

Abstract
High-grade gliomas (HGGs) are aggressive primary brain tumors with local invasive growth and poor clinical prognosis in both adult and pediatric patients. Clinical response is compounded by resistance to standard frontline antineoplastic agents, an absence of novel therapeutics, and poor in vitro models to evaluate these. We screened a range of recently identified anticancer compounds in conventional adult, pediatric, and new biopsy-derived HGG models. These in vitro lines showed a range of sensitivity to standard chemotherapeutics, with varying expression levels of the prognostic markers hypoxia-induced factor (HIF) 1α and p53. Our evaluation of lead DIVERSet library compounds identified that JAG-6A, a compound that was significantly more potent than temozolomide or etoposide, was effective against HGG models in two-dimensional and three-dimensional systems; mediated this response by the potent inhibition of topoisomerase Iiα; remained effective under normoxic and hypoxic conditions; and displayed limited toxicity to non-neoplastic astrocytes. These data suggest that JAG-6A could be an alternative topoisomerase IIα inhibitor and used for the treatment of HGG.
Introduction
In 2015 alone, 22,850 adults (12,630 men and 10,280 women) were diagnosed with brain and other central nervous system (CNS) cancers in the United States, with 15,320 resulting in patient death [1]. High-grade gliomas (HGGs) account for 52% of all primary brain tumors and in children occur at a frequency of 5.7 cases per 100,000 [2]. Adult HGG (aHGG) treatment comprises a multidisciplinary approach including surgical resection and combined radio- and chemotherapy [3]. Despite this, mean patient survival is less than 15 months [4], [5]. The current standard chemotherapeutic for aHGG treatment is the DNA alkylating drug temozolomide (Temodal) (TMZ) [6], [7]. The addition of TMZ to the surgical and radiotherapy regimen (the “Stupp protocol”) demonstrated a significant patient survival benefit with a median increase in survival of 2.5 months [8]. However, this protocol yields a 24-month progression-free survival of just 14%, where a discernible TMZ response was only noted in tumors exhibiting O6-methylguanine DNA methyltransferase (MGMT) promoter methylation [9]. Irrespective of MGMT promoter methylation, almost all patients demonstrate disease relapse and eventual progressive disease. Pediatric HGG (pHGG) is significantly different from adult disease, where recent substantive genomic and epigenomic studies has resulted in an almost complete reclassification of childhood brain tumors, incorporating diverse histological and molecular-phenotype differences [10], [11], [12]. Both adult and pediatric HGGs present with areas of attenuated vascularization forming hypoxic regions (areas of insufficient oxygenation) [13], [14], [15]. This environment drives the modulation and stabilization of hypoxia induced factor (HIF)-1α, a transcription factor implicated in oncogenesis, angiogenesis, proliferation, and invasion. HIF-1α expression correlates with poor clinical prognosis, in part due to co-inhibition of the p53 mediated proapoptotic network in HGG [16], [17]. While TP53 mutations have not been associated with aHGG patient prognosis, approximately 40% of pHGGs are associated with TP53 mutations [18] [12], and this correlated with patient prognosis [19], [20], [21].
Similar to aHGG patients, pHGG treatment consists of an aggressive multidisciplinary approach that incorporates surgery (if possible), radiotherapy, and chemotherapy [22]. Prognosis remains dismal with 5-year progression-free survival between 10% and 30% [23], [24], [25], [26]. In contrast to aHGG where TMZ treatment has a clear therapeutic benefit, within the pHGG patient population, TMZ regimens revealed no impact on patient outcome [27], [28]. Consequently, pHGG chemotherapy regimens incorporate procarbazine, lomustine, etoposide, cisplatin, and vincristine [3], [27], [29], [30], [31]. The incorporation of etoposide (in combination with cisplatin) represents a common treatment modality for patients and is a compelling therapeutic agent. Etoposide, a topoisomerase II poison, induces double-strand DNA breaks by increasing the amount of cleavable topoisomerase II:DNA complexes. The level of these complexes is topoisomerase II concentration dependent, where it is far higher in rapidly dividing cancer cells versus non-neoplastic cells [32]. Topoisomerase II inhibitors have been extensively tested against a number of cancer types and have shown promise against aHGG [33], [34]. One of the critical limitations for topoisomerase II poisons relates to their pharmacokinetic profile and poor blood–brain barrier permissiveness. Despite these limitations, there have been a number of approaches to enhance the delivery of these agents into the CNS. This includes convection enhanced delivery [35]. Importantly, the identification of novel therapeutics, particularly topoisomerase II poisons with a significantly lower molecular weight (with increase solubility), would be of significant interest to the field.
In silico analysis using the DIVERSet compound library from Chembridge (San Diego, CA) identified a number of lead anticancer agents for further analysis [36]. Here we performed in vitro two-dimensional (2D) and three-dimensional (3D) analysis of conventional and recently obtained, biopsy-derived aHGG cells as well as established pHGG cells following exposure to each lead agent. We evaluated cell viability posttreatment with each compound and evaluated the effect of these compared to TMZ, etoposide, vincristine, and gemcitabine. We revealed that the lead agents were potent inhibitors of topoisomerase II and, consistent with this cellular target, examined the HIF-1α and p53 signaling networks in key HGG lines following exposure to these compounds.
Methods and Methods
Tumor Specimens and Primary Tumor Cultures
Following informed consent and in accordance with the LREC review board (11/SC/0048), HGG samples were obtained from patients undergoing biopsy surgery at Kings College Hospital NHS Foundation Trust (London, UK).Tumors were classified based on WHO criteria after examination by neuropathologists. The tumor mass was mechanically dissociated into explant clumps, allowing neoplastic cells to colonize the flask. Biopsies were cultured in DMEM with 10% heat-inactivated FCS. Upon establishment of cell cultures, a combined STR profile was conducted for each adult HGG (Agilent Bioscience). All experimental protocols were approved by the University of Portsmouth, Faculty of Research.
Chemotherapeutics and Cell Culture
TMZ (T2577) and vincristine (V0400000) were from Sigma-Aldrich. Gemcitabine (S1714) was from SelleckChem. JAG-6A, CC-I, JAG-32, and JAG-79 were provided by Opal Oncology (Cambridge, UK). U87MG was obtained from the ATCC. KNS42 and SF188 cells were obtained from Professor Chris Jones (Institute of Cancer Research, London, UK). UP-029, SEBTA-003, SEBTA-023, and SEBTA-025 were cultured in DMEM (61965 Fisher Scientific) supplemented with 10% HIFBS (F7524-500ML Sigma-Alrich). SC1800 and CC2565 non-neoplastic astrocytes were purchased from Lonza and maintained in astrocyte growth medium supplemented with SingleQuots (CC-3187 Lonza) including (CC-4123) rhEGF, insulin, ascorbic acid, and L-glutamine. Cells were cultured under normoxic (21%) or hypoxic (1%) O2.
Spheroid Formation and Staining Assay
We modified the Sirenko and Montenegro et al. protocol [37], [38]. Cells were seeded in ultra-low adherence plates and treated with compounds after 12 hours. Spheroids were stained in 4 μM Calcein AM (Fisher Scientific, C1430), 50 μg/ml propidium iodide (PI) (Fisher Scientific, P3566), and 33 μM Hoechst 33342 (Fisher Scientific, H1399) in phenol-free, serum-free DMEM. Imaging was conducted on InCell 6000 at 10× magnification, 4 fields with 15% overlap, 13 z-stacks, and 20 μM per step. Z-stacks were compressed with maximum projection, extended focus algorithm, to a single in-focus image. Four fields were stitched to a single image. Each individual stained image is included in Supplemental Figure 1.
MTS Cell Viability Assay
Cells were seeded in triplicate in a 96-well plate. Twenty-four hours postseeding, cell lines were treated at varying dosages of each therapeutic agent. MTS assay (G3580 Promega) was conducted at indicated time points following the manufactures guide. Absorbance at 490 nm was recorded on BMG Labtech Polarstar.
Annexin V/propidium Iodide Apoptosis Assay
Cells after the treatment with each agent were collected, and the cell concentration was determined using a Countess II FL (Thermo Fisher, UK). Cells were centrifuged at 400g for 5 minutes, and the pellet was resuspended in 300 μl PBS. Cells were centrifuged again at 400g for 5 minutes, and the pellet was resuspended in 100 μl Annexin V binding buffer. Annexin V-CF488A conjugate was added to the cells including Hoechst (final concentration: 10 μg/ml) and mixed by pipetting. Cells were incubated at 37°C for 15 minutes. Following incubation, cells were centrifuged at 400g for 5 minutes. The cell pellet was resuspended in Annexin V binding buffer and centrifuged again, and the pellet was resuspended in Annexin V supplemented with 10 μg/ml PI. Samples were run and analyzed using a NucleoCounter NC-3000 (Chemometec, Denmark).
Western Blot Analysis
Total protein was harvested using RIPA buffer (89900, Thermo-Fisher) and 1× protease inhibitor cocktail (78442 Thermo-Fisher). Primary antibodies HIF-1α (ABE279 Millipore), p53 (DO1, sc-126 SCBT), or β-actin (sc-47778 SCBT) were added to the membrane overnight. Secondary antibody was added (LICOR) at 1:10,000 dilution for 1 hour. Membranes were imaged on Odyssey CLX (Licor). All full-length gels and blots are included in our Supplemental Information file (Supplemental Figures 2-4).
Quantitative Real-Time PCR
Total RNA was extracted (RNAeasy, 74104 Qiagen) and quantified using RNA6000 chip arrays (Agilent Bioscience). Real-time PCR was performed per sample in triplicate on a Roche LightCycler 96. Primers are as follows: PUMA (fwd 5-gacctcaacgcacagtacga-3 and rev 5-tgggtaagggcaggagtc-3), Bax (fwd 5-ctgacggcaacttcaactg-3, rev 5-cactgtgacctgctccagaa-3). p21 (fwd 5-ggaagaccatgtggacctgt-3, rev 5-aagatgtagagcgggccttt-3). Aldolase c, (fwd 5-tctctcaacctcaat-3, rev 5-agtacatagc-3). HEK2 (fwd 5-tcgcatctgcttgcctacttc-3, rev 5-cttctggagcccattgtccgt-3), and GAPDH (fwd 5-gagtcaacggatttggtcgt-3, rev 5-ttgattttggagggatctcg-3). Data analysis was carried out using the 2−∆∆CT method [39].
Topoisomerase IIα Decatenation Assay
Kinetoplast DNA (kDNA) decatenation assay was performed using a Topopoisomerase II assay kit (TopoGEN, Inc., Port Orange, FL) according to the manufacturer’s instructions. Topoisomerase IIα decatenates kDNA which consists of highly catenated networks of circular DNA in an ATP-dependent reaction to yield individual minicircles of DNA. In brief, for topoisomerase IIa–mediated kDNA decatenation assay, the 20-μl reaction mixture contains the following components: 50 mM Tris–HCl, pH 8.0, 150 mM NaCl, 10 mM MgCl2, 0.5 mM dithiothreitol, 30 mg/ml bovine serum albumin, 2 mM ATP, 260 ng of kDNA, several concentrations of compounds, and 4 U of human topoisomerase IIα. The final concentration of 0.5% (v/v) DMSO was used.
Molecular Modeling Study
The molecular modeling studies were based on the X-ray crystal structure of human topoisomerase IIα (5BTD). Calculations were performed using the program Molecular Operating Environment (Chemical Computing Group Inc., Montreal, Canada). Ligand binding energies in kcal/mol were calculated using force field refinement (Amber12EHT) following initial placement via the “Triangle Matcher” placement methodology.
Scanning Electron Microscopy
Spheroids were fixed with 2.5% glutaraldehyde and 2% paraformaldehyde in phosphate buffer (0.1 M, pH 7.4) and treated with 1.5% potassium ferrocyanide and 1% osmium tetroxide, then with 1% osmium tetroxide and a final 1% aqueous uranyl acetate. Spheroids were imaged at 80 kV on Zeiss EVO MA10 SEM.
Data Analysis and Statistics
In vitro experiments were analyzed (GraphPad Prism) and are represented as mean values ± SD, indicating the number of experiments carried out for each assay. Statistical significance has been calculated using Student’s t test, (*P ≤ .05), two-tailed ANOVA analysis, or the log-rank test for Kaplan-Meier survival analyses.
Results
Conventional Chemotherapeutics Significantly Reduce aHGG Cell Viability
We obtained aHGG-derived biopsy material and isolated in vitro cell lines designated UP-029, SEBTA-023, SEBTA-025, and SEBTA-003. Each was DNA fingerprinted using a combined short tandem repeat and fragment-length amplification [40]. We included the human glioma cell line U87MG in our studies. While the U87MG model has been questioned, U87MG is HGG in origin and is a widely used in vitro model for preclinical testing [41], [42]. U87MG cells were exposed to TMZ, gemcitabine, etoposide, or vincristine for 96 hours under normoxic conditions (Figure 1A). U87MG cells were refractory to TMZ (EC50 = 15.8 μM), gemcitabine (EC50 = 103.6 μM), and etoposide (76.4 μM), although they were sensitive to vincristine (EC50 = 0.315 nM). We questioned if any of our biopsy-derived aHGG cell lines were sensitive to these standard frontline chemotherapeutics (Figure 1, B-E). The most potent chemotherapeutic tested was vincristine. The UP-029 aHGG cells displayed sensitivity to TMZ (Figure 1B), whereas both SEBTA-025 (Figure 1C) and SEBTA-003 (Figure 1D) aHGG cell lines were refractory to this chemotherapeutic. In contrast, the SEBTA-023 aHGG cells were highly resistant to all chemotherapeutics except vincristine (Figure 1E).
Figure 1.

Novel biopsy-derived aHGG cell lines display varying sensitivity to the chemotherapeutics temozolomide, gemcitabine, and vincristine. (A) U87MG, (B) UP-029, (C) SEBTA-025, (D) SEBTA-003, and (E) SEBTA-023 were seeded in 96-well plates overnight. The next day, cells were treated with temozolomide (≤128 mM), gemcitabine (≤330 mM), or vincristine (≤1 mM) in DMEM. Ninety-six hours posttreatment, loss of cell viability was assessed by CellTiter 96 AQueous One Solution Cell Proliferation Assay (Promega). Data presented as mean average of n = 3 ± SD.
Novel Anticancer Agents Mediate a Potent Cytotoxic Response in Adult HGG
We next questioned if any of our lead DIVERSet anticancer agents [termed JAG-6A, CC-I (JAG-31), JAG-32, and JAG-79] (Figure 2A) would demonstrate an anti-HGG effect in the U87MG model (Figure 2B) or in our novel biopsy lines (Figure 2, C-F). The U87MG cells showed sensitivity to the JAG agents, in particular JAG-6A (EC50 3.6 μM) and CC-I (EC50 7.4 μM). Similarly, JAG-6A was the most potent therapeutic in our novel in vitro models. We next compared the average EC50 value at 96 hours posttreatment following the single exposure of each cell line to TMZ, etoposide, or JAG-agent (Figure 2G). For each in vitro cell line, JAG-6A was significantly more effective than TMZ or etoposide. The biopsy-derived HGG cell lines were significantly more resistant to these fronttline chemotherapeutics when compared to the widely utilized U87MG cell line. The SEBTA-023 aHGG model was highly resistant to all compounds tested.
Figure 2.

Novel JAG anticancer agents demonstrate a differential and significant anti-aHGG response. (A) Chemical structure of each JAG agent used in this study. (B) U87MG cells (5×103 per well) were seeded overnight and treated the following day with JAG-6A (black), CC-I (blue), JAG-32 (green), or JAG-79 (purple) at ≤1000 μM. Ninety-six hours posttreatment, loss of cell viability was assessed by CellTiter 96 AQueous One Solution Cell Proliferation Assay (Promega). Data presented as average of n = 3 independent studies ± SD. (C-F) JAG agent susceptibility was determined for each novel aHGG model. (G) Average EC50 values (±SD) for TMZ and each JAG agent were determined. Two-tailed ANOVAs was conducted, and P values are shown for each comparison; n = 3. (H) Representative microscopy (×10) images of UP-029 cells. UP-029 aHGG cells were seeded (1×104). The next day, cells were treated with indicated agents (10 mM TMZ or 30 μM per JAG agent) and imaged (up to 24 hours) at 37°C under normoxic conditions. Images were recorded using EVOS FL Auto (Life Technologies). (I) Apoptosis assays were conducted for each indicated cell line 24 hours post 30 μM treatment with each JAG agent. Annexin V/PI staining was conducted and apoptosis status was determined using an NC-3000 counter; n = 3 ± SD.
Following exposure to each agent, we examined cell morphology (Figure 2H). Twenty-four hours following JAG-6A treatment, each biopsy-derived aHGG cell line displayed morphology changes, including rounding up, membrane blebbing, and monolayer detachment (Figure 2H panel 3). This phenotype was not as pronounced 24 hours posttreatment with CC-I or JAG-79 (Figure 2H panel 4 and 6). No cell morphology changes were noted following JAG-32 exposure (Figure 2H panel 5). Consistent with our viability studies, some rounding up, membrane detachment, and cell death were noted post-TMZ exposure. We questioned the mechanism of cell death following exposure to each JAG agent. Our aHGG lines were treated with TMZ or each JAG agent up to 96 hours. Annexin V/PI staining was conducted, and in agreement with our previous data, we noted a significant increase in Annexin V and Annexin V/PI staining posttreatment with each compound (Figure 2I). For each in vitro model, Annexin V and dual Annexin V/PI staining was highest following JAG-6A treatment. TMZ, CC-I, and JAG-32 exposure each induced limited Annexin V and Annexin V/PI staining.
JAG Anticancer Agents Are Effective Against Pediatric HGG In Vitro Models
Having observed that some JAG agents were effective in various aHGG models, we questioned if these agents were effective against pHGG. KNS42 or SF188 (both grade IV) cells were treated with JAG agents (Figure 3, A-B). Similar to our aHGG cells, KNS42 cells displayed varying sensitivity post-JAG agent treatment, with JAG-6A being the most potent compound tested. SF188 cells showed notable resistance to these therapeutics, exhibiting sensitivity at only the very highest tested drug concentrations. We observed that pHGG cell morphology changed following JAG agent exposure consistent with cell viability (Figure 3, C and D). JAG-6A induced the most prominent change in KNS42 cell morphology 24 hours posttreatment, characterized by extensive rounding up and monolayer detachment. We conducted Annexin V/PI staining in our pHGG cells following treatment with each JAG-agent (Figure 3D). We noted that, 24 hours posttreatment, there was a significant increase in both early and late apoptotic cell populations. As we would predict from our previous data, limited apoptosis was detected in JAG agent–treated SF188 cells.
Figure 3.

JAG agents mediate a potent anti-pHGG response with limited toxicity to non-neoplastic astrocytes. (A) KNS42 and (B) SF188 pHGG cells treated with each JAG agent. After 96 hours, loss of cell viability was determined by CellTiter 96 AQueous One Solution Cell Proliferation Assay (Promega). Representative microscopy (×10) images of (C) KNS42 and SF188 cells. Cells were treated with indicated agents (30 μM per JAG agent) and imaged (up to 24 hours) at 37°C under normoxic conditions. Images and films were recorded using EVOS FL Auto (Life Technologies). (D) Apoptosis assays were conducted for each indicated cell line 24 hours post 30 μM treatment with each JAG agent. Annexin V/PI staining was conducted, and apoptosis status was determined using an NC-3000 counter; n = 3 ± SD. (E and F) Dose-response curves for non-neoplastic CC2565 cells following exposure to TMZ (≤128 mM), vincristine (≤1 mM), or individual JAG agents ≤1000 μM in complete astrocyte growth medium. Data presented as average of n = 3. Error bars indicate ±SD.
JAG Anticancer Agents Are Well Tolerated by Non-Neoplastic Astrocytes
A critical consideration for any therapeutic is toxicity to non-neoplastic cells. We questioned the sensitivity of the non-neoplastic astrocyte cell line CC2565 to each conventional therapeutic (Figure 3E) and JAG agent (Figure 3F). TMZ and JAG agents were well tolerated; however, CC2565 cells displayed significant sensitivity to vincristine.
JAG Anticancer Agents Remain Effective Under Hypoxic Conditions
In addition to non-neoplastic cell tolerance, any HGG therapeutic must remain effective under hypoxic (1%) O2 conditions. To address this question, we examined cell viability posttreatment under both normoxic (21%) and hypoxic (1%) O2 conditions. Taking this into consideration, we determined that there was no significant difference in EC50 value for each JAG agent under hypoxic conditions. Both JAG-6A and CC-I remained effective under this condition (Figure 4, A-B). Supporting our previous data, SF188 cells showed little to no response to each JAG agent irrespective of oxygenation levels (Figure 4C). For each of our tested cell lines, there was no statistical difference between EC50 values for any of the tested agents under either hypoxic or normoxic conditions.
Figure 4.

