

Abstract
Motivation
Glycosylation is one of the most heterogeneous and complex protein post-translational modifications. Liquid chromatography coupled mass spectrometry (LC-MS) is a common high throughput method for analyzing complex biological samples. Accurate study of glycans require high resolution mass spectrometry. Mass spectrometry data contains intricate sub-structures that encode mass and abundance, requiring several transformations before it can be used to identify biological molecules, requiring automated tools to analyze samples in a high throughput setting. Existing tools for interpreting the resulting data do not take into account related glycans when evaluating individual observations, limiting their sensitivity.
Results
We developed an algorithm for assigning glycan compositions from LC-MS data by exploring biosynthetic network relationships among glycans. Our algorithm optimizes a set of likelihood scoring functions based on glycan chemical properties but uses network Laplacian regularization and optionally prior information about expected glycan families to smooth the likelihood and thus achieve a consistent and more representative solution. Our method was able to identify as many, or more glycan compositions compared to previous approaches, and demonstrated greater sensitivity with regularization. Our network definition was tailored to N-glycans but the method may be applied to glycomics data from other glycan families like O-glycans or heparan sulfate where the relationships between compositions can be expressed as a graph.
Availability and implementation Built Executable
http://www.bumc.bu.edu/msr/glycresoft/ and Source Code: https://github.com/BostonUniversityCBMS/glycresoft.
Supplementary information
Supplementary data are available at Bioinformatics online.
1 Introduction
Glycosylation modulates the structures and functions of proteins and lipids in a broad class of biological processes (Varki, 2017). Accurate mass measurement defines monosaccharide composition given assumptions regarding glycan class and biosynthesis (Zaia, 2008). For unseparated mixtures, mass spectrometry analysis determines the mass-to-charge ratio values for only the most abundant glycans; dynamic range for detection of glycans is poor because of ion suppression (Peltoniemi et al., 2013). By contrast, online separations coupled with mass spectrometry improve dynamic range and reproducibility of glycan analysis, at the cost of increased analysis time and workflow complexity.
There are many tools for interpreting glycan mass spectral datasets (Ceroni et al., 2008; Frank and Schloissnig, 2010; Goldberg et al., 2009; Kronewitter et al., 2014; Maxwell et al., 2012; Peltoniemi et al., 2013; Yu et al., 2013) for both unseparated and separated experimental protocols. These programs address instrument-specific signal processing requirements. For example SysBioWare (Frank and Schloissnig, 2010) performs sophisticated baseline removal prior to fitting peaks, while GlyQ-IQ (Kronewitter et al., 2014) was written for cleaner Fourier Transform MS (FTMS) that does not require such a baseline removal step. Tools that build on the THRASH implementation from Decon2LS (Jaitly et al., 2009; Maxwell et al., 2012; Yu et al., 2013) are unable to deal with variable baseline noise or extreme dynamic range.
Each tool also has its own format for defining glycan structures or compositions, some even bundling a large database with their software to remove the burden from the user to build a list of candidates themselves (Goldberg et al., 2009; Kronewitter et al., 2014; Yu et al., 2013) while others define methods for building glycan databases as part of the program (Ceroni et al., 2008; Maxwell et al., 2012). Many of these tools are designed for specific glycan subclass such as N-glycans or glycosaminoglycans and/or organisms, limiting their vocabulary of possible monosaccharides to just those commonly found in that subgroup (Goldberg et al., 2009; Kronewitter et al., 2014; Peltoniemi et al., 2013; Yu et al., 2013). Often, these tools are tailored for analysis of a particular derivatization state, adduction conditions, or neutral loss pattern (Maxwell et al., 2012; Peltoniemi et al., 2013; Yu et al., 2013). Work has been done to construct a standardized namespace and representation for glycans, glySpace, including both structures and compositions (Campbell et al., 2014; Tiemeyer et al., 2017). This data is publicly accessible, including a programmatic query interface using SPARQL over HTTPS (Aoki-Kinoshita et al., 2015). Tools that can communicate with these services have the potential to lead researchers to find deeper connections from cross-referenced information, and other researchers can more readily find and use their work.
These spectral processing and glycan library properties are reflected in the scoring function that each program uses to discriminate glycan signal from the background noise and contaminants. Several methods have been developed using different facets of the observed data. Yu et al. (2013) used the isotopic pattern goodness-of-fit while Peltoniemi et al. (2013) used intensity features of associated MSnscans to evaluate partial structure and composition match quality. Kronewitter et al. (2014) combined several features of the MS1 evidence, including elution profile peak shape goodness-of-fit, isotopic fit, mass accuracy, scan count and in-source fragmentation correlation. Some of these methods are well-defined and invariant from instrument to instrument in this era of high resolution mass spectrometry, but others are tightly coupled to the experimental equipment. Missing from this list are methods to target a glycan’s intrinsic properties, such as charge state distribution or facility in acquiring adducts, which can increase the number of spurious assignments if not considered. We propose a new scoring function which is able to combine those properties which are independent of experimental setup with these glycan-aware features.
As observed by Goldberg et al. (2009), there is also value in including related glycan composition identifications in how much confidence one assigns to a given glycan composition assignment. They used a method to exploit the known biosynthetic rules of N-glycans to connect peaks in a MALDI mass spectrum assigned to a particular N-glycan by intact mass alone. Their method using the maximum weighted subgraph of the biosynthetic network had demonstrably better performance than chance with their expert system annotation method. Kronewitter et al. (2014) considered a similar idea with more emphasis on handling in-source fragmentation observed in LC-MS and LC-MS/MS experiments.
We extend this notion of a glycan family to cover more sectors of the biosynthetic landscape which we term ‘neighborhoods’, and present an algorithm for learning the importance of each neighborhood from observed data, which can in turn be used to improve glycan composition assignment performance. We also apply our method using three different glycan composition search spaces to show how the underlying database can influence results. We present our method on typical N-glycans in humans, though our method can be applied to any variety of glycan composition whose monosaccharides can be described using IUPAC trivial names or whose components can be described in terms of chemical formulae.
This method is implemented as part of, GlycReSoft, a collection of open source tools for interpretation of glycans and glycopeptides from LC-MS or LC-MS/MS data. It includes programs to construct glycan databases from either a text file enumerating all compositions, combinatorial constraints describing the space of glycan compositions, and by querying GlyTouCan. These glycans can be combined with arbitrary reduction and derivatization modifications. GlycReSoft can also combine these glycan databases with peptides to produce glycopeptide databases. GlycReSoft also contains a deconvolution program to convert raw mass spectra stored in mzML or mzXML from an LC-MS or LC-MS/MS experiment files into a mzML file containing monoisotopic peak and charge states for each observed isotopic pattern. The deconvolution algorithm is able to handle both the noise commonly found in TOF spectra as well as the isotopic pattern truncation characteristic of Orbitrap spectra. Lastly it includes a search engine for identifying these fitted neutral masses as either glycans or glycopeptides from a database with combinatorial composition shifts to detect adducts, neutral losses, or other chemical modifications not already part of the database. We further incorporate adduction into the scoring process, treating it as another feature of the data as it is unavoidable, rather than treating it purely as a confounding factor. These features make the program more flexible and robust than the previously cited works (Kronewitter et al., 2014; Peltoniemi et al., 2013; Yu et al., 2013) and we demonstrate these abilities by interpreting native, permethylated, reduced and deutero-reduced samples from both QTOF and Orbitrap instruments. GlycReSoft is composed of a set of command line tools and provides a GUI that composes them. The GUI is powered by a web server which can be deployed on a network to allow multiple users to access its features and can leverage multiple CPUs. The tools are all written in Python and C, licensed under the Apache2 Common License. For more details, please see the documentation at http://www.bumc.bu.edu/msr/glycresoft/
2 Materials and methods
2.1 Glycan hypothesis generation
In eukaryotes, a 14 monosaccharide N-glycan of composition HexNAc2 Hex12 is transferred to a newly synthesized protein in the endoplasmic reticulum by the oligosaccharyl transferase protein complex. This glycan is trimmed to HexNAc2 Hex9 during protein folding and quality control. As the glycoprotein transits the Golgi apparatus, N-glycans are trimmed to HexNAc2 Hex5 before being elaborated into hybrid and complex N-glycan classes (Stanley et al., 2009). Glycan structures are refined by a series of reactions that yield over a million possible N-glycan topologies, as shown in Akune et al. (2016). These topologies define the glycan’s geometry and protein binding properties. Neither MS1 nor collisional tandem MS of glycans can capture the full tree or graph structure of an N-glycan, so we reduced the topology to a count of each type of residue, a composition.
Starting with the core motif HexNAc2 Hex3, we generated all combinations of monosaccharides ranging between the limits in Table 1 to build a human N-glycan composition database, which produced 1240 distinct compositions. These rules are able to efficiently generate all glycan compositions from canonical branching patterns and lactosamine extensions, as well as rarer constructs such as LacdiNAc Goldberg et al. (2009) at the cost of including some wholly improbable compositions. To perform a side-by-side comparison we also extracted the glycan list from Yu et al. (2013) derived from the biosynthetic rules in Krambeck and Betenbaugh (2005) with 319 compositions, and another database using all curated N-glycans from glySpace via GlyTouCan (Tiemeyer et al., 2017) containing only [Hex, HexNAc, Fuc, Neu5Ac, sulfate], with 275 distinct compositions. As previous analysis of Influenza A virus samples detected sulfated N-glycans (Khatri et al., 2016), we also created a combinatorial database with up to one sulfate included, for a total of 2480 compositions. As our algorithm treats HexNAc and HexNAc(S) as distinct entities, for all monosaccharides with post-attachment substituents such as sulfate and phosphate, we detached the substituent from the core monosaccharide. Our implementation is able to interpret IUPAC trivial names and compositions thereof with standard substituent and unambiguous backbone modifications, permitting a wide range of possible glycan compositions.
Table 1.
Human N-glycan composition bounds (Stanley et al., 2009)
| Monosaccharide | Lower limit | Upper limit | Constraints |
|---|---|---|---|
| HexNAc | 2 | 9 | |
| Hex | 3 | 10 | |
| Fuc | 0 | 4 | |
| NeuAc | 0 | 5 |
2.2 LC-MS data preprocessing
We analyzed samples from several sources, including both Quadrupole Time-of-Flight (QTOF) and Orbitrap instruments as shown in Supplementary Table S1. For details on sample preparation and data acquisition, please see the source citations in the referenced table. We converted all datasets to mzML format (Martens et al., 2011) using Proteowizard (Kessner et al., 2008) without any data transforming filters. We applied a background reduction method based upon (Kaur and O'Connor, 2006), using a window length of 2 m/z. Next, we picked peaks using a Gaussian model and iteratively charge state deconvoluted and deisotoped using an averagine (Senko et al., 1995) formula appropriate to the molecule under study. For native glycans, the formula was H 1.690 C 1.0 O 0.738 N 0.071, for permethylated glycans, the formula was H 1.819 C 1.0 O 0.431 N 0.042. We used an iterative approach which combines aspects of the dependence graph method (Liu et al., 2010) and with subtraction. All samples were processed using a minimum isotopic fit score of 20 with an isotopic strictness penalty of 2.
2.3 Chromatogram aggregation
We clustered peaks whose neutral masses were within parts-per-million error (PPM) of each other. When there were multiple candidate clusters for a single peak, we used the cluster with the lowest mass error. Next, we sorted each cluster by time, creating a list of aggregated chromatograms. To account for small mass differences, we found all chromatograms which were within PPM of each other and which overlap in time and merge them. These mass tolerances were selected empirically, and can be adjusted as needed by the user.
2.4 Glycan composition matching
For each chromatogram, we searched each glycan database for compositions whose masses were within PPM for QTOF data, 5 PPM for FTMS data. These values are commonly used for data from these instruments based upon information from their manufacturers. We merged all chromatograms matching the same composition. Then, for each mass shift combination expected for each sample, we searched each glycan database for compositions whose neutral mass were within δmass of the observed neutral mass—mass shift combination mass, followed by another round of merging chromatograms with the same assigned composition. We reduced the data by splitting each feature where the time between sequential observation was greater than min and removed chromatograms with fewer than k = 5 data points. The same chromatogram may be given multiple assignments and designated multiple mass shifts, and chromatograms without glycan assignments may use chromatograms with glycan assignments as mass shifted components. This ambiguity information was propagated through each merge and split step. We termed these remaining assigned and unassigned chromatograms candidate features.
2.5 Feature evaluation
We computed several metrics to estimate how distinguishable each candidate feature was from random noise. The metrics are mentioned in List 1, but for more information see Supplementary Section S3.
List 1: Chromatographic feature metrics
Goodness-of-fit of chromatographic peak shape to a model function (Kronewitter et al., 2014; Yu and Peng, 2010).
Goodness-of-fit of isotopic pattern to glycan composition weighted by peak abundance (Maxwell et al., 2012).
Observed charge states with respect to glycan composition and mass.
Time gap between MS1 observations detecting missing peaks and interference.
Adduction states with respect to glycan composition and mass.
These metrics are bounded in . Any observation for which any metric was observed below a feature specific threshold was discarded as having insufficient evidence for consideration. The observed score s for each candidate feature is the sum of the logit-transformation of these metrics. This produces a single value bounded in , whose distribution we assume is asymptotically normal. A value of s < 8 reflects a low confidence match, with confidence increasing as s does. As these metrics are tied to reliable detection of the glycan by the mass spectrometer, they depend upon glycan abundance, sample quality and mass spectrometer resolution.
2.6 Glycan composition network smoothing
Ideally, each glycan present in a sample under analysis would produce sufficient experimental evidence that they can be identified. In practice, glycan compositions with lower abundances may not present strong evidence, leading to those glycan compositions being discarded. Others have demonstrated that it is advantageous to use relationships between glycans based on biosynthetic or structural rules to adjust the score of a single glycan assignment (Goldberg et al., 2009; Kronewitter et al., 2014). To improve performance, we propose a method based on Laplacian regularized least squares (Belkin et al., 2006) to use evidence from glycan compositions related over a network to smooth its evaluation of glycan composition feature matching.
Previous approaches to using information regarding identification of one glycan composition to increase the confidence in another have been proposed by Goldberg et al. (2009) and Kronewitter et al. (2014) using different techniques. Goldberg et al. used random walks along the biosynthetic network between identified glycan compositions to increase the confidence of those connected compositions. This method works well but requires that the parameters of the random walk be properly tuned for the biosynthetic network being used. Laplacian regularized least squares is more robust to small changes to the network and is able to use the entire network. Kronwitter et al. included a term in their criterion for detection requiring the presence of another glycan composition with one more or one less monosaccharide to permit identification. This puts substantial weight on a Boolean term, giving it the ability to overrule other experimental evidence. Similar methods could be devised using methods like ant colony optimization to traverse the biosynthetic graph, or a database-specific belief network, but these methods would require considerable manual tuning for each new database to be tested.
2.6.1 Glycan composition graph
For each database of theoretical glycan compositions we create, we define each composition to be a coordinate vector in a space where c is the number of components in any glycan composition, and represented by a node in an undirected glycan composition graph . Under this interpretation, we can compute the L1-distance between two glycan compositions, representing the biosynthetic distance between the two compositions, an analog for the number of enzymatic steps needed to go from one glycan to the other. For any two glycan compositions gu, gv, if we add an edge connecting gu and gv to with weight w = 1.
2.6.2 Neighborhood definition
Our definition of distance connects glycan compositions which differ by a single monosaccharide, but we can assert how larger collections of glycan compositions are related. To this end, we extend the definition of neighborhoods for N-glycans using intervals over monosaccharide counts shown in Table 2. These neighborhoods are arranged to span particular epitopes or biosynthetically related subtypes of N-glycans, such as sialylation state or branching pattern. Neighborhoods overlap sets of glycan compositions which are also biosynthetically related. Each neighborhood spans the eponymous class of glycan compositions, as well as the preceding class and proceeding class. For example, the Tri-Antennary neighborhood spans Bi-Antennary and Tetra-Antennary compositions. This helps to channel the estimation of among related groups. The Hybrid, Bi-Antennary and Asialo-Bi-Antennary neighborhoods introduce complications because they are biosynthetically close to each other. For the simplicity, we chose to include all of Hybrid in Asialo-Bi-Antennary and permit up to one NeuAc in its members.
Table 2.
N-glycan neighborhood definitions
| Name | HexNAC |
Hex |
NeuAc |
Size | |||
|---|---|---|---|---|---|---|---|
| Min | Max | Min | Max | Min | Max | ||
| High mannose | 2 | 2 | 3 | 10 | 0 | 0 | 16 |
| Hybrid | 2 | 4 | 2 | 6 | 0 | 2 | 80 |
| Bi-antennary | 3 | 5 | 3 | 6 | 1 | 3 | 104 |
| Asialo-bi-antennary | 3 | 5 | 3 | 6 | 0 | 1 | 96 |
| Tri-antennary | 4 | 6 | 4 | 7 | 1 | 4 | 172 |
| Asialo-tri-antennary | 4 | 6 | 4 | 7 | 0 | 0 | 56 |
| Tetra-antennary | 5 | 7 | 5 | 8 | 1 | 5 | 240 |
| Asialo-tetra-antennary | 5 | 7 | 5 | 8 | 0 | 0 | 60 |
| Penta-antennary | 6 | 8 | 6 | 9 | 1 | 5 | 280 |
| Asialo-penta-antennary | 6 | 8 | 6 | 9 | 0 | 0 | 60 |
| Hexa-antennary | 7 | 9 | 7 | 10 | 1 | 6 | 300 |
| Asialo-hexa-antennary | 7 | 9 | 7 | 10 | 0 | 0 | 60 |
| Hepta-antennary | 8 | 10 | 8 | 11 | 1 | 7 | 150 |
| Asialo-hepta-antennary | 8 | 10 | 8 | 11 | 0 | 0 | 30 |
Note: These define the ranges of monosaccharides which will be used to classify a glycan composition as being a member of each neighborhood, and the number of combinatorial N-glycan compositions in each neighborhood.
Glycan compositions may belong to zero or more neighborhoods, as there are unusual glycan compositions which do not satisfy any neighborhood’s rules, and several neighborhoods intentionally overlap to express broad relationships between groups.
We define a matrix A as an n × k matrix where is the degree to which gi belongs kth neighborhood:
| (1) |
To reduce the impact of neighborhood size on the elements of A, the columns of A are first normalized to sum to 1, and then the rows of A are normalized to sum to 1. We assume that members of the same neighborhood will share a central tendency .
2.6.3 Laplacian regularization
To accomplish our goal, we can use Laplacian regularized least squares to find a new score , based upon s and relationships among the observed glycans described by our biosynthetic graph . These relationships can be directed to move towards some central tendency τ using the Laplacian of and some definitions of broad groups in .
We combine the observed score s and the structure of to estimate a smoothed score that combines the evidence for each individual glycan composition as well as its relatives. As s is the size of the set of observed glycan composition p while is of size n, we partition into a block vector with dimensions .
Let L be the weighted Laplacian matrix of , which is an n × n matrix. To ensure L is invertible, we add to L. We partition L into blocks . We also partition A into and , .
We find the that minimizes the expression
| (2) |
| (3) |
where λ controls how much weight is placed on the network structure and τ.
To obtain the optimal , we take the partial derivative of w.r.t :
| (4) |
| (5) |
and w.r.t.
| (6) |
| (7) |
To use this method, we must provide values for λ and . While these values could be chosen based on the expectations of the user for a given experiment, we provide an algorithm for selecting their values in Supplementary Section S5. These methods use the topology of the glycan composition graph and the distribution of observed scores, and cannot fully capture boundary cases or related but disconnected parts of the graph.
3 Results
We demonstrated the performance of our algorithm using released influenza hemagglutinin dataset 20141103–02-Phil-BS and a serum glycan dataset Perm-BS-070111-04-Serum. Please refer to Supplementary Section S7 for all other datasets. For each comparison, the unregularized case is not smoothed, effectively λ = 0, the partially regularized case uses the grid search fitted values of but uses a fixed , and the fully regularized case uses the grid search fitted values of both and λ.
3.1 Chromatogram assignment performance for 20141103-02-Phil-BS
The fitted parameters for the network constructed for 20141103-02-Phil-BS are shown in Table 3. The assigned chromatograms are shown in Figure 1. We observe up to seven branch structures in this sample, consistent with these N-glycans being derived from an avian context (Khatri et al., 2016; Stanley et al., 2009).
Table 3.
Estimated values of smoothing parameters τ, λ and γ for each dataset and database
| Neighborhood τ |
Phil-BS |
Serum |
||||
|---|---|---|---|---|---|---|
| Combinatorial + Sulfate | glySpace | Krambeck | Combinatorial | glySpace | Krambeck | |
| High-mannose | 18.008 | 15.061 | 17.089 | 20.328 | 19.392 | 19.720 |
| Hybrid | 13.440 | 12.435 | 12.503 | 20.997 | 18.610 | 20.056 |
| Bi-antennary | 0.000 | 0.000 | 0.000 | 15.901 | 16.826 | 17.593 |
| Asialo-bi-antennary | 14.078 | 10.916 | 13.591 | 22.585 | 21.563 | 21.827 |
| Tri-antennary | 0.000 | 0.000 | 0.000 | 26.420 | 19.605 | 23.644 |
| Asialo-tri-antennary | 14.538 | 6.565 | 11.952 | 20.025 | 21.128 | 19.764 |
| Tetra-antennary | 0.000 | 0.000 | 0.000 | 19.508 | 18.542 | 17.674 |
| Asialo-tetra-antennary | 14.331 | 4.842 | 12.373 | 2.472 | 7.180 | 2.568 |
| Penta-antennary | 0.000 | 0.000 | 0.000 | 11.878 | 15.035 | 11.682 |
| Asialo-penta-antennary | 11.588 | 1.255 | 9.784 | 0.000 | 0.000 | 0.000 |
| Hexa-antennary | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 |
| Asialo-hexa-antennary | 11.094 | 3.883 | 13.223 | 0.000 | 0.000 | 0.000 |
| Hepta-antennary | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 |
| Asialo-hepta-antennary | 3.117 | 1.529 | 2.703 | 0.000 | 0.000 | 0.000 |
| 0.99 | 0.69 | 0.99 | 0.99 | 0.99 | 0.99 | |
| 11.39 | 14.60 | 10.42 | 20.57 | 18.42 | 20.72 | |
Fig. 1.
