

Abstract
Microglia are the immune cells of the brain, they are activated in the brain of Alzheimer's disease (AD) patients and mouse models of AD, and they express the innate immune receptor toll-like receptor 2 (TLR2). The present study investigated role of this receptor in the progression of AD-like pathologies. Here we show that amyloid β (Aβ) stimulates TLR2 expression in a small proportion of microglia. We then generated triple transgenic mice that are deficient in TLR2 from mice that harbor a mutant human presenelin 1 and a chimeric mouse/human amyloid precursor protein (APP) genes. TLR2 deficiency accelerated spatial and contextual memory impairments, which correlated with increased levels of Aβ1–42 and transforming growth factor β1 in the brain. NMDA receptors 1 and 2A expression levels were also lower in the hippocampus of APP–TLR2−/− mice. Gene therapy in cells of the bone marrow using lentivirus constructs expressing TLR2 rescued the cognitive impairment of APP–TLR2−/− mice. Indeed, lenti-green fluorescent protein/TLR2 treatment had beneficial effects by restoring the memory consolidation process disrupted by TLR2 deficiency in APP mice. These data suggest that TLR2 acts as an endogenous receptor for the clearance of toxic Aβ by bone-marrow-derived immune cells. The cognitive decline is markedly accelerated in a context of TLR2 deficiency. Upregulating this innate immune receptor may then be considered as a potential new powerful therapeutic approach for AD.
Keywords: bone marrow stem cells, inflammation, innate immunity microglia, neuroprotection, postsynaptic receptors, transforming growth factor β1
Introduction
Alzheimer's disease (AD) is characterized by memory loss and increasingly severe dementia (Hardy and Selkoe, 2002). The neuropathological changes are manifested by accumulation of plaques containing the amyloid β (Aβ) protein, intracellular neurofibrillary tangles, activated microglia and astrocytes, and degenerating neurons. Early in the disease process, Aβ accumulation reduces synapse density of cortical and hippocampal neurons, which strongly correlates with memory impairments (Selkoe, 2002). Aβ of 40 and 42 amino acids are highly toxic for the synaptic connections, and their levels are elevated in the brain of AD patients. These peptides are produced from the cleavage of amyloid precursor protein (APP) by enzymatic complexes known as β- and γ-secretase. Many mutations in APP and presenelin (PS1) genes are believed to increase Aβ in the brain of patients (Hardy and Selkoe, 2002), and expression of these mutated genes induces AD-like pathologies in mice (Hsiao et al., 1996; Borchelt et al., 1997).
Microglia are the immune cells of the brain, and they are attracted to amyloid deposits both in human samples and in rodent transgenic models that develop this disease. The precise role of microglia in AD is still under intensive debate. Microglia are activated by Aβ and secrete neurotoxic molecules, but they have neuroprotective actions by secreting neurotrophic agents and eliminating toxic Aβ by phagocytosis (Akiyama et al., 2000; Verdier et al., 2004). The dual role of microglia may also depend on their origin. Bone-marrow-derived cells are able to cross the blood–brain barrier and differentiate into fully competent microglia (Hess et al., 2004; Simard and Rivest, 2004; Malm et al., 2005; Massengale et al., 2005). These new differentiated cells express higher levels of proteins that are required for antigen presentation and are more efficient phagocytes than resident microglia (Simard and Rivest, 2004; Simard et al., 2006). However, they seem to lose these properties while the disease is progressing (Simard et al., 2006), and they may not be efficient immunologically to clear toxic Aβ in AD patients and mouse models of this disease.
Toll-like receptors (TLRs) are type I transmembrane pattern recognition receptors that act as endogenous sensors for specific elements called the pathogen-associated molecular patterns (PAMPs) (Akira et al., 2006). PAMPs from Gram-positive bacteria are recognized by TLR2, although few proteins of the host are believed to act as endogenous ligands for this innate immune receptor. There is a robust transcriptional expression of TLR2 in microglia during infection, brain injuries, and diseases (Glezer et al., 2007). Actually, TLR2 mRNA induction is a reliable marker of activated microglia, but its physiological significance has yet to be uncovered (Laflamme et al., 2001). Increased levels of TLR2 mRNA have been found in microglia isolated from AD patients (Bsibsi et al., 2002), and TLR2 activation increases Aβ clearance from the brain (Tahara et al., 2006). The main objective of this study was to follow the progression of the disease in a context of TLR2 gene deficiency and overexpression in bone marrow stem cells. We have identified a novel innate immune mechanism of neuroprotection.
Materials and Methods
Transgenic mouse lines.
APP transgenic mice harboring the human presenelin I (A246E variant) and the chimeric mouse/human Aβ precursor protein (APP695swe) under the control of independent mouse prion protein promoter elements [B6C3-Tg(APP695)3Dbo Tg(PSEN1)5Dbo/J] were bred with the TLR2−/− mouse strain (B6.129-Tlr2tm1Kir/J) for at least three generations to generate APP–TLR2 triple transgenic animals. All newborn pups were genotyped as described in supplemental Table 1 (available at www.jneurosci.org as supplemental material). All mouse strains were purchased from The Jackson Laboratory and maintained in a C57BL/6J background. All protocols were conducted according to the Canadian Council on Animal Care guidelines, as administered by the Laval University Animal Welfare Committee.
To collect brain tissues, mice were deeply anesthetized via an intraperitoneal injection of a mixture of ketamine hydrochloride and xylazine and then perfused intracardially with ice-cold 0.9% saline, followed by 4% paraformaldehyde (PFA) in a 0.1 m borax buffer, pH 9.5, at 4°C. Brains were rapidly removed from skulls, postfixed in PFA 1–3 d at 4°C, and cryoprotected in 10% sucrose diluted in PFA overnight. The frozen brains were sectioned into 25-μm-thick coronal sections using a microtome (Reichert-Jung, Cambridge Instruments Company), and slices were collected in a cold cryoprotectant solution (0.05 m sodium phosphate buffer, pH 7.3, 30% ethylene glycol, and 20% glycerol, stored at −20°C).
For protein analysis, mice were anesthetized with isoflurane, and blood was removed from organs by intracardiac punctures. Brains were rapidly frozen in liquid nitrogen and stored at −80°C.
Aβ1–42 intracerebