

Abstract
Glutamate is one of the most important neurotransmitters in the process of signal transduction in the CNS. Excessive amounts of this neurotransmitter lead to glutamate excitotoxicity, which is accountable for neuronal death in acute neurological disorders, including stroke and trauma, and in neurodegenerative diseases. Inorganic polyphosphate (PolyP) plays multiple roles in the mammalian brain, including function as a calcium-dependent gliotransmitter mediating communication between astrocytes, while its role in the regulation of neuronal activity is unknown. Here we studied the effect of PolyP on glutamate-induced calcium signal in primary rat neurons in both physiological and pathological conditions. We found that preincubation of primary neurons with PolyP reduced glutamate-induced and AMPA-induced but not the NMDA-induced calcium signal. However, in rat hippocampal acute slices, PolyP reduced ion flux through NMDA and AMPA receptors in native neurons. The effect of PolyP on glutamate and specifically on the AMPA receptors was dependent on the presence of P2Y1 but not of P2X receptor inhibitors and also could be mimicked by P2Y1 agonist 2MeSADP. Preincubation of cortical neurons with PolyP significantly reduced the initial calcium peak as well as the number of neurons with delayed calcium deregulation in response to high concentrations of glutamate and resulted in protection of neurons against glutamate-induced cell death. As a result, activation of P2Y1 receptors by PolyP reduced calcium signal acting through AMPA receptors, thus protecting neurons against glutamate excitotoxicity by reduction of the calcium overload and restoration of mitochondrial function.
SIGNIFICANCE STATEMENT One of the oldest polymers in the evolution of living matter is the inorganic polyphosphate (PolyP). It is shown to play a role of gliotransmitter in the brain; however, the role of polyphosphate in neuronal signaling is not clear. Here we demonstrate that inorganic polyphosphate is able to reduce calcium signaling induced by physiological or high concentrations of glutamate. The effect of polyphosphate on glutamate-induced calcium signal in neurons is due to the effect of this polymer on the AMPA receptors. The effect of PolyP on glutamate-induced and AMPA-induced calcium signal is dependent on P2Y receptor antagonist. The ability of PolyP to restrict the glutamate-induced calcium signal lies in the basis of its protection of neurons against glutamate excitotoxicity.
Keywords: AMPA, excitotoxicity, glutamate, inorganic polyphosphate, neuron, NMDA
Introduction
Signal transduction in the CNS is a well balanced process that includes multiple players and factors. In astrocytes, signals propagate predominantly through calcium signaling, and in neurons it is mostly a change in plasma membrane potential and calcium signal (Fellin, 2009; Bazargani and Attwell, 2016). Neurons are capable of the release of multiple and very diverse neurotransmitters and neuromodulators. Astrocytes release gliotransmitters, which can also have an effect on neurons (Gundersen et al., 2015). One of the major neurotransmitters is glutamate, which can act as an excitatory transmitter in the vast majority of the neurons and play a role of a signaling molecule for astrocytes (Hertz and Zielke, 2004). Synaptically released glutamate activates ionotropic or metabotropic glutamate receptors, which change the electrical activity of neurons and trigger the Ca2+ signal. Prolonged stimulation of neurons with a high concentration of glutamate, as seen in many pathologies, leads to excitotoxicity, and is believed to be one of the major reasons behind neuronal loss in stroke, epilepsy, and some neurodegenerative diseases (Olney, 1969; Choi, 1985). Both excitatory and inhibitory types of neurotransmission are mediated in large part by glutamate and GABA; however, some other neurotransmitters and neuromodulators, including dopamine and those of the purinergic system, could modify the glutamate signal (Krnjević, 1970; Vaarmann et al., 2013).
One of the oldest polymers in the evolution of the living matter is the inorganic polyphosphate (PolyP). It plays an essential role in bacteria, yeast, and protozoa, but is also presented in relatively high concentration in the mammalian brain, up to 50 μm, where it acts as a gliotransmitter (Lorenz et al., 1997; Holmström et al., 2013; Angelova et al., 2014, 2018). As seen by the relatively small number of publications in this field to date, the function of this polymer is largely underestimated, especially for the brain. Potentially, PolyP can play multiple roles in the CNS (Kumble and Kornberg, 1995; Angelova et al., 2016; Cremers et al., 2016; Müller et al., 2017). For example, in astrocytes it is localized in mitochondria, lysosomes, and vesicular nucleotide transporter (VNUT)-containing vesicles (Abramov et al., 2007; Pavlov et al., 2010; Holmström et al., 2013; Angelova et al., 2018). PolyP is released from lysosomes or VNUT vesicles by exocytosis, which leads to the development of calcium waves (Holmström et al., 2013; Angelova et al., 2018). Structurally, PolyP consists of long chains of only orthophosphate residues, but despite such a simple composition it is able to activate P2Y1 receptors, stimulating the activation of phospholipase C and inducing the release of Ca2+ from the endoplasmic reticulum via the IP3 receptors (Holmström et al., 2013; Dinarvand et al., 2014). Moreover, PolyP directly induces calcium signal in only ∼3% of the neurons, and it has been found positioned to the synapses (Holmström et al., 2013; Stotz et al., 2014). Furthermore, it has been shown to modulate the activity of ion channels (i.e., TRPM8 channels; Zakharian et al., 2009). However, PolyP significantly changes neuronal activity in vivo (Holmström et al., 2013), and this effect can be induced both directly, by the effect of PolyP on neurons, or indirectly, through the activation of astrocytes. Here, we study whether PolyP is as well able to reduce the glutamate-induced calcium signal and in particular the signal mediated by AMPA and NMDA receptors (NMDARs). We then tested whether the effect of PolyP on the calcium signal stimulated by glutamate involves the purinergic system. Finally, we tested the ability of PolyP to influence the glutamate-induced calcium signal and effectively protect neurons from the delayed calcium deregulation (DCD) and consequently from glutamate-induced cell death.
Materials and Methods
Cell culture.
Cocultures of cortical neurons and astrocytes were prepared from Sprague Dawley rat pups of either sex 2–4 d postpartum (from a UCL breeding colony) as described previously (Angelova et al., 2019). Brain cortices were removed into ice-cold HBSS (Ca2+, Mg2+-free; Invitrogen). The tissue was minced and trypsinized (0.1% for 15 min at 37°C), triturated, plated on poly-d-lysine-coated coverslips, and cultured in Neurobasal Medium (Thermo Fisher Scientific) supplemented with B-27 (Thermo Fisher Scientific) and 2 mm l-glutamine. The cultures were maintained at 37°C (5% CO2), and the media changed twice a week. To avoid the age dependence on the DCD, cells were used after 12–15 d in vitro in all experiments.
[Ca2+]c and [Δψm] imaging.
Hippocampal or cortical neurons were loaded for 30 min at room temperature with 5 μm fura-FF AM (Molecular Probes) or 5 μm fura-2 AM (Molecular Probes) and 0.005% Pluronic acid in an HBSS: 156 NaCl, 3 KCl, 2 MgSO4, 1.25 KH2PO4, 2 CaCl2, 10 glucose, and 10 HEPES, pH adjusted to 7.35. For simultaneous measurement of [Ca2+]c and mitochondrial membrane potential (Δψm), Rh123 (1 μm, Thermo Fisher Scientific) was added into the cultures during the last 15 min of the fura-2 or fura-FF loading period, and the cells were then washed three to five times before an experiment. Fluorescence measurements were obtained on an epifluorescence inverted Olympus microscope equipped with a 20× fluorite objective. [Ca2+]c and Δψm were monitored in single cells using excitation light provided by a xenon arc lamp, with the beam passing sequentially through 10 nm bandpass filters centered at 340, 380, and 490 nm housed in a computer-controlled filter wheel (Cairn Research). Emitted fluorescence light was reflected through a 515 nm long-pass filter to a cooled CCD camera (Retiga, QImaging). The fura-2 or fura-FF data have not been calibrated in terms of [Ca2+]c because of the uncertainty arising from the use of different calibration techniques. For ΔFura ratio calculation, we used the maximal value of fluorescence obtained during glutamate exposure and, as the baseline, the mean of fluorescence recorded during the time preceding the stimulation. For Δψm measurements, we have normalized the fluorescence signals between resting level (set to 0) and a maximal signal (which correspond to full mitochondrial depolarization) generated in response to the protonophore FCCP (carbonyl cyanide-p-trifluoromethoxyphenylhydrazone; 1 μm; set to 100%). Changes in Rh123 fluorescence were then expressed as the difference between peak values (at 15 min) attained during glutamate stimulation and basal values. All imaging data were collected and analyzed using iQ3 software from (Andor).
PolyP standards (sodium salt).
Short-length PolyP (S-PolyP) 14 ortophosphates, medium-length (M-PolyP) 60 ortophosphates, and long-length PolyP (L-PolyP) 130 ortophosphates were provided by Dr. T. Shiba (RegeneTiss).
Neuronal toxicity experiment.
For toxicity assay, cortical neurons were sequentially exposed to 20 μm PolyP for 10 min and to 100 μm glutamate for a further 30 min. When P2Y and P2X inhibitors were tested, compounds were added 20 min before PolyP exposure. Then, cells were cultured with fresh media without polyp and glutamate, and cell viability was assessed 24 h after treating neurons with 20 μm propidium iodide (PI) and 4.5 μm Hoechst 33342 stain (Thermo Fisher Scientific) for 30 min before imaging. The PI is excluded from viable cells and exhibits a red fluorescence (excitation, 530 nm; emission, >560 nm) following a loss of membrane integrity, while the Hoechst 33342 stain labels all nuclei blue (excitation, 405 nm; emission, 460 nm). This allows subtraction of the number of dead (red-stained) cells from the total number of cells counted (blue-stained nuclei). Using phase contrast optics, a bright-field image allowed the identification of neurons, which are morphologically different from the flatter glial component and lie in a different focal plane, above the glial layer. A total number of 100–300 neurons or glial cells was counted in five to six fields of each coverslip. Each experiment was repeated three or more times, using different neuronal preparations.
Electrophysiology.
Transverse hippocampal slices (300–400 μm thick) were used for in vitro electrophysiological recordings. Slices were prepared from 3- to 5-week-old Sprague Dawley rats. Animals were killed by cervical dislocation after being anesthetized by an overdose of isoflurane according to the United Kingdom Animals (Scientific Procedures) Act of 1986. After decapitation, brains were rapidly removed and dissected, and whole-brain sagittal slices were prepared with a Leica VT1200S Vibratome in an ice-cold sucrose-based solution containing the following (in mm): 70 sucrose, 80 NaCl, 2.5 KCl, 7 MgCl2, 0.5 CaCl2, 25 NaHCO3, 1.25 NaH2PO4, and 22 glucose, equilibrated with 95% O2 plus 5% CO2, at pH 7.35 and 315–330 mOsm. Slices were equilibrated to room temperature and then placed to recover in continuously oxygenated sucrose-ACSF chamber for at least 1 h before recording. After recovering, individual slices were transferred into a recording chamber. The perfusion solution contained the following (in mm): 119 NaCl, 2.5 KCl, 1.3 MgSO4, 2.5 CaCl2, 26.2 NaHCO3, 1 NaH2PO4, and 22 glucose and was continuously gassed with 95%O2 and 5%CO2, at pH 7.35 and 290–298 mOsm. For the whole-cell patch-clamp recordings, we used borosilicate glass electrodes of 4–6 MΩ impedance. Recordings were performed with a Multiclamp 700B Amplifier running under pClamp 11x software (Molecular Devices). The intracellular pipette solution contained the following (in mm): 120.5 CsCl, 10 KOH-HEPES, 2 EGTA, 8 NaCl, 5 QX-314 Br− salt, 2 Mg-ATP, and 0.3 Na-GTP. Voltage-clamp recordings of NMDARs activity in hippocampal dentate gyrus granule cells (DGCs) were performed at a holding potential of +30 mV. To isolate the NMDAR response pharmacologically, we added to the perfusion solution the following antagonists (in μm): picrotoxin 50, GGP-55645 1, strychnine 1, NBQX 20, and S-MCPG 200. Voltage-clamp recordings of AMPA receptor (AMPAR) activity in DGCs were performed at a −70 mV holding potential. To isolate the AMPAR response pharmacologically, we added to the perfusion solution the following antagonists (in μm): picrotoxin 50, GGP-55645 1, strychnine 1, APV 50, and S-MCPG 200. For both NMDAR and AMPAR recordings, the response amplitude after polyphosphate application was normalized to that of control recording from the same cell. Synaptic responses were evoked by bipolar tungsten electrode placed in perforant path and connected to DS2A constant-voltage stimulator (Digitimer). Electrical stimuli were applied with a 200 ms time interval once per 15 s.
Statistical and data analysis.
Statistical analysis and data analysis were performed using Origin 9 software (Microcal Software). Statistical analysis was performed using an unpaired or paired two-tailed Student's t test, as indicated. A test for outliers was not conducted on the data. Results are expressed as the mean ± SEM. Differences were considered to be significantly different at p < 0.05.
Results
PolyP decreases glutamate-induced calcium signal
The application of the physiological concentration of glutamate (5 μm) to the primary cortical coculture of neurons and astrocytes induced a fast and transient increase in the [Ca2+]c of neurons (by 1.57 ± 0.04 fura-2 ratio; n = 120; Fig. 1A,C). Stimulation of neurons with S-PolyP (5–15 orthophosphates; 20 μm) induced only minor changes in the [Ca2+]c of neurons (Fig. 1B). However, the consequent addition of 5 μm glutamate results in a significantly lower calcium signal in these neurons compared with control [fura-2 ratio from 1.56 ± 0.04 (n = 120) to 0.98 ± 0.04 (n = 63); p < 0.0001; Fig. 1B,C]. The ability of PolyP to reduce glutamate-induced calcium signal was dependent on the length of polymer: it was less effective with elongation of the chain (Fig. 1C).
Figure 1.
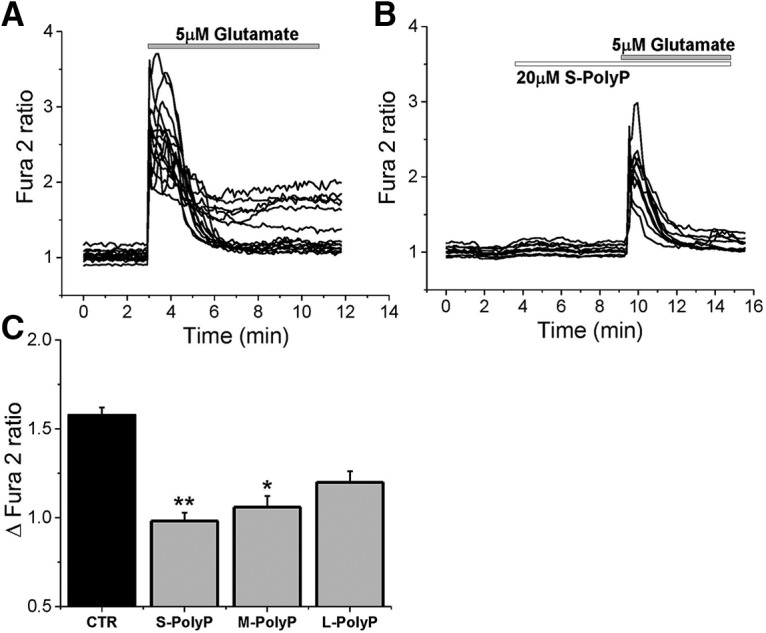
PolyP suppresses the effect of low glutamate concentrations (5 μm) on neuronal calcium signal. A, Application of 5 μm glutamate to cortical neurons induced a rise in [Ca2+]c (fura-2 ratio). B, Pretreatment of 20 μm S-PolyP significantly reduced the glutamate-induced calcium signal. C, Histogram representing the effect of different lengths of PolyP on the glutamate-induced calcium signals in cortical neurons as a Δfura-2 ratio between the basal level of [Ca2+]c and the maximal peak after glutamate (5 μm) application. *p < 0.05, **p < 0.001.
High and toxic concentrations of glutamate induce initial calcium influx followed by DCD, which triggers excitotoxicity (Fig. 2A). Preincubation of neurons with 20 μm S-PolyP (p = 0.0157; Fig. 2B) and M-PolyP (p = 0.0250; Fig. 2C) both significantly reduced the number of cells with delayed calcium deregulation. Importantly, S-PolyP was more effective in the overall protection of the neurons against the occurrence of DCD compared with M-PolyP and L-PolyP (Fig. 2E), but, interestingly, all types of PolyP significantly decreased the amplitude of glutamate-induced (100 μm) calcium signal (from 0.64 ± 0.03, n = 178 in control to the following: 0.32 ± 0.04, n = 64, p < 0.0001 for S-PolyP; 0.45 ± 0.03, n = 146, p < 0.0001 for M-PolyP; and 0.45 ± 0.03, n = 97, p < 0.0001 for L-PolyP, respectively; Fig. 2F). Thus, both S-PolyP and M-PolyP are more effective in suppressing the glutamate-induced calcium signal that results in protection of the neurons against excitotoxic DCD compared with L-PolyP.
Figure 2.

PolyP reduces the number of neurons with DCD induced by high concentration of glutamate. A, Kinetic measurements of the changes in [Ca2+]c (fura-FF ratio) of single neurons in response to 100 μm glutamate in primary cortical cocultures. B–D, Pretreatment (6 min) of cortical neurons with respectively short, medium, and long PolyP reduced [Ca2+]c responses to glutamate (100 μm). E, Changes in the percentage of neurons responding to glutamate (100 μm)-induced DCD after pretreatment with different lengths of PolyP. F, Histogram showing the effect of different length of PolyP on total glutamate-induced calcium increase (Δfura-FF ratio between basal and maximal amplitude) in cortical neurons.*p < 0.05, ***p < 0.0001.
PolyP reduces glutamate-induced mitochondrial depolarization
Glutamate-induced DCD is associated with profound mitochondrial depolarization (Vergun et al., 1999; Abramov and Duchen, 2008). In our experiments, the exposure of neurons to 100 μm glutamate induced simultaneous DCD and mitochondrial depolarization (the Rh123 signal rose by 38 ± 12% in 7 min after glutamate addition, n = 81; Fig. 3A,D). Preincubation of cortical neurons with 20 μm S-PolyP did not change basal mitochondrial potential but significantly reduced the effect of high glutamate exposure on Δψm (Fig. 3B–D). Thus, Rh123 fluorescence rose only to 17 ± 5% (n = 55, p = 0.0279), which correlates with the decrease of the number of cells with DCD in cultures treated with S-PolyP.
Figure 3.

PolyP mitigates the glutamate-induced mitochondrial membrane depolarization. A, Application of toxic (100 μm) concentrations of glutamate induced a decrease in Δψm (represented as an increase in Rhodamine123 signal, maximal depolarization after 1 μm FCCP was taken as 100%). B, Pretreatment of cortical neurons with S-PolyP reduced the effect of high concentration of glutamate on mitochondrial membrane potential. C, Average traces from single representative experiments showing kinetic changes in Δψm of control and PolyP-pretreated neurons in response to 100 μm glutamate. D, Summary of the effect of S-PolyP on the glutamate-induced mitochondrial depolarization in cortical neurons. *p < 0.05.
PolyP reduces glutamate-stimulated calcium signal via P2Y receptors
PolyP can stimulate P2Y receptors and induce the release of vesicles with transmitters that potentially can have an effect on glutamate signal (Holmström et al., 2013; Angelova et al., 2018). To assess the potential effect of the purinergic system in the PolyP-induced reduction of glutamate response, we used inhibitors of P2Y and P2X receptors, suramin, and TNP (2,4,6-trinitrophenol)-ATP, respectively. In agreement with the data presented in Figure 1, short PolyP induced a significant reduction of the glutamate-induced calcium signal compared with untreated cells (from 1.57 ± 0.05, n = 120 in control to 0.98 ± 0.05 with S-PolyP, n = 91; p < 0.0001; Fig. 4A,B,F). Preincubation of the cortical coculture with 20 μm suramin significantly mitigated the effects of S-PolyP on calcium signal induced by 5 μm glutamate [from in 0.98 ± 0.05 with S-PolyP (n = 91) to 1.27 ± 0.07 (n = 63); p = 0.0002; Fig. 4C,F]. Importantly, preincubation of the cortical coculture with the more specific inhibitor for P2X receptors (20 μm TNP-ATP) had no significant effect on the action of PolyP on the glutamate-induced rise in [Ca2+]c of cortical neurons (n = 76; Fig. 4D,F). Another inhibitor of P2 (more specific for P2X) receptors, PPADS (pyridoxalphosphate-6-azophenyl-2′,4′-disulfonic acid tetrasodium salt), also had no effect on the ability of PolyP to reduce the calcium signal under application of 5 μm glutamate (n = 44; Fig. 4E,F).
Figure 4.

Modulation of the glutamate-induced calcium signal by S-PolyP is P2Y receptor mediated. A–E, Kinetic measurement of the changes in [Ca2+]c from single neurons in response to 5 μm glutamate in cortical cultures with (B–E) and without (A) S-PolyP, in the presence of P2 receptors antagonists 20 μm suramin (C), 20 μm TNP-ATP (D), and 20 μm PPADS (E). F, Summary of the effects of P2 receptor antagonists on the S-PolyP action on glutamate-induced calcium signal. *p < 0.05.
Stimulation of P2 receptors by PolyP reduce the number of cells with DCD
The ability of PolyP to reduce DCD in neurons under exposure of the 100 μm glutamate was also dependent on the presence of the inhibitor of the P2Y receptor suramin (n = 48, p = 0.0007; Fig. 5C,F) and PPADS (n = 32, p = 0.0208; Fig. 5E,F), but not TNP-ATP (n = 36, p = 0.0676; Fig. 5D,F). Thus, PolyP acts more likely through P2Y receptors to reduce calcium responses to physiological (5 μm) and excitotoxic (100 μm) concentrations of glutamate.
Figure 5.

Effect of PolyP on [Ca2+]c rise in response to toxic concentration of glutamate (100 μm) in the presence of P2 receptor antagonists. A, B, One hundred micromolar glutamate induced profound elevation of [Ca2+]c in primary cortical neurons (A), which could be reduced by pretreatment of the cells with 20 μm PolyP (B). C–E, Preincubation of primary cortical coculture with P2 receptors blockers 20 μm suramin (C), 20 μm TNP-ATP (D), and 20 μm PPADS (E) modified the effect of S-PolyP on glutamate-induced calcium signal. F, Summary of the effects of P2 antagonists on the reduction of glutamate-induced calcium signal by PolyP (Δfura-FF ratio between basal calcium level and maximal calcium amplitude after application of 100 μm glutamate). *p < 0.05, ***p < 0.0001.