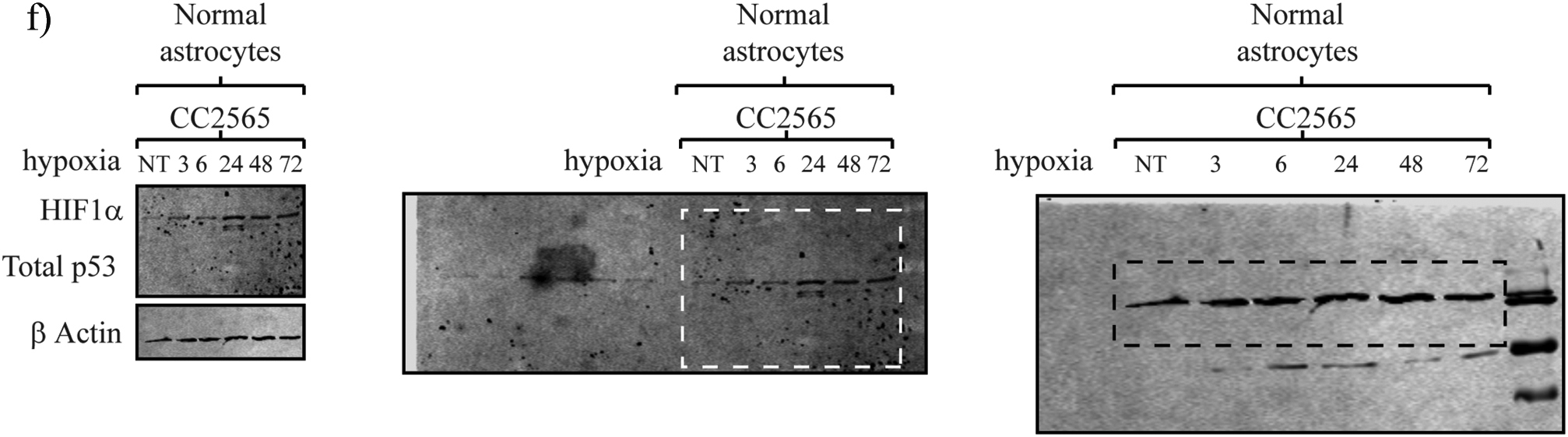
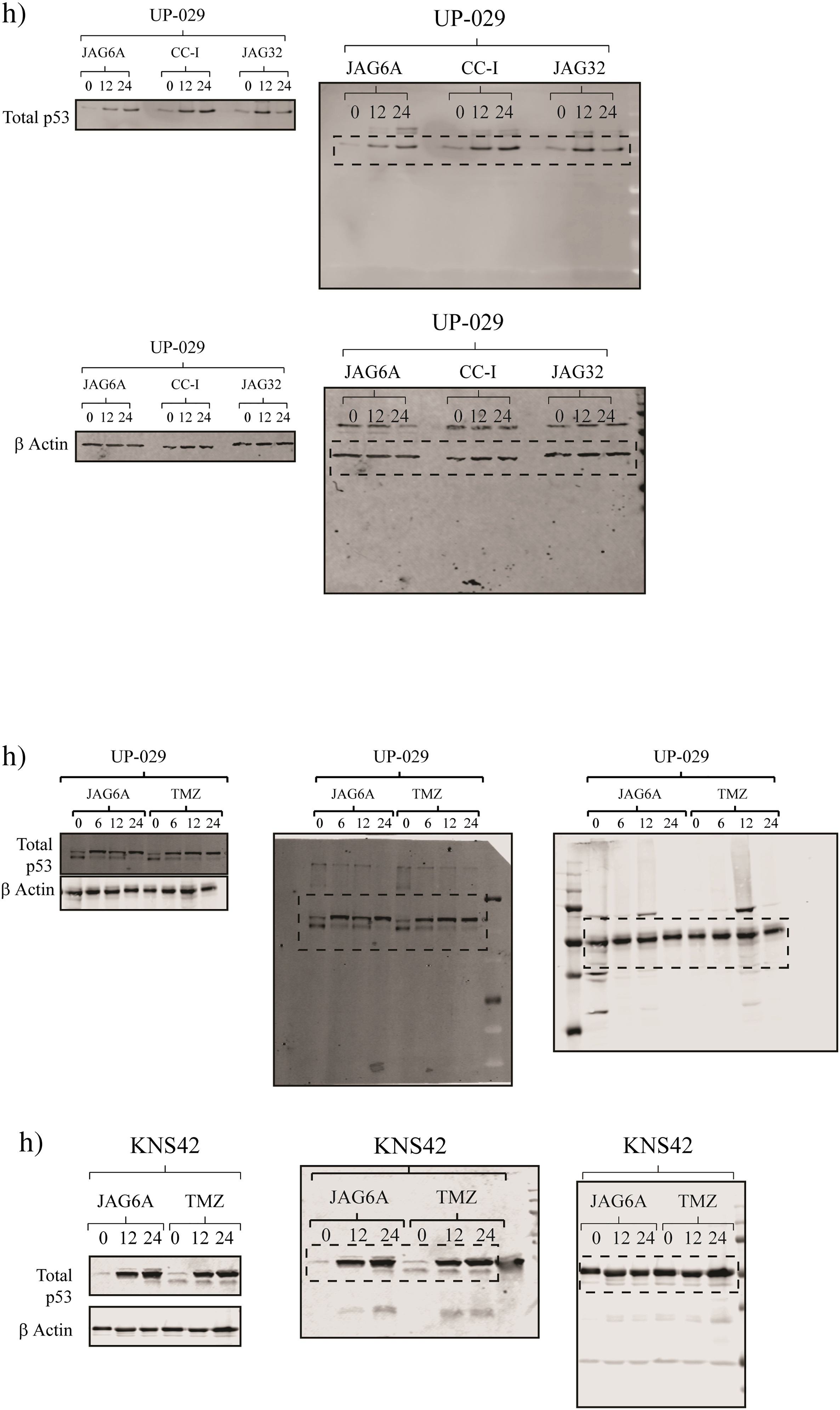
JAG agents are effective under hypoxic conditions, while novel aHGG models express differential levels of HIF1α and p53. Average EC50 values (±SD) at 96 hours posttreatment for TMZ and each JAG agent were determined under normoxic (21% O2) or hypoxic (1% O2) conditions. (A) UP-029 aHGG, (B) KNS42, and (C) SF188 pHGG cells; n = 3. Two-tailed ANOVA was conducted for each pair; all comparisons were not significant. (D) Novel aHGG cells, (E) pHGG cells, and (F) non-neoplastic astrocytes were cultured under normoxic (NT) or hypoxic (1% O2) conditions for up to 72 hours, and total protein lysates were analyzed by Western blotting. (G) Indicated cell lines were cultured under either normoxic (21% O2) or hypoxic (1% O2) conditions for 24 hours. Cells were collected at the end of the incubation. RNA was extracted and qRT-PCR for aldolase c and HEK2 mRNA was analyzed; n = 3. Shown are fold change in aldolase c or HEK2 relative to GAPDH. (H) (left) UP-029 aHGG cells were cultured under normoxic conditions and exposed to 10 μM JAG-6A, CC-I, JAG-32, or TMZ up to 24 hours. (Middle) UP-029 cells were treated with 10 μM JAG-6A or TMZ up to 24 hours. (Right) KNS42 cells were treated with 10 μM JAG-6A or TMZ up to 24 hours. Total protein lysates were analyzed by Western blotting for total p53 or β actin. (I) UP-029 aHGG cells were cultured under normoxic conditions (21% O2) and exposed to 10 μM of each JAG agent up to 12 hours. Expression of p21, PUMA, and Bax mRNA was measured; n = 3. Shown are fold change in each gene relative to GAPDH mRNA of chemotherapy-treated versus mock-treated cells normalized to 1.0. (J) UP-029 aHGG cells were cultured under normoxic conditions (21% O2) and exposed to 10 mM of TMZ or 30 μM gemcitabine up to 12 hours. Cells were collected at the end of each incubation. RNA was extracted and qRT-PCR for PUMA and Bax mRNA was analyzed. n = 3, error bars indicate ±SD.
Differential Prognostic Protein Expression Occurs in Adult and Pediatric HGG Models
Based on these hypoxia studies, we next assessed HIF-1α protein expression under both normoxic and hypoxic conditions (Figure 4D). Under normoxic conditions, UP-029, SEBTA-023, and SEBTA-003 aHGG cells demonstrated high HIF-1α protein expression. KNS42 and SF188 pHGG cells also had detectable HIF-1α protein expression under normoxic conditions, a characteristic indicative of poor clinical prognosis (Figure 4E). Non-neoplastic CC2565 cells showed little HIF-1α protein expression under normoxic conditions, although after hypoxic incubation, they showed protein accumulation (Figure 4F). We questioned if this accumulation correlated with HIF-1α–dependent gene transcription. Consistent with our HIF-1α data, there was expression of aldolase c and HEK2 under normoxic and hypoxic conditions in HGG (Figure 4G). In contrast to aHGG cells, a significant increase in aldolase c and HEK2 transcription in non-neoplastic astrocytes was observed after hypoxic incubation.
Under normoxic conditions, there was significant variation in the total p53 protein level in our cell lines (Figure 4, D and E). There was detectable total p53 in the UP-029 cells, while the SEBTA-023 cell line had little to no detectable total p53. SF188 cells showed an appreciably lower, albeit specific, band. We questioned if our HGGs demonstrate p53 accumulation and/or activation posttreatment with each JAG agent or TMZ (Figure 4H). We observed the accumulation of total p53 after JAG-6A, CC-I, or JAG-32 exposure in the UP-029 aHGG cells. Both SEBTA-023 and SF188 showed no detectable total p53 and suggest, with our previous data, that there was a p53 dependency for these anticancer agents. We questioned if there were any changes in p53-dependent gene expression, in particular, those that direct apoptosis or cell cycle arrest (Figure 4I).
We found that there was little to no induction of p21 transcription but significant expression of PUMA and Bax consistent with the apoptosis and cell death previously described. Similarly, we detected PUMA and Bax transcription following TMZ or etoposide exposure (Figure 4J).
JAG-6A Mediates a Potent Cytotoxic Response in Adult and Pediatric HGG 3-D Spheroids
Having revealed a strong anti-aHGG response post-JAG-6A treatment, we questioned if the effectiveness of this compound when used to treat 2D cell cultures was conserved in 3D models. We established two 3D HGG spheroid models: one adult, (UP-029) and one pediatric (KNS42). Both formed robust spheroids, observed by confocal (Figure 5A) and scanning electron microscopy (Figure 5B). Spheroids were established and, 12 hours postdevelopment, exposed to JAG agents or TMZ (Figure 5C). The percentage of live cells in mock treated KNS42 and UP-029 spheroids remained high (87% ±5.26% and 96.0% ±1.957%) up to 72 hours (Figure 5D). A significant increase in the percentage of dead cells post JAG-6A treatment (UP-029, 44.321% ± 6.254% and KNS42, 88.1% ±4.379%, respectively) was noted. The remaining JAG agents showed only a modest increase in cell death up to 72 hours. As we observed in our 2D studies, JAG-6A–treated spheres displayed a significantly increased total sum of dead cells (Figure 5E). Based on these data, we concluded that JAG-6A retained efficacy in 3D HGG models. In contrast to our 2D studies where CC-I instigated a significant reduction in the viability of UP-029 cells, in UP-092 3D spheroids, CC-I effectiveness was significantly attenuated. These data raised the hypothesis that the reduced surface area of the spheroid or the diffusion of CC-I through the cell mass was diminished. UP-029 aHGG showed a significant increase of PUMA and Bax transcription post JAG-6A treatment of the spheroids and a significant increase in Bax transcription post TMZ exposure (Figure 5F). Consistent with our viability studies, CC-I exposure did not induce the transcription of either PUMA or Bax in our 3D spheroids.
Figure 5.

Novel aHGG models form spheroids. Adult/pediatric HGG spheroids are sensitive to specific JAG anticancer agents. (A) A total of 5×103 UP-029 aHGG or KNS42 pHGG cells were cultured under ultra-low adherent conditions. Twenty-four hours later, both in vitro lines formed dense, highly viable spheroids. (B) SEM of each spheroid model; left panel scale bar: 50 μm, right panel scale bar. (C) UP-029 or KNS42 spheroids were treated with TMZ (10 mM) or JAG agents (30 μM). Seventy-two hours posttreatment, triple staining for nuclear, live, and dead cells was conducted (see Methods). (D) Percentage of PI-positive (dead) cells are presented as mean ± SD. (E) Sum (total number) of dead cells (per spheroid/micron2) is presented as mean ± SD. n = 6. (F) UP-029 adult GBM cells were cultured in ultra-low adherence conditions at 21% O2 and then exposed to 10 mM of TMZ or 30 μM of each JAG agent up to 12 hours. Spheroids were collected at the end of each incubation. RNA was extracted, and p21, PUMA, and Bax expression was analyzed. n = 3. For all, two-tailed ANOVA was conducted, and P values are shown for each comparison. (G) Structure of each agent docked into topoisomerase IIα. The binding cavity of topoisomerase IIα is shown in yellow. Topoisomerase is shown as the brown-colored ribbon with residues on the binding site. (H) Binding energy (kcal/mol−1) for each JAG agent and etoposide. Note, the most energy-stable binding for JAG79 is outside of the topoisomerase IIα cavity. (I) The JAG agent concentration-dependent inhibition of human topoisomerase IIα–mediated kDNA decatenation. All experiments were carried out according to instructions from the Topogen kit (Port Orange, FL). Reactions contained 4 U of enzyme, 0.26 mg of DNA substrate, and the indicated concentration of each agent dissolved in DMSO (0.5% final concentration (v/v)). Different topological forms exhibited different mobility as indicated. Etoposide was included as a positive control.
Our data revealed that JAG-6A directed a clear anti-HGG response in both 2D and 3D models independent of the environmental oxygenation conditions. However, it remained elusive as to how JAG-6A could elicit this response. To answer this question, we conducted molecular modeling studies. We noted that, with the exception of JAG-79, each JAG agent bound to human topoisomerase IIα. The predicted binding location for JAG6A, CCI, and JAG32 was inside the topoisomerase IIα cavity, where we hypothesized that they may function as an inhibitor (Figure 5G). Etoposide (a known topoisomerase Iiα) poison and chemotherapeutic agent also bound in this cavity. Whereas the best binding energies (kcal/mol−1) for JAG6A, CCI, and JAG32 were found to be inside the same region of space within topoisomerase IIα (Figure 5H), the JAG79 molecule was outside of this region. The modeling profile and location for JAG-79 in part suggest a compelling reason why the effectiveness of this compound was significantly worse than all of the other JAG agents examined. We performed kDNA decatenation assays to determine the ability of these compounds to inhibit topoisomerase IIα enzyme activity. As we predicted, etoposide potently inhibited topoisomerase IIα. Strikingly, we observed that JAG6A, CC-I, and JAG32 inhibited topoisomerase IIα activity in a dose-dependent manner. At concentrations ≈50 μM, CC-I–inhibited topoisomerase IIα catalyzed kDNA decatenation, but noticeably, both JAG6A and JAG32 inhibited topoisomerase IIα kDNA decatenation at 5 μM (Figure 5I). We note that JAG79 had no effect on topoisomerase IIα kDNA decatenation, consistent with the predicted inability to bind within the topoisomerase IIα cavity. These data indicated that the anti-HGG effect mediated by these agents, in particular JAG6A, is by the potent inhibition of topoisomerase IIα.
Discussion
In both adult and pediatric patients, HGG treatment efficacy is extremely limited due to widespread resistance to conventional chemotherapeutics. Novel agents that are more potent than those currently available (including TMZ and etoposide) are urgently needed, in particular, agents that are less toxic to non-neoplastic cells.
Here we have compared a number of novel anticancer agents to various conventional frontline chemotherapeutics. We conducted this evaluation using a range of classic and new patient-derived aHGG cell lines. Furthermore, we included two pediatric HGG in vitro models. We examined the anticancer effectiveness of each compound including the level (and activation) of HIF-1α and p53. Using molecular modeling and enzymatic assays, we identified that topoisomerase IIα was the cellular target of our novel agents and that the lead JAG compound (JAG-6A) was a potent inhibitor of this enzyme. Consistent with this inhibition, JAG-6A directed the strongest anti-HGG response, and this response was not diminished when cells were cultured and then treated under hypoxic (1% O2) conditions. Consistent with this efficacy under hypoxic conditions, when adult or pediatric HGG lines were cultured in 3D spheroids, JAG-6A mediated a potent anti-HGG response. Interestingly, JAG-6A had a significantly lower EC50 compared to TMZ when UP-029 or KNS42 3D spheroids were treated. JAG-6A exposure triggered a significant induction of the p53-dependent target genes PUMA and Bax with a concomitant increase in both proteins up to 24 hours. We did not observe an increase of p21 transcription, consistent with the induction of apoptosis we report and the known prosurvival role of p21 [43] [44], [45].