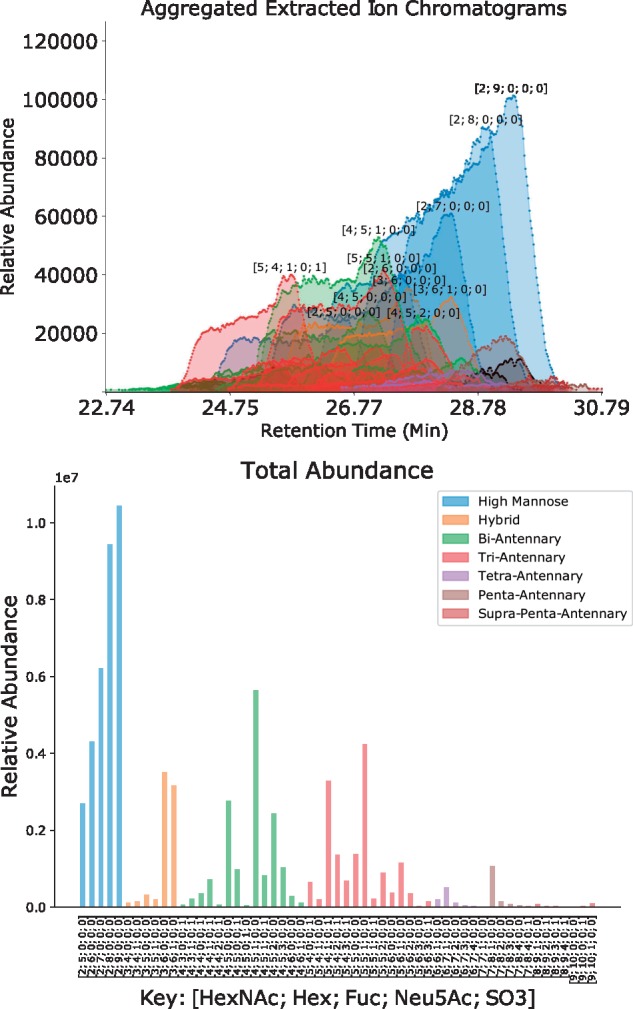
Chromatogram Assignments and Quantification for 20141103-02-Phil-BS Using the Combinatorial + Sulfate database. The Retention Time (Min) axis shows the experimental retention time in minutes, and the Relative Abundance axis shows the intensity of the signal from each aggregated ion species. The identified glycan compositions are labeled with a tuple describing the number of each component of the form [HexNAc, Hex, Fuc, NeuAc, SO3] (Color version of this figure is available at Bioinformatics online.)
The comparison of assignment performance with differing degrees of smoothing for each database are shown in Figure 2 and Table 4. We used the Receiver Operator Characteristic (ROC) Area Under the Curve (AUC) to measure performance, using manually validated compositions as ground truth. We observed the greatest number of assignments using the Combinatorial + Sulfate database, and the greatest ROC AUC in the partially regularized condition.
Fig. 2.
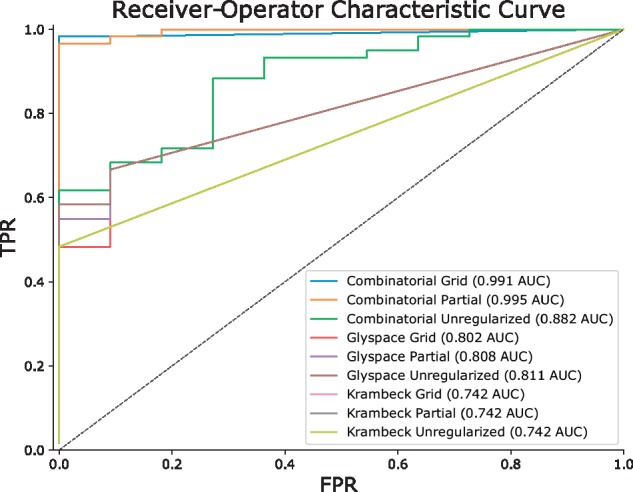
Performance Comparison with and without Network Smoothing for 20141103-02-Phil-BS. The Receiver Operator Characteristic Curve (ROC) comparing True Positive Rate (TPR) to False Positive Rate (FPR) shows how each database performed under different regularization conditions, summarized with the Area Under the Curve (AUC) in the legend. The Combinatorial + Sulfate database showed the best performance, and improved with regularization (Color version of this figure is available at Bioinformatics online.)
Table 4.
Performance comparison for 20141103-02-Phil-BS using receiver operator characteristic (ROC) area under the curve (AUC)
| Name | ROC AUC | True matchesa |
|---|---|---|
| Combinatorial unregularized | 0.882 | 56 |
| Combinatorial partial | 0.995 | 57 |
| Combinatorial grid | 0.991 | 57 |
| GlySpace unregularized | 0.811 | 40 |
| GlySpace partial | 0.808 | 38 |
| GlySpace grid | 0.802 | 31 |
| Krambeck unregularized | 0.742 | 28 |
| Krambeck partial | 0.742 | 29 |
| Krambeck grid | 0.742 | 29 |
| Khatri et al. (2016) | – | 46 |
Note: The Combinatorial Partial Regularization approach performed best.
