, Metcalf KJ, Hackel BJ, Chan JR,&Shusta EV (2014). Directed evolution of brain-derived neurotrophic factor for improved folding and expression in Saccharomyces cerevisiae. Applied and Environmental Microbiology, 80, 5732–5742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang DS, Hsu E, Hottinger DG, & Cohen SP (2016). Anti-nerve growth factor in pain management: Current evidence. Journal of Pain Research, 9, 373–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chao G, Cochran JR, & Dane Wittrup K (2004). Fine epitope mapping of anti-Epidermal growth factor receptor antibodies through random mutagenesis and yeast surface display. Journal of Molecular Biology, 342, 539–550. [DOI] [PubMed] [Google Scholar]
- Chao G, Lau WL, Hackel BJ, Sazinsky SL, Lippow SM, & Wittrup KD (2006). Isolating and engineering human antibodies using yeast surface display. Nature Protocols, 1, 755–768. [DOI] [PubMed] [Google Scholar]
- Doolan KM, & Colby DW (2015). Conformation-Dependent epitopes recognized by prion protein antibodies probed using mutational scanning and deep sequencing. Journal of Molecular Biology, 427, 328–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellgaard L, & Helenius A (2003). Quality control in the endoplasmic reticulum. Nature Reviews. Molecular Cell Biology, 4, 181–191. [DOI] [PubMed] [Google Scholar]
- Feng D, Kim T, Ozkan E, Light M, Torkin R, Teng KK,... Garcia KC (2010). Molecular and Structural Insight into proNGF Engagement of p75NTR and Sortilin. Journal of Molecular Biology, 396, 967–984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gai SA, & Wittrup KD (2007). Yeast surface display for protein engineering and characterization. Current Opinion in Structural Biology, 17,467–473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hauburger A, Kliemannel M, Madsen P, Rudolph R, & Schwarz E (2007). Oxidative folding of nerve growth factor can be mediated by the pro-peptide of neurotrophin-3. FEBS Letters, 581, 4159–4164. [DOI] [PubMed] [Google Scholar]
- Huang EJ, & Reichardt LF (2001). Neurotrophins: Roles in neuronal development and function. Annual Review of Neuroscience, 24, 677–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitamura T, Tange T, Terasawa T, Chiba S, Kuwaki T, Miyagawa K, ... Takaku (1989). Establishment and characterization of a unique human cell line that proliferates dependently on GM-CSF, IL-3, or erythropoietin. Journal of Cellular Physiology, 140, 323–334. [DOI] [PubMed] [Google Scholar]
- Klesmith JR, Bacik J-P, Wrenbeck EE, Michalczyk R, & Whitehead TA (2017). Trade-offs between enzyme fitness and solubility illuminated by deep mutational scanning. Proceedings of the National Academy of Sciences of the United States of America, 114, 2265–2270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kliemannel M, Golbik R, Rudolph R, Schwarz E, & Lilie H (2007). The pro-peptide of proNGF: Structure formation and intramolecular association with NGF. Protein Science, 16, 411–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kliemannel M, Rattenholl A,Golbik R, Balbach J, Lilie H, Rudolph R, & Schwarz E (2004). The mature part of proNGF induces the structure of its pro-peptide. FEBS Letters, 566, 207–212. [DOI] [PubMed] [Google Scholar]
- Kowalsky CA, Klesmith JR, Stapleton JA, Kelly V, Reichkitzer N, & Whitehead TA (2015). High-Resolution sequence-Function mapping of full-Length proteins. ed. gideon schreiber. PLoS ONE, 10, e0118193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kowalsky CA, & Whitehead TA (2016). Determination of binding affinity upon mutation for type I dockerin-cohesin complexes from C lostridium thermocellum and C lostridium cellulolyticum using deep sequencing. Proteins: Structure, Function, and Bioinformatics, 84, 1914–1928. [DOI] [PubMed] [Google Scholar]