Within our small adult cohort, all patients underwent the Stupp protocol post biopsy surgery. Patients with biopsy lines that responded poorly (or not at all) to JAG-6A (as well as TMZ) had a significantly worse clinical outcome. UP-029 and SEBTA-025 showed a potent response to JAG-6A (and TMZ). SEBTA-003 (with unmethylated MGMT) was resistant to TMZ, although it was sensitive to JAG-6A, highlighting a divergent mechanism of action (a topoisomerase Iiα inhibitor compared to a DNA alkylating agent). SEBTA-023 was highly resistant to conventional treatments and JAG-6A, likely in part to the p53 status of this model, a finding consistent with reports demonstrating that topoisomerase IIα inhibitors direct p53-dependent cell death [46], [47], [48]. There are a number of processes that might modulate cancer susceptibility to etoposide, including the level of topoisomerase II expression (at the level of both transcription and translation), the DNA binding of topoisomerase II, its activity following DNA binding, and topoisomerase II posttranslational modifications. Mutations in the Kelch-like ECH-associated protein 1 (KEAP1), the SWI/SNF complex (a nucleosome remodeling complex), and the methyltransferase EZH2 influence topoisomerase II at a transcriptional level, modifying susceptibility to topoisomerase poisons [49], [50], [51], [52]. An extensive evaluation of cell line sensitivity to etoposide highlighted that for many classical high-grade glioma lines, their response were comparable to that of small cell lung cancer and myeloma, both cancers that have traditionally been treated with etoposide [35]. At present, there are a number of investigations of topoisomerase poisons in combination with novel delivery mechanisms. This includes pediatric brain tumors, where laser interstitial thermal therapy is applied to disrupt the blood–brain barrier and enhance the delivery of this (and other) chemotherapeutic agents (ClinicalTrials.govNCT02372409). Another glioma clinical trial is investigating etoposide in combination with sodium thiosulfate in order to determine if the addition of sodium thiosulfate can protect against thrombocytopenia (low blood platelet count) noted post etoposide administration (ClinicalTrials.gov-NCT00075387) [49].
A personalized medicine approach to chemotherapy for brain tumors can potentially enhance the efficacy of many treatments and minimize unnecessary exposure to toxic agents that will not benefit the patient. A critical caveat for the use of topoisomerase II inhibitors (such as etoposide) relates to delivery effectiveness. Novel delivery techniques may allow therapeutic intratumoral concentrations of topoisomerase II inhibitors to be reached and minimize the systemic toxicity commonly associated with these agents that so far has limited their effectiveness. Our data presented here suggest that JAG-6A (a significantly smaller molecular weight compound than etoposide) potently inhibits topoisomerase II and remains effective independent of oxygenation in both 2D and 3D models. Together, these data warrant further investigation of JAG-6A for the treatment of high-grade gliomas.
Acknowledgments
Acknowledgements
We thank all patients and staff in procuring samples and Kings College Hospital NHS Foundation Trust. Dr. Val Millar (Target Discovery Institute, University of Oxford) provided assistance with high content image analysis.
Funding
Research is core funded by Brain Tumour Research. A. H. is supported by the Ollie Young Foundation. P. A. M. is funded by FCT Investigator contract from the Foundation for Science and Technology (FCT), Portugal (ref: IF/00614/2014) and FCT exploratory grant, ref.: IF/00614/2014/CP12340006. C. B. M. R. is financed by FCT Research Center Grant ref.: UID/BIM/04773/2013CBMR1334.
Author Contributions
A. H., C. S., N. K., K. M., S. C., P. M., P. C., and R. H. performed the experiments; G. S. and A. K. obtained and provided clinical material and data; R. H. designed the study; S. Y. L. and A. M. conducted the DIVERset library screen; A. H., N. K., G. P., A. M., P. C., and R. H. interpreted the data. R. H. wrote the manuscript, and all authors checked and revised the final paper.
Competing Interests
Opal Oncology Ltd. (A. Morley) has filed a patent relating to JAG agents described in this work to the Intellectual Property Office (United Kingdom). All other authors declare no potential conflict of interest.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.tranon.2019.07.007.
Appendix A. Supplementary data
The following are the Supplementary data to this article:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
References
- 1.Dolecek TA, Propp JM, Stroup NE, Kruchko C. CBTRUS statistical report: primary brain and central nervous system tumors diagnosed in the United States in 2005-2009. Neuro Oncol. 2012;14(Suppl 5):v1–v49. doi: 10.1093/neuonc/nos218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ostrom QT, Gittleman H, Fulop J, Liu M, Blanda R, Kromer C, Wolinsky Y, Kruchko C, Barnholtz-Sloan JS. CBTRUS statistical report: primary brain and central nervous system tumors diagnosed in the United States in 2008-2012. Neuro Oncol. 2015;17(Suppl 4):iv1–iv62. doi: 10.1093/neuonc/nov189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ajaz M, Jefferies S, Brazil L, Watts C, Chalmers A. Current and investigational drug strategies for glioblastoma. Clin Oncol. 2014;26:419–430. doi: 10.1016/j.clon.2014.03.012. [DOI] [PubMed] [Google Scholar]
- 4.Wilson T, Karajannis M, Harter D. Glioblastoma multiforme: state of the art and future therapeutics. Surg Neurol Int. 2014;5:64. doi: 10.4103/2152-7806.132138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pace A, Dirven L, Koekkoek JAF, Golla H, Fleming J, Rudà R, Marosi C, Le Rhun E, Grant R, Oliver K. European Association of Neuro-Oncology palliative care task force, European Association for Neuro-Oncology (EANO) guidelines for palliative care in adults with glioma. Lancet Oncol. 2017;18:e330–e340. doi: 10.1016/S1470-2045(17)30345-5. [DOI] [PubMed] [Google Scholar]
- 6.Yung WK, Albright RE, Olson J, Fredericks R, Fink K, Prados MD, Brada M, Spence A, Hohl RJ, Shapiro W. A phase II study of temozolomide vs. procarbazine in patients with glioblastoma multiforme at first relapse. Br J Cancer. 2000;83:588–593. doi: 10.1054/bjoc.2000.1316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Stupp R, Gander M, Leyvraz S, Newlands E. Current and future developments in the use of temozolomide for the treatment of brain tumours. Lancet Oncol. 2001;2:552–560. doi: 10.1016/S1470-2045(01)00489-2. [DOI] [PubMed] [Google Scholar]