Selected at .
3.2 Chromatogram assignment performance for Perm-BS-070111-04-Serum
The fitted parameters for the network constructed for Perm-BS-070111-04-Serum are shown in Table 3. The assigned chromatograms are shown in Figure 4.
Fig. 4.
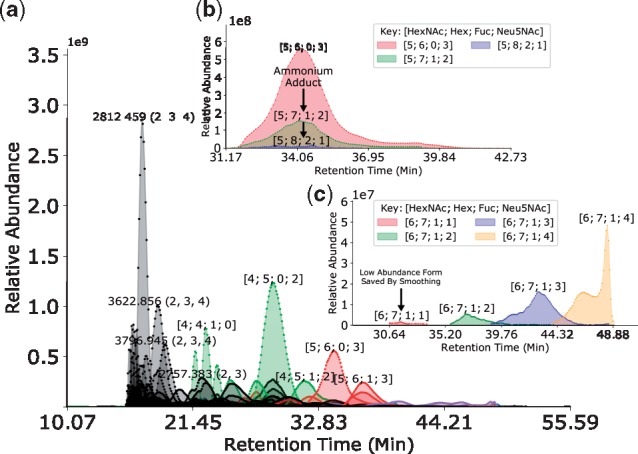
Chromatogram Assignments for Perm-BS-070111-04-Serum. In all panels, the Retention Time (Min) axis shows the experimental retention time in minutes, and the Relative Abundance axis shows the intensity of the signal from each aggregated ion species. The identified glycan compositions are labeled with a tuple describing the number of each component of the form [HexNAc, Hex, Fuc, NeuAc]. (a) Features Assigned After Grid Regularization of Perm-BS-070111-04-Serum. (b) This sample contains heavy ammonium adduction which introduces ambiguity in intact mass based assignments. (c) Low scoring features which may be discarded based on individual evidence alone may be more reasonable to accept given evidence from related composition, such as our network smoothing method (Color version of this figure is available at Bioinformatics online.)
The comparison of assignment performance with differing degrees of smoothing is shown in Figure 3. We observe the greatest number of total true identifications using the partially regularized Combinatorial database. However, the Combinatorial database also has many more false positives, with a ROC AUC of 0.816. These false positives do not appear in the biosynthetically constrained Krambeck database which maximizes its ROC AUC in the partially regularized condition at 0.883. After removing all ambiguous matches, the Krambeck database also has nearly the same number of true matches as the Combinatorial database.
Fig. 3.
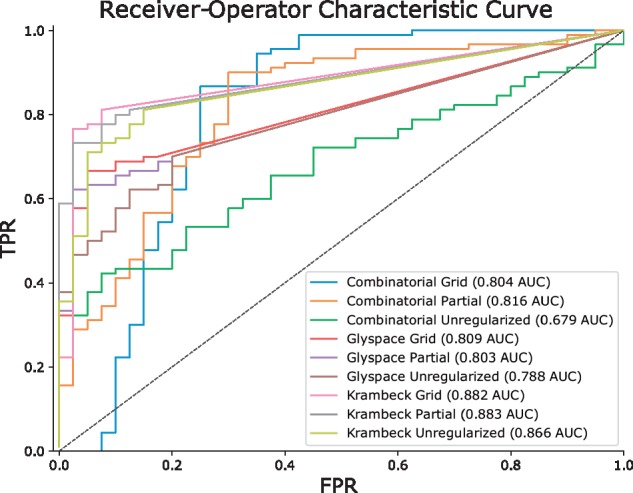
Performance Comparison with and without Network Smoothing for Perm-BS-070111-04-Serum. The Receiver Operator Characteristic Curve (ROC) comparing True Positive Rate (TPR) to False Positive Rate (FPR) shows how each database performed under different regularization conditions, summarized with the Area Under the Curve (AUC) in the legend (Color version of this figure is available at Bioinformatics online.)
4 Discussion
We demonstrated that the regularization method improved the sensitivity and specificity of glycan composition assignment for LC-MS based experiments. The method used similar assumptions about the importance of common substructural elements of N-glycans to Goldberg et al. (2009), but we extend this concept with the addition of a procedure for learning the relationship strengths and use broader groups of structures.
The experimental results from the original analysis of 20141103-02-Phil-BS and 20141031-07-Phil-82 82 demonstrated that while both strains expressed predominantly high-mannose glycosylation, 20141103-02-Phil-BS expressed more larger complex-type structures (Khatri et al., 2016). In our findings shown in Figure 1, we recapitulate these results while reducing the number of false assignments (Table 4). There are substantial differences in both the mass spectral processing and scoring schemes which contribute to these results, but the regularization procedure is responsible for recovering many low abundance features from this comparison. As these samples are derived from chicken eggs, we have observed larger branching patterns than are observed in normal mammalian tissue (Stanley et al., 2009). There is evidence for this in the 20141103-02-Phil-BS with HexNAc9 Hex10-based compositions suggesting a seven branch pattern, though this cannot be determined without high quality MSn data. The fit for Phil-BS (shown) and Phil-82 (supplement) have smaller values in the neighborhoods of their largest glycan compositions as these features tended to be low in abundance and not high scoring in their own right, but were partially supported by the overlap with the next largest neighborhood, as expected. We observed the best performance with the Combinatorial + Sulfate database, which produced more than half-again as many true matches than the other two databases. It produced several false matches as well, but the smoothing process removed these while boosting the score of other low abundance matches which were consistent with higher scoring matches.