- Kowalsky CA, Faber MS, Nath A, Dann HE, Kelly VW, Liu L, ... Whitehead TA. (2015). Rapid fine conformational epitope mapping using comprehensive mutagenesis and deep sequencing. The Journal of Biological Chemistry, 290, 26457–26470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar V, & Mahal BA (2012). NGF - the TrkA to successful pain treatment. Journal of Pain Research, 5, 279–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- La Porte SL, Eigenbrot C, Ultsch M, Ho W-H, Foletti D, Forgie A, ... Pons J (2014). Generation of a high-fidelity antibody against nerve growth factor using library scanning mutagenesis and validation with structures of the initial and optimized Fab-antigen complexes. MAbs, 6,1059–1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lessmann V, Gottmann K, & Malcangio M (2003). Neurotrophin secretion: Current facts and future prospects. Progress in Neurobiology, 69, 341–374. [DOI] [PubMed] [Google Scholar]
- Mata-Fink J, Kriegsman B, Yu HX, Zhu H, Hanson MC, Irvine DJ, & Wittrup KD (2013). Rapid conformational epitope mapping of anti-gp120 antibodies with a designed mutant panel displayed on yeast. Journal of Molecular Biology, 425, 444–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medina-Cucurella AV, & Whitehead TA (2018). Characterizing protein-Protein interactions using deep sequencing coupled to yeast surface display. Methods in Molecular Biology, 1764, 101–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nomoto H, Takaiwa M, Mouri A, & Furukawa S (2007). Pro-region of neurotrophins determines the processing efficiency. Biochemical and Biophysical Research Communications, 356, 919–924. [DOI] [PubMed] [Google Scholar]
- Pagadala PC, Dvorak LA, & Neet KE (2006). Construction of a mutated pro-nerve growth factor resistant to degradation and suitable for biophysical and cellular utilization. Proceedings of the National Academy of Sciences of the United States of America, 103,17939–17943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park H, & Poo M (2013). Neurotrophin regulation of neural circuit development and function. Nature Reviews Neuroscience, 14, 7–23. [DOI] [PubMed] [Google Scholar]
- Park S, Xu Y, Stowell XF, Gai F, Saven JG, & Boder ET (2006). Limitations of yeast surface display in engineering proteins of high thermostability. Protein Engineering, Design and Selection, 19, 211–217. [DOI] [PubMed] [Google Scholar]
- Rattenholl A, Lilie H, Grossmann A, Stern A, Schwarz E, & Rudolph R (2001). The pro-sequence facilitates folding of human nerve growth factor from Escherichia coli inclusion bodies. European Journal of Biochemistry, 268, 3296–3303. [DOI] [PubMed] [Google Scholar]
- Rattenholl A, Ruoppolo M, Flagiello A, Monti M, Vinci F, Marino G,... Rudolph R (2001). Pro-sequence assisted folding and disulfide bond formation of human nerve growth factor. Journal of Molecular Biology, 305, 523–533. [DOI] [PubMed] [Google Scholar]
- Slosky LM, Largent-Milnes TM,&Vanderah TW (2015). Use of animal models in understanding cancer-induced bone pain. Cancer Growth Metastasis, 8, 47–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suter U, Heymach JV, Shooter EM, & Shooter EM (1991). Two conserved domains in the NGF propeptide are necessary and sufficient for the biosynthesis of correctly processed and biologically active NGF. EMBO Journal, 10, 2395–2400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson JJ, Allen SJ, & Dawbarn D (2008). Targeting nerve growth factor in pain. BioDrugs, 22, 349–359. [DOI] [PubMed] [Google Scholar]
- Whitehead T, Chevalier A, Song Y, & Dreyfus C (2012). Optimization of affinity, specificity and function of designed influenza inhibitors using deep sequencing. Nature Biotechnology, 30, 543–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wrenbeck EE, Faber MS, & Whitehead TA (2017). Deep sequencing methods for protein engineering and design. Current Opinion in Structural Biology, 45, 36–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wrenbeck EE, Klesmith JR, Stapleton JA, Adeniran A, Tyo KEJ, & Whitehead TA (2016). Plasmid-based one-pot saturation mutagenesis. Nature Methods, 13, 928–930. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Full datasets including normalized fitness metrics, pre- and post-selection read counts, and raw log base two enrichment scores for each variant can be found in Supplementary Data S1–S8. Raw sequencing reads for this work have been deposited in the SRA(SAMN07693504-SAMN07693526).