- 8.Stupp R, Hegi ME, Mason WP, van den Bent MJ, Taphoorn MJ, Janzer RC, Ludwin SK, Allgeier A, Fisher B, Belanger K. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 2009;10:459–466. doi: 10.1016/S1470-2045(09)70025-7. [DOI] [PubMed] [Google Scholar]
- 9.Felsberg J, Thon N, Eigenbrod S, Hentschel B, Sabel MC, Westphal M, Schackert G, Kreth FW, Pietsch T, Löffler M. Promoter methylation and expression of MGMT and the DNA mismatch repair genes MLH1, MSH2, MSH6 and PMS2 in paired primary and recurrent glioblastomas. Int J Cancer. 2011;129:659–670. doi: 10.1002/ijc.26083. [DOI] [PubMed] [Google Scholar]
- 10.Northcott PA, Pfister SM, Jones DTW. Next-generation (epi)genetic drivers of childhood brain tumours and the outlook for targeted therapies. Lancet Oncol. 2015;16:e293–e302. doi: 10.1016/S1470-2045(14)71206-9. [DOI] [PubMed] [Google Scholar]
- 11.Louis DN, Perry A, Reifenberger G, von DA, Figarella-Branger D, Cavenee WK, Ohgaki H, Wiestler OD, Kleihues P, Ellison DW. 2016. The 2016 World Health Organization classification of tumors of the central nervous system: a summary. [DOI] [PubMed] [Google Scholar]
- 12.Mackay A, Burford A, Carvalho D, Izquierdo E, Fazal-Salom J, Taylor KR, Bjerke L, Clarke M, Vinci M, Nandhabalan M. Integrated molecular meta-analysis of 1,000 pediatric high-grade and diffuse intrinsic pontine glioma. Cancer Cell. 2017;32:520–537.e5. doi: 10.1016/j.ccell.2017.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Joseph JV, Conroy S, Pavlov K, Sontakke P, Tomar T, Eggens-Meijer E, Balasubramaniyan V, Wagemakers M, den Dunnen WFA, Kruyt FAE. Hypoxia enhances migration and invasion in glioblastoma by promoting a mesenchymal shift mediated by the HIF1α-ZEB1 axis. Cancer Lett. 2015;359:107–116. doi: 10.1016/j.canlet.2015.01.010. [DOI] [PubMed] [Google Scholar]
- 14.Bristow RG, Hill RP. Hypoxia and metabolism. Hypoxia, DNA repair and genetic instability. Nat Rev Cancer. 2008;8:180–192. doi: 10.1038/nrc2344. [DOI] [PubMed] [Google Scholar]
- 15.Uribe D, Torres Á, Rocha JD, Niechi I, Oyarzún C, Sobrevia L, San Martín R, Quezada C. Multidrug resistance in glioblastoma stem-like cells: Role of the hypoxic microenvironment and adenosine signaling. Mol Aspects Med. 2017;55:140–151. doi: 10.1016/j.mam.2017.01.009. [DOI] [PubMed] [Google Scholar]
- 16.Merighi S, Benini A, Mirandola P, Gessi S, Varani K, Leung E, Maclennan S, Baraldi PG, Borea PA. Hypoxia inhibits paclitaxel-induced apoptosis through adenosine-mediated phosphorylation of bad in glioblastoma cells. Mol Pharmacol. 2007;72:162–172. doi: 10.1124/mol.106.031849. [DOI] [PubMed] [Google Scholar]
- 17.Hsieh C-H, Lin Y-J, Wu C-P, Lee H-T, Shyu W-C, Wang C-C. Livin contributes to tumor hypoxia-induced resistance to cytotoxic therapies in glioblastoma multiforme. Clin Cancer Res. 2015;21:460–470. doi: 10.1158/1078-0432.CCR-14-0618. [DOI] [PubMed] [Google Scholar]
- 18.Pollack IF, Finkelstein SD, Woods J, Burnham J, Holmes EJ, Hamilton RL, Yates AJ, Boyett JM, Finlay JL, Sposto R. Expression of p53 and prognosis in children with malignant gliomas. N Engl J Med. 2002;346:420–427. doi: 10.1056/NEJMoa012224. [DOI] [PubMed] [Google Scholar]
- 19.Pollack IF, Hamilton RL, Sobol RW, Nikiforova MN, Lyons-Weiler MA, LaFramboise WA, Burger PC, Brat DJ, Rosenblum MK, Holmes EJ. IDH1 mutations are common in malignant gliomas arising in adolescents: a report from the Children’s Oncology Group. Childs Nerv Syst. 2011;27:87–94. doi: 10.1007/s00381-010-1264-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pollack IF, Finkelstein SD, Burnham J, Holmes EJ, Hamilton RL, Yates AJ, Finlay JL, Sposto R, Children’s Cancer Group Age and TP53 mutation frequency in childhood malignant gliomas: results in a multi-institutional cohort. Cancer Res. 2001;61:7404–7407. http://www.ncbi.nlm.nih.gov/pubmed/11606370 [PubMed] [Google Scholar]
- 21.Sure U, Rüedi D, Tachibana O, Yonekawa Y, Ohgaki H, Kleihues P, Hegi ME. Determination of p53 mutations, EGFR overexpression, and loss of p16 expression in pediatric glioblastomas. J Neuropathol Exp Neurol. 1997;56:782–789. http://www.ncbi.nlm.nih.gov/pubmed/9210874 [PubMed] [Google Scholar]
- 22.Broniscer A, Gajjar A. Supratentorial high-grade astrocytoma and diffuse brainstem glioma: two challenges for the pediatric oncologist. Oncologist. 2004;9:197–206. doi: 10.1634/theoncologist.9-2-197. http://www.ncbi.nlm.nih.gov/pubmed/15047924 [DOI] [PubMed] [Google Scholar]
- 23.Cohen KJ, Pollack IF, Zhou T, Buxton A, Holmes EJ, Burger PC, Brat DJ, Rosenblum MK, Hamilton RL, Lavey RS. Temozolomide in the treatment of high-grade gliomas in children: a report from the Children’s Oncology Group. Neuro Oncol. 2011;13:317–323. doi: 10.1093/neuonc/noq191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cohen KJ, Heideman RL, Zhou T, Holmes EJ, Lavey RS, Bouffet E, Pollack IF. Temozolomide in the treatment of children with newly diagnosed diffuse intrinsic pontine gliomas: a report from the Children’s Oncology Group. Neuro Oncol. 2011;13:410–416. doi: 10.1093/neuonc/noq205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Korones DN, Fisher PG, Kretschmar C, Zhou T, Chen Z, Kepner J, Freeman C. Treatment of children with diffuse intrinsic brain stem glioma with radiotherapy, vincristine and oral VP-16: a Children’s Oncology Group phase II study. Pediatr Blood Cancer. 2008;50:227–230. doi: 10.1002/pbc.21154. [DOI] [PubMed] [Google Scholar]
- 26.Pollack IF, Jakacki RI, Butterfield LH, Hamilton RL, Panigrahy A, Normolle DP, Connelly AK, Dibridge S, Mason G, Whiteside TL. Antigen-specific immunoreactivity and clinical outcome following vaccination with glioma-associated antigen peptides in children with recurrent high-grade gliomas: results of a pilot study. J Neurooncol. 2016;130:517–527. doi: 10.1007/s11060-016-2245-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Prados MD, Scott C, Curran WJ, Nelson DF, Leibel S, Kramer S. Procarbazine, lomustine, and vincristine (PCV) chemotherapy for anaplastic astrocytoma: a retrospective review of radiation therapy oncology group protocols comparing survival with carmustine or PCV adjuvant chemotherapy. J Clin Oncol. 1999;17:3389–3395. doi: 10.1200/JCO.1999.17.11.3389. [DOI] [PubMed] [Google Scholar]
- 28.Broniscer A, Chintagumpala M, Fouladi M, Krasin MJ, Kocak M, Bowers DC, Iacono LC, Merchant TE, Stewart CF, Houghton PJ. Temozolomide after radiotherapy for newly diagnosed high-grade glioma and unfavorable low-grade glioma in children. J Neurooncol. 2006;76:313–319. doi: 10.1007/s11060-005-7409-5. [DOI] [PubMed] [Google Scholar]