The Krambeck database performed identically in all smoothing conditions as it was only able to match the common species, not including cases that were multiply fucosylated or sulfated. It had no false matches ranked alongside its true matches so smoothing could not change its performance. The glySpace-derived database produced more true matches, but also lacked some of these more fucosylated and complex compositions. Some of the compositions included by the glySpace-derived database were lower scoring, but the chosen value of γ for that database was greater than 18, causing the fitted values of to omit the larger, less abundant complex-type N-glycans. This caused smoothing to lower the scores of these real matches rather than raise them, as with the Combinatorial + Sulfate database.
As we show in Figure 3, regularization improves the predictive performance of the identification algorithm on Perm-BS-070111-04-Serum for all databases. We reproduce the majority of the glycan assignments from Yu et al. (2013), but the ambiguity caused by ammonium adduction as shown in Figure 4 makes a direct comparison of composition assignment lists difficult. Our algorithm requires a minimum amount of MS1 information in order to compute a score, which some of the assignments in the original published results do not possess, and are omitted from the count in Table 5. After accounting for ambiguity, we were able to assign all of the compositions previously reported using the Krambeck database, which was used by Yu et al. (2013), and with the combinatorial database. The glySpace-derived database did not contain all of these compositions, but performed competitively with the combinatorial database’s ROC AUC. The combinatorial database matched a small number of glycan compositions which were not in Krambeck but which were consistent with other glycan compositions observed nearby in retention time. The combinatorial database also benefited most substantially from smoothing, discarding many false positives while retaining many more true positives at the same false positive rate compared to the other databases. These invalid glycan compositions can match LC-MS features at any point in the elution profile, though in this dataset the majority of these matches appear to be in the time range between 10 and 22 min, and similar glycan compositions that are biosynthetically valid elute later on in the experiment. Therefore a for a retention-time aware approach to evaluating glycan composition assignments, as described in Hu et al. (2016) could also be useful, but this is likely dependent upon the experimental workup and separation technique used.
Table 5.
Performance comparison for Perm-BS-070111-04-Serum using receiver operator characteristic (ROC) area under the curver (AUC) and number of non-ambiguous matches
| Name | ROC AUC | True matchesa | Non-ambiguous matches |
|---|---|---|---|
| Combinatorial unregularized | 0.679 | 86 | 61 |
| Combinatorial partial | 0.816 | 87 | 62 |
| Combinatorial grid | 0.804 | 86 | 61 |
| GlySpace unregularized | 0.788 | 59 | 51 |
| GlySpace partial | 0.803 | 60 | 52 |
| GlySpace grid | 0.809 | 60 | 52 |
| Krambeck unregularized | 0.866 | 70 | 60 |
| Krambeck partial | 0.883 | 70 | 60 |
| Krambeck grid | 0.882 | 69 | 59 |
| Yu et al. (2013) | – | 72b | 59 |
Note: While the Krambeck database had a better ROC AUC, the Combinatorial database had more true matches.
Selected at .
Only includes cases with sufficient MS1 scans available for comparison.
While the biosynthetically constrained Krambeck database performed better on Perm-BS-070111-04-Serum, it did not contain all of the reasonably assignable glycan compositions, and it performed poorly on 20141103-02-Phil-BS with a false negative rate of 50% compared to the combinatorial database. This is because the necessary enzymatic pathways were either not considered in the original authors’ model because either the enzyme was excluded for simplicity (Krambeck et al., 2009) or because the particular enzymes used were not within the scope of the model used (Ichimiya et al., 2014; Spiro and Spiro, 2000). This highlights the importance of selecting a good reference database, though a post-processing step such as we described here can help mitigate using too large a database, but not a too small one.
In this work, we used the same network neighborhood imposed over different underlying sets of composition nodes, and the connectivity of those networks did not take into account the constraints of the biosynthetic process. It may be possible to obtain better performance by defining network connectivity according to concrete enzymatic relationships. This may also alter how the neighborhoods are defined and how A is parameterized, and in turn how is learned. Similarly, this procedure depends upon the scoring functions used, so selecting another set of functions for the data to fit may lead to different parameter values.
Lastly, while these case studies have demonstrated the algorithm’s ability to learn network parameters from the data, an expert can define and A themselves or obtain a model fitted on related data and apply it directly without a fitting step. An expert could use this model specification to impose prior beliefs on the evaluation process, and adjust λ to control the importance of these beliefs. Similarly, one could also use the derivation of to estimate the score for an unobserved glycan composition, given A and .
We used our glycoinformatics toolkit to produce a richer abstraction of glycans and monosaccharides, including producing standard-compliant textual representations of these structures and compositions. We produced a text file containing all of the glycan compositions found in the Krambeck and Combinatorial database but not the glySpace-derived database in the above samples (see Supplementary Section S9), and have submit it to GlyTouCan (Tiemeyer et al., 2017) for registration so that future researchers can use these structures.
5 Conclusions
In this study, we demonstrated the advantages of our application of Laplacian Regularization to smooth LC-MS assignments of glycan compositions across multiple experimental protocols (Hu and Mechref, 2012; Khatri et al., 2016). Our algorithm’s performance is competitive with existing tools for analyzing the same type of data, with the added benefit of more flexible evaluation process and broader range of understood monosaccharides. Our tools integrate with glySpace and allow users to leverage existing glycomics repositories to build databases where applicable.