PolyP stimulates P2Y1 receptor and reduces calcium signal through AMPA receptors
To identify which type of glutamate receptor is affected by PolyP, we used the application of receptor agonists AMPA and NMDA to fura-2-loaded cortical neurons. The addition of 20 μm NMDA induced a rise in the [Ca2+]c of cortical neurons (n = 88; Fig. 6A,J). The exposure of the neurons to 20 μm short PolyP did not significantly change the amplitude or the shape of the calcium signal in response to NMDA (n = 76; Fig. 6B,J). However, short PolyP significantly reduced AMPA-induced calcium signal in neurons compared with control cells [from 2.3 ± 0.142 (n = 208) to 0.3045 ± 0.0469 (n = 161); p < 0.0001; Fig. 6C,D,J]. Importantly, suramin suppressed the action of S-PolyP on AMPA-induced calcium signal in the same way as it did for glutamate [from 0.3045 ± 0.0469 (n = 161) to 2.459 ± 0.1806 (n = 132); p = 0.0004; Fig. 6E,J]. However, considering that none of the following P2 receptor antagonists used is purely selective for P2X or P2Y (suramin, PPADS, or TNP-ATP), we used a selective antagonist for the P2Y1 receptor MRS2179 (2′-deoxy-N6-methyladenosine 3′,5′-bisphosphate tetrasodium salt). Preincubation of primary cortical neurons with 50 μm MRS2179 completely prevented the effect of short PolyP on AMPA-induced calcium signal [from 0.3045 ± 0.0469 (n = 161) to 2.409 ± 0.1765 (n = 157); p = 0.0016; Fig. 6F,J]. Thus, PolyP activates P2Y1 receptors, which reduce calcium signal through AMPA receptors in response to AMPA or glutamate. Application of another classical agonist of P2 receptors, ATP, at 100 μm, induced a typical calcium signal as expected only in astrocytes (data not shown), but not in neurons (Fig. 6G). Interestingly, prestimulation of neurons with 100 μm ATP did not reach statistical significance in the change of AMPA-induced calcium signal in these neurons (n = 82; p = 0.0977; Fig. 6G,J). However, when reduced to a concentration of 10 μm ATP induced a primary response in cortical neurons and significantly reduced the AMPA-evoked calcium signal from 2.3 ± 0.142 (n = 208) to 0.7 ± 0.055 (n = 54; p < 0.0001; Fig. 6H,J). Additionally, the application of another P2Y agonist 2MeSADP (2-methylthio-adenosine-5′-diphosphate; 1 μm; a P2Y1, P2Y12, and P2Y13 receptor, moderately potent aa P2Y11 receptors), was able to reduce the AMPA-induced calcium signal in neurons similar to the level of reduction by 20 μm short-chain PolyP (0.3045 ± 0.0469, n = 161, for AMPA+S PolyP; compared with 0.796 ± 0.095, n = 77, for AMPA+2MeSADP; p < 0.0001; Fig. 6I,J).
Figure 6.

Effects of PolyP on the AMPA- and NMDA-induced calcium signals. A, The 20 μm NMDA-induced calcium signal in cortical neurons (fura-2 ratio). B, Preapplication of short PolyP (20 μm) had no significant effect on NMDA-induced increase in the fura-2 ratio. C, D, Effect of AMPA (20 μm) on [Ca2+]c of cortical neurons (C) can be reduced by pretreatment of cells with 20 μm S-PolyP (D). E, F, J, P2 antagonist suramin (20 μm; E, J) or P2Y1 blocker MRS2179 (50 μm; F) completely abolished the effect of S-PolyP on the AMPA-induced calcium signal. G, H, Agonist of P2 receptor ATP-induced calcium rise in astrocytes did change neuronal [Ca2+]c in response to 20 μm AMPA significantly when applied at 10 μm (H), but not at 100 μm (G) concentration. I, A more specific agonist of the P2Y1, P2Y12, and P2Y13 receptor 2MeSADP, at 1 μm, mimicked the effect of S-PolyP on the AMPA receptors. J, Summary of the effects of P2 agonists and antagonists on the S-PolyP effect on the AMPA-induced calcium signal. ***p < 0.0001.
PolyP downregulates electrophysiological neural response in living tissue
Considering the role that has been given to the P2Y receptors in the literature for activation of electrical activity of glutamate receptors and to test the effects of PolyP in a system where intercellular interaction is more pronounced, we studied the effect of stimulation with PolyP in ex vivo hippocampal slices. To test whether PolyP affects electrical charge transfer through NMDARs in native DGCs, we performed electrophysiological recording of pharmacologically isolated NMDAR and AMPAR response evoked by electrical stimulation (Fig. 7A,B). We found that both long-chain and short-chain 20 μm PolyP, when added to perfusion solution, decreased the NMDAR response amplitude significantly (L-PolyP: to 0.66 ± 0.09 of control value; n = 5; p < 0.05, paired Student's t test; S-PolyP: to 0.67 ± 0.08 of control; n = 8; p < 0.01, paired Student's t test). However, neither L-PolyP nor S-PolyP significantly affected the paired-pulse ratio (PPR) of paired responses (for L-PolyP: 0.97 ± 0.07; n = 5; p < 0.05, paired Student's t test; for S-PolyP: 0.94 ± 0.38; n = 8; p < 0.05, paired Student's t test.
Figure 7.

Polyphosphate suppresses the glutamate receptor synaptic response. A, Representative traces illustrating the impact of 20 μm PolyP on the response generated by isolated NMDARs from DGCs from the hippocampus. Top, L-PolyP (L-PP); bottom, S-PolyP (S-PP). B, Summary of the effects depicted in A: first response amplitudes and PPR. C, Representative traces illustrating the impact of 20 μm PolyP on the response generated by isolated AMPARs in DGCs. Top, L-PolyP (L-PP); bottom, S-PolyP (S-PP). D, Statistical summary of the effects of PolyP in C on the first response amplitudes and PPR. Data were normalized to values generated under control; asterisks denote significance of difference from unity. *p < 0.05, **p < 0.01.
Similarly, PolyP species affected the response amplitude, but not the PPR, of AMPARs (Fig. 7C,D). Both the long-chain and short-chain 20 μm PolyP downregulated the amplitude of pharmacologically isolated AMPAR response to 0.72 ± 0.068 and 0.58 ± 0.092 of control, respectively (n = 8; in both cases, p < 0.01, paired Student's t test). The PPR of the AMPAR response was calculated to be 1.12 ± 0.11 of control with L-PolyP and 0.97 ± 0.083 of control with S-PolyP (n = 8; p < 0.05 for both cases, paired Student's t test).
PolyP protects neurons against glutamate excitotoxicity via P2Y receptors
The application of 100 μm glutamate to cortical coculture neurons and astrocytes induced significant cell death measured 24 h after stimulation (36.6 ± 5.4, n = 250; Fig. 8B). High concentrations of long PolyP also can be toxic for neurons (Angelova et al., 2016). However, preincubation (20 μm, 10 min) of the cortical cocultures with long, medium, or short PolyP significantly decreased the number of glutamate-induced dead cells (to 10.47 ± 0.95, 12.54 ± 1.75, and 12.62 ± 2.33, respectively, for both; n = 250; p < 0.0001; Fig. 8A,B). Importantly, the inhibitor of P2Y receptors suramin (20 μm added 20 min before S-PolyP and glutamate) significantly reduced the protective effect of 20 μm short PolyP against glutamate-induced excitotoxicity (from 26.48 ± 2.38 to 10.18 ± 0.89, for both; n = 250; p < 0.0001; Fig. 8C), while a more selective inhibitor of P2X receptor TNP-ATP (20 μm, 20 min before PolyP and glutamate) had no effect on neuronal viability. This strongly suggests that PolyP protects neurons against glutamate-induced toxicity via stimulation of P2Y receptors.
Figure 8.

PolyP protects neurons against glutamate-induced cell death. A, Representative images depicting the effect of PolyP of different length on cell death induced by 100 μm glutamate using PI fluorescence. B, Histogram represents the effect of different lengths of PolyP on the glutamate-induced cell death in cortical neurons. C, Histogram summarizing the effect of PolyP on glutamate-induced toxicity in the presence of P2 receptor antagonists. The number of dead cells was counted with respect to the total number of cells identified by staining the nuclei with Hoechst 33342 stain. The percentage of death cells shown in the histogram was calculated as the sum of average number of dead neurons obtained for each field of view. Each experiment was repeated three or more times using three or more different neuronal preparations. ***p < 0.0001.
Discussion
In the present study, we found that PolyP is able to reduce the physiological calcium signal induced by glutamate. This effect was dependent on the length of the polymer, and activity was reduced with elongation of the polyphosphate chain. The ability of PolyP to reduce Ca2+ influx through glutamate receptors is more likely to be a basis for significant reduction of the number of neurons with DCD underexposure of the cells with a toxic concentration of glutamate (100 μm). DCD is induced by multiple factors (Vergun et al., 1999; Khodorov, 2004; Abramov and Duchen, 2010) but is initiated by massive ion flux through glutamate receptors. Reduction of the calcium influx or activation of Ca2+-ATPases protects neurons against DCD induced by high concentrations of glutamate in the same way as PolyP (Suwanjang et al., 2013; Vaarmann et al., 2013). Although PolyP is shown to play an important role in mitochondria (Abramov et al., 2007; Pavlov et al., 2010), and in our experiments PolyP significantly reduced glutamate-induced depolarization (Fig. 4), the protective effect of PolyP is less likely due to the direct effect on mitochondria. The effect of this polymer on Δψm under glutamate exposure is more likely due to the reduction of [Ca2+]c that leads to lower calcium uptake into mitochondria. Interestingly, the inhibition or sequestration of mitochondrial calcium uptake by mitochondria leads to protection against glutamate-induced depolarization (Abramov and Duchen, 2008; Angelova et al., 2019).
Most of the effects that we observed here (reduction of the calcium signal upon stimulation with low or high concentrations of glutamate by PolyP and protection of the cells against glutamate toxicity) were dependent of the presence of P2Y (but not P2X) antagonist suramin. Previously, we and others have shown that PolyP activates calcium signal in astrocytes (and in 3% neurons) via stimulation of P2Y1 receptor (Holmström et al., 2013; Dinarvand et al., 2014). Purinergic receptors (P2X and P2Y) can be activated by purines (Burnstock, 2013) and induced calcium signal in astrocytes, that depends on the activation of phospholipase C (Domijan et al., 2014). Activation of both ionotropic (P2X) and metabotropic (P2Y) receptors suppresses AMPA and NMDA and protects against glutamate excitotoxicity (Ortega et al., 2011; Pougnet et al., 2016). PolyP suppresses glutamate-induced calcium signal and glutamate toxicity via P2Y1, which can be proven by experiments with the highly selective inhibitor MRS2179 and less likely through P2X due to weaker effects of TNP-ATP and PPADS in our experiments.
Clear downregulation of NMDA receptor response amplitude without significant changes in paired-pulse ratio suggests the suppression of NMDA receptor conductance due to postsynaptic mechanisms (i.e., impact on biochemical regulatory networks) rather than presynaptic mechanisms (i.e., modulation of glutamate release from synaptic bouton).
The Absence of significant PolyP-generated suppression of NMDA-induced Ca2+ waves (Fig. 7B) and the presence of such a suppression of NMDAR electrophysiological response (Fig. 7A,B) looks to be, to some extent, counterintuitive. However, this apparent discrepancy has two potential explanations. First, NMDA-induced Ca2+ elevation may not be only due to Ca2+ transport through NMDA receptor channels; Ca2+-induced Ca2+ release from endoplasmic reticulum in response to NMDA application, which has long been established for DGCs (Lazarewicz et al., 1998), provides a good alternative source. Hence, a Ca2+ wave after glutamate receptor activation may be generated mostly by Ca2+ release from endoplasmic reticulum, whereas, to start such a wave, NMDARs at the cytoplasmic membrane should provide just an small initial amount of Ca2+, which works as a trigger; therefore, the reduction of Ca2+ ion transfer through NMDARs by PolyP leaves enough Ca2+ to initiate release from endoplasmic reticulum, and thus does not cause a significant impact on the overall Ca2+ rise in cytoplasm.
Second, the permeability of NMDARs to Ca2+ depends on NMDAR subunit composition (Evans et al., 2012). Therefore, PolyP may suppress selectively NMDARs of subunit composition with low permeability to Ca2+: this would reduce the overall charge transfer (Fig. 6), but not the overall Ca2+ influx (Fig. 6).
On other side, PolyP can directly act on P2Y1 receptors in neurons producing (or not producing) a much smaller calcium signal due to much smaller calcium ER pool compared with astrocytes. This effect results in suppression of glutamate receptors. However, we cannot exclude the fact that PolyP induces the activation of astrocytic P2Y1 receptors (Holmström et al., 2013) and stimulates the release of neuromodulators which in turn reduces the conductance of glutamate receptors for Ca2+, one example of which could be underlying neuron–glia interaction (Angelova and Abramov, 2014).
Despite the fact that the P2 agonist ATP was shown to be able to decrease the conductance of glutamate receptors (Ortega et al., 2011; Pougnet et al., 2016), in our experiments 100 μm ATP had no effect on AMPA-induced calcium signal (Fig. 6G). This could possibly be explained by the higher selectivity of PolyP to P2Y1 receptor compared with 100 μm ATP, which is possibly more selective to other types of P2 receptors. When we tested a lower concentration, 10 μm ATP, we obtained a similar effect to those of short PolyP. Importantly, it also can be proven with the P2Y1-specific agonist 2MeSADP.
Footnotes
This work was supported by an Engineering and Physical Sciences Research Council grant (A.Y.A., P.R.A.), and by The Rosetrees Trust (S.S.) and Wellcome Trust (S.S.). We thank Dr. Toshikazu Shiba for providing polyphosphates.
The authors declare no competing financial interests.
References
- Abramov AY, Duchen MR (2008) Mechanisms underlying the loss of mitochondrial membrane potential in glutamate excitotoxicity. Biochim Biophys Acta 1777:953–964. 10.1016/j.bbabio.2008.04.017 [DOI] [PubMed] [Google Scholar]
- Abramov AY, Duchen MR (2010) Impaired mitochondrial bioenergetics determines glutamate-induced delayed calcium deregulation in neurons. Biochim Biophys Acta 1800:297–304. 10.1016/j.bbagen.2009.08.002 [DOI] [PubMed] [Google Scholar]
- Abramov AY, Fraley C, Diao CT, Winkfein R, Colicos MA, Duchen MR, French RJ, Pavlov E (2007) Targeted polyphosphatase expression alters mitochondrial metabolism and inhibits calcium-dependent cell death. Proc Natl Acad Sci U S A 104:18091–18096. 10.1073/pnas.0708959104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angelova PR, Abramov AY (2014) Interaction of neurons and astrocytes underlies the mechanism of abeta-induced neurotoxicity. Biochem Soc Trans 42:1286–1290. 10.1042/BST20140153 [DOI] [PubMed] [Google Scholar]
- Angelova PR, Agrawalla BK, Elustondo PA, Gordon J, Shiba T, Abramov AY, Chang YT, Pavlov EV (2014) In situ investigation of mammalian inorganic polyphosphate localization using novel selective fluorescent probes JC-D7 and JC-D8. ACS Chem Biol 9:2101–2110. 10.1021/cb5000696 [DOI] [PubMed] [Google Scholar]
- Angelova PR, Baev AY, Berezhnov AV, Abramov AY (2016) Role of inorganic polyphosphate in mammalian cells: from signal transduction and mitochondrial metabolism to cell death. Biochem Soc Trans 44:40–45. 10.1042/BST20150223 [DOI] [PubMed] [Google Scholar]
- Angelova PR, Iversen KZ, Teschemacher AG, Kasparov S, Gourine AV, Abramov AY (2018) Signal transduction in astrocytes: localization and release of inorganic polyphosphate. Glia 66:2126–2136. 10.1002/glia.23466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angelova PR, Vinogradova D, Neganova ME, Serkova TP, Sokolov VV, Bachurin SO, Shevtsova EF, Abramov AY (2019) Pharmacological sequestration of mitochondrial calcium uptake protects neurons against glutamate excitotoxicity. Mol Neurobiol 56:2244–2255. 10.1007/s12035-018-1204-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bazargani N, Attwell D (2016) Astrocyte calcium signaling: the third wave. Nat Neurosci 19:182–189. 10.1038/nn.4201 [DOI] [PubMed] [Google Scholar]
- Burnstock G. (2013) Introduction to purinergic signalling in the brain. Adv Exp Med Biol 986:1–12. 10.1007/978-94-007-4719-7_1 [DOI] [PubMed] [Google Scholar]
- Choi DW. (1985) Glutamate neurotoxicity in cortical cell culture is calcium dependent. Neurosci Lett 58:293–297. 10.1016/0304-3940(85)90069-2 [DOI] [PubMed] [Google Scholar]
- Cremers CM, Knoefler D, Gates S, Martin N, Dahl JU, Lempart J, Xie L, Chapman MR, Galvan V, Southworth DR, Jakob U (2016) Polyphosphate: a conserved modifier of amyloidogenic processes. Mol Cell 63:768–780. 10.1016/j.molcel.2016.07.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dinarvand P, Hassanian SM, Qureshi SH, Manithody C, Eissenberg JC, Yang L, Rezaie AR (2014) Polyphosphate amplifies proinflammatory responses of nuclear proteins through interaction with receptor for advanced glycation end products and P2Y1 purinergic receptor. Blood 123:935–945. 10.1182/blood-2013-09-529602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Domijan AM, Kovac S, Abramov AY (2014) Lipid peroxidation is essential for phospholipase C activity and the inositol-trisphosphate-related Ca(2) (+) signal. J Cell Sci 127:21–26. 10.1242/jcs.138370 [DOI] [PubMed] [Google Scholar]
- Evans RC, Morera-Herreras T, Cui Y, Du K, Sheehan T, Kotaleski JH, Venance L, Blackwell KT (2012) The effects of NMDA subunit composition on calcium influx and spike timing-dependent plasticity in striatal medium spiny neurons. PLoS Comput Biol 8:e1002493. 10.1371/journal.pcbi.1002493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fellin T. (2009) Communication between neurons and astrocytes: relevance to the modulation of synaptic and network activity. J Neurochem 108:533–544. 10.1111/j.1471-4159.2008.05830.x [DOI] [PubMed] [Google Scholar]
- Gundersen V, Storm-Mathisen J, Bergersen LH (2015) Neuroglial transmission. Physiol Rev 95:695–726. 10.1152/physrev.00024.2014 [DOI] [PubMed] [Google Scholar]
- Hertz L, Zielke HR (2004) Astrocytic control of glutamatergic activity: astrocytes as stars of the show. Trends Neurosci 27:735–743. 10.1016/j.tins.2004.10.008 [DOI] [PubMed] [Google Scholar]
- Holmström KM, Marina N, Baev AY, Wood NW, Gourine AV, Abramov AY (2013) Signalling properties of inorganic polyphosphate in the mammalian brain. Nat Commun 4:1362. 10.1038/ncomms2364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khodorov B. (2004) Glutamate-induced deregulation of calcium homeostasis and mitochondrial dysfunction in mammalian central neurones. Prog Biophys Mol Biol 86:279–351. 10.1016/j.pbiomolbio.2003.10.002 [DOI] [PubMed] [Google Scholar]
- Krnjević K. (1970) Glutamate and gamma-aminobutyric acid in brain. Nature 228:119–124. 10.1038/228119a0 [DOI] [PubMed] [Google Scholar]
- Kumble KD, Kornberg A (1995) Inorganic polyphosphate in mammalian cells and tissues. J Biol Chem 270:5818–5822. 10.1074/jbc.270.11.5818 [DOI] [PubMed] [Google Scholar]
- Lazarewicz JW, Rybkowski W, Sadowski M, Ziembowicz A, Alaraj M, Wegiel J, Wisniewski HM (1998) N-methyl-D-aspartate receptor-mediated, calcium-induced calcium release in rat dentate gyrus/CA4 in vivo. J Neurosci Res 51:76–84. [DOI] [PubMed] [Google Scholar]
- Lorenz B, Münkner J, Oliveira MP, Kuusksalu A, Leitão JM, Müller WE, Schröder HC (1997) Changes in metabolism of inorganic polyphosphate in rat tissues and human cells during development and apoptosis. Biochim Biophys Acta 1335:51–60. 10.1016/S0304-4165(96)00121-3 [DOI] [PubMed] [Google Scholar]
- Müller WEG, Wang S, Neufurth M, Kokkinopoulou M, Feng Q, Schröder HC, Wang X (2017) Polyphosphate as a donor of high-energy phosphate for the synthesis of ADP and ATP. J Cell Sci 130:2747–2756. 10.1242/jcs.204941 [DOI] [PubMed] [Google Scholar]
- Olney JW. (1969) Brain lesions, obesity, and other disturbances in mice treated with monosodium glutamate. Science 164:719–721. 10.1126/science.164.3880.719 [DOI] [PubMed] [Google Scholar]
- Ortega F, Pérez-Sen R, Delicado EG, Teresa Miras-Portugal M (2011) ERK1/2 activation is involved in the neuroprotective action of P2Y13 and P2X7 receptors against glutamate excitotoxicity in cerebellar granule neurons. Neuropharmacology 61:1210–1221. 10.1016/j.neuropharm.2011.07.010 [DOI] [PubMed] [Google Scholar]
- Pavlov E, Aschar-Sobbi R, Campanella M, Turner RJ, Gómez-García MR, Abramov AY (2010) Inorganic polyphosphate and energy metabolism in mammalian cells. J Biol Chem 285:9420–9428. 10.1074/jbc.M109.013011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pougnet JT, Compans B, Martinez A, Choquet D, Hosy E, Boué-Grabot E (2016) P2X-mediated AMPA receptor internalization and synaptic depression is controlled by two CaMKII phosphorylation sites on GluA1 in hippocampal neurons. Sci Rep 6:31836. 10.1038/srep31836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stotz SC, Scott LO, Drummond-Main C, Avchalumov Y, Girotto F, Davidsen J, Gómez-Gárcia MR, Rho JM, Pavlov EV, Colicos MA (2014) Inorganic polyphosphate regulates neuronal excitability through modulation of voltage-gated channels. Mol Brain 7:42. 10.1186/1756-6606-7-42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suwanjang W, Holmström KM, Chetsawang B, Abramov AY (2013) Glucocorticoids reduce intracellular calcium concentration and protects neurons against glutamate toxicity. Cell Calcium 53:256–263. 10.1016/j.ceca.2012.12.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaarmann A, Kovac S, Holmström KM, Gandhi S, Abramov AY (2013) Dopamine protects neurons against glutamate-induced excitotoxicity. Cell Death Dis 4:e455. 10.1038/cddis.2012.194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vergun O, Keelan J, Khodorov BI, Duchen MR (1999) Glutamate-induced mitochondrial depolarisation and perturbation of calcium homeostasis in cultured rat hippocampal neurones. J Physiol 519:451–466. 10.1111/j.1469-7793.1999.0451m.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zakharian E, Thyagarajan B, French RJ, Pavlov E, Rohacs T (2009) Inorganic polyphosphate modulates TRPM8 channels. PLoS One 4:e5404. 10.1371/journal.pone.0005404 [DOI] [PMC free article] [PubMed] [Google Scholar]