- 29.Prados MD, Seiferheld W, Sandler HM, Buckner JC, Phillips T, Schultz C, Urtasun R, Davis R, Gutin P, Cascino TL. Phase III randomized study of radiotherapy plus procarbazine, lomustine, and vincristine with or without BUdR for treatment of anaplastic astrocytoma: final report of RTOG 9404. Int J Radiat Oncol. 2004;58:1147–1152. doi: 10.1016/j.ijrobp.2003.08.024. [DOI] [PubMed] [Google Scholar]
- 30.Shaw EG, Wang M, Coons SW, Brachman DG, Buckner JC, Stelzer KJ, Barger GR, Brown PD, Gilbert MR, Mehta MP. Randomized trial of radiation therapy plus procarbazine, lomustine, and vincristine chemotherapy for supratentorial adult low-grade glioma: initial results of RTOG 9802. J Clin Oncol. 2012;30:3065–3070. doi: 10.1200/JCO.2011.35.8598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.van den Bent MJ, Brandes AA, Taphoorn MJB, Kros JM, Kouwenhoven MCM, Delattre J-Y, Bernsen HJJA, Frenay M, Tijssen CC, Grisold W. Adjuvant procarbazine, lomustine, and vincristine chemotherapy in newly diagnosed anaplastic oligodendroglioma: long-term follow-up of EORTC brain tumor group study 26951. J Clin Oncol. 2013;31:344–350. doi: 10.1200/JCO.2012.43.2229. [DOI] [PubMed] [Google Scholar]
- 32.Froelich-Ammon SJ, Osheroff N. Topoisomerase poisons: harnessing the dark side of enzyme mechanism. J Biol Chem. 1995;270:21429–21432. doi: 10.1074/jbc.270.37.21429. [DOI] [PubMed] [Google Scholar]
- 33.Mehta A, Awah CU, Sonabend AM. Topoisomerase II poisons for glioblastoma; existing challenges and opportunities to personalize therapy. Front Neurol. 2018;9:459. doi: 10.3389/fneur.2018.00459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Feun L, Savaraj N. Topoisomerase I inhibitors for the treatment of brain tumors. Expert Rev Anticancer Ther. 2008;8:707–716. doi: 10.1586/14737140.8.5.707. [DOI] [PubMed] [Google Scholar]
- 35.Sonabend AM, Carminucci AS, Amendolara B, Bansal M, Leung R, Lei L, Realubit R, Li H, Karan C, Yun J. Convection-enhanced delivery of etoposide is effective against murine proneural glioblastoma. Neuro Oncol. 2014;16:1210–1219. doi: 10.1093/neuonc/nou026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lee SY, Slagle-Webb B, Rizk E, Patel A, Miller PA, Sung S-S, Connor JR. Characterization of a novel anti-cancer compound for astrocytomas. PLoS One. 2014;9 doi: 10.1371/journal.pone.0108166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Montenegro RC, Clark PGK, Howarth A, Wan X, Ceroni A, Siejka P, Nunez-Alonso GA, Monteiro O, Rogers C, Gamble V. BET inhibition as a new strategy for the treatment of gastric cancer. Oncotarget. 2016;7:43997–44012. doi: 10.18632/oncotarget.9766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sirenko O, Mitlo T, Hesley J, Luke S, Owens W, Cromwell EF. High-content assays for characterizing the viability and morphology of 3D cancer spheroid cultures. Assay Drug Dev Technol. 2015;13:402–414. doi: 10.1089/adt.2015.655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(−Delta Delta C(T)) Method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 40.An Q, Fillmore HL, Vouri M, Pilkington GJ. Brain tumor cell line authentication, an efficient alternative to capillary electrophoresis by using a microfluidics-based system. Neuro Oncol. 2014;16:265–273. doi: 10.1093/neuonc/not202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dolgin E. Venerable brain-cancer cell line faces identity crisis. Nature. 2016;537:149–150. doi: 10.1038/nature.2016.20515. [DOI] [PubMed] [Google Scholar]
- 42.Allen M, Bjerke M, Edlund H, Nelander S, Westermark B. Origin of the U87MG glioma cell line: Good news and bad news. Sci Transl Med. 2016;8 doi: 10.1126/scitranslmed.aaf6853. [DOI] [PubMed] [Google Scholar]
- 43.Hill R, Madureira PA, Waisman DM, Lee PWK. DNA-PKCS binding to p53 on the p21WAF1/CIP1 promoter blocks transcription resulting in cell death. Oncotarget. 2011;2:1094–1108. doi: 10.18632/oncotarget.378. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 44.Waldman T, Zhang Y, Dillehay L, Yu J, Kinzler K, Vogelstein B, Williams J. Cell-cycle arrest versus cell death in cancer therapy. Nat Med. 1997;3:1034–1036. doi: 10.1038/nm0997-1034. http://www.ncbi.nlm.nih.gov/pubmed/9288734 [DOI] [PubMed] [Google Scholar]
- 45.Kreis N-N, Louwen F, Zimmer B, Yuan J. Loss of p21<sup>Cip1/CDKN1A</sup> renders cancer cells susceptible to Polo-like kinase 1 inhibition. Oncotarget. 2015;6:6611–6626. doi: 10.18632/oncotarget.2844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wu S-Y, Pan S-L, Xiao Z-Y, Hsu J-L, Chen M-C, Lee K-H, Teng C-M. NPRL-Z-1, as a new topoisomerase II poison, induces cell apoptosis and ROS generation in human renal carcinoma cells. PLoS One. 2014;9 doi: 10.1371/journal.pone.0112220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gewirtz DA, Sundaram S, Magnet KJ. Influence of topoisomerase II inhibitors and ionizing radiation on growth arrest and cell death pathways in the breast tumor cell. Cell Biochem Biophys. 2000;33:19–31. doi: 10.1385/CBB:33:1:19. [DOI] [PubMed] [Google Scholar]
- 48.Demoulin B, Hermant M, Castrogiovanni C, Staudt C, Dumont P. Resveratrol induces DNA damage in colon cancer cells by poisoning topoisomerase II and activates the ATM kinase to trigger p53-dependent apoptosis. Toxicol Vitro. 2015;29:1156–1165. doi: 10.1016/j.tiv.2015.04.015. [DOI] [PubMed] [Google Scholar]
- 49.Mehta A, Awah CU, Sonabend AM. Topoisomerase II poisons for glioblastoma; existing challenges and opportunities to personalize therapy. Front Neurol. 2018;9 doi: 10.3389/fneur.2018.00459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wijdeven RH, Pang B, van der Zanden SY, Qiao X, Blomen V, Hoogstraat M, Lips EH, Janssen L, Wessels L, Brummelkamp TR. Genome-wide identification and characterization of novel factors conferring resistance to topoisomerase II poisons in cancer. Cancer Res. 2015;75:4176–4187. doi: 10.1158/0008-5472.CAN-15-0380. [DOI] [PubMed] [Google Scholar]
- 51.Fillmore CM, Xu C, Desai PT, Berry JM, Rowbotham SP, Lin Y-J, Zhang H, Marquez VE, Hammerman PS, Wong K-K. EZH2 inhibition sensitizes BRG1 and EGFR mutant lung tumours to TopoII inhibitors. Nature. 2015;520:239–242. doi: 10.1038/nature14122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Dubey R, Lebensohn AM, Bahrami-Nejad Z, Marceau C, Champion M, Gevaert O, Sikic BI, Carette JE, Rohatgi R. Chromatin-remodeling complex SWI/SNF controls multidrug resistance by transcriptionally regulating the drug efflux pump ABCB1. Cancer Res. 2016;76:5810–5821. doi: 10.1158/0008-5472.CAN-16-0716. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.