All of the methods demonstrated in this paper are available as part of the open source, cross-platform glycomics and glycoproteomics software GlycReSoft, freely available at http://www.bumc.bu.edu/msr/glycresoft/.
Funding
This work was supported by National Institute of Health U01CA221234.
Conflict of Interest: none declared.
Supplementary Material
References
- Akune Y. et al. (2016) Comprehensive analysis of the N-glycan biosynthetic pathway using bioinformatics to generate UniCorn: a theoretical N-glycan structure database. Carbohydrate Res., 431, 56–63. [DOI] [PubMed] [Google Scholar]
- Aoki-Kinoshita K. et al. (2015) GlyTouCan 1.0 – the international glycan structure repository. Nucleic Acids Res., 44, D1237–D1242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belkin M. et al. (2006) Manifold regularization: a geometric framework for learning from labeled and unlabeled examples. J. Mach. Learn. Res., 7, 2399–2434. [Google Scholar]
- Campbell M.P. et al. (2014) UniCarbKB: building a knowledge platform for glycoproteomics. Nucleic Acids Res., 42, D215–D221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ceroni A. et al. (2008) GlycoWorkbench: a tool for the computer-assisted annotation of mass spectra of glycans. J. Proteome Res., 7, 1650–1659. [DOI] [PubMed] [Google Scholar]
- Frank M., Schloissnig S. (2010) Bioinformatics and molecular modeling in glycobiology. Cell. Mol. Life Sci., 67, 2749–2772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldberg D. et al. (2009) Glycan family analysis for deducing N-glycan topology from single MS. Bioinformatics, 25, 365–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Y., Mechref Y. (2012) Comparing MALDI-MS, RP-LC-MALDI-MS and RP-LC-ESI-MS glycomic profiles of permethylated N-glycans derived from model glycoproteins and human blood serum. Electrophoresis, 33, 1768–1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Y. et al. (2016) LC-MS/MS of permethylated N-glycans derived from model and human blood serum glycoproteins. Electrophoresis, 37, 1498–1505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ichimiya T. et al. (2014) Frequent glycan structure mining of influenza virus data revealed a sulfated glycan motif that increased viral infection. Bioinformatics, 30, 706–711. [DOI] [PubMed] [Google Scholar]
- Jaitly N. et al. (2009) Decon2LS: an open-source software package for automated processing and visualization of high resolution mass spectrometry data. BMC Bioinformatics, 10, 87.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaur P., O'Connor P.B. (2006) Algorithms for automatic interpretation of high resolution mass spectra. J. Am. Soc. Mass Spectrometry, 17, 459–468. [DOI] [PubMed] [Google Scholar]
- Kessner D. et al. (2008) ProteoWizard: open source software for rapid proteomics tools development. Bioinformatics, 24, 2534–2536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khatri K. et al. (2016) Integrated omics and computational glycobiology reveal structural basis for Influenza A virus glycan microheterogeneity and host interactions. Mol. Cell. Proteomics, 15, 1895.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krambeck F.J., Betenbaugh M.J. (2005) A mathematical model of N-linked glycosylation. Biotechnol. Bioeng., 92, 711–728. [DOI] [PubMed] [Google Scholar]
- Krambeck F.J. et al. (2009) A mathematical model to derive N-glycan structures and cellular enzyme activities from mass spectrometric data. Glycobiology, 19, 1163–1175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kronewitter S.R. et al. (2014) GlyQ-IQ: glycomics quintavariate-informed quantification with high-performance computing and glycogrid 4D visualization. Anal. Chem., 86, 6268–6276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X. et al. (2010) Deconvolution and database search of complex tandem mass spectra of intact proteins. a combinatorial approach. Mol. Cell. Proteomics, 9, 2772–2782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martens L. et al. (2011) mzML–a community standard for mass spectrometry data. Mol. Cell. Proteomics, 10, R110.000133.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maxwell E. et al. (2012) GlycReSoft: a software package for automated recognition of glycans from LC/MS data. PLoS One, 7, e45474.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peltoniemi H. et al. (2013) Novel data analysis tool for semiquantitative LC-MS-MS2 profiling of N-glycans. Glycoconjugate J., 30, 159–170. [DOI] [PubMed] [Google Scholar]
- Senko M.W. et al. (1995) Determination of monoisotopic masses and ion populations for large biomolecules from resolved isotopic distributions. J. Am. Soc. Mass Spectrometry, 6, 229–233. [DOI] [PubMed] [Google Scholar]
- Spiro M.J., Spiro R.G. (2000) Sulfation of the N-linked oligosaccharides of influenza virus hemagglutinin: temporal relationships and localization of sulfotransferases. Glycobiology, 10, 1235–1242. [DOI] [PubMed] [Google Scholar]
- Stanley P. et al. (2009) N-Glycans. Cold Spring Harbor Laboratory Press, Long Island, New York. [PubMed] [Google Scholar]
- Tiemeyer M. et al. (2017) GlyTouCan: an accessible glycan structure repository. Glycobiology, 27, 915–919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varki A. (2017) Biological roles of glycans. Glycobiology, 27, 3–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu C.-Y.C.-Y. et al. (2013) Automated annotation and quantification of glycans using liquid chromatography-mass spectrometry. Bioinformatics, 29, 1706–1707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu T., Peng H. (2010) Quantification and deconvolution of asymmetric LC-MS peaks using the bi-Gaussian mixture model and statistical model selection. BMC Bioinformatics, 11, 559.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaia J. (2008) Mass spectrometry and the emerging field of glycomics. Chem. Biol., 15, 881–892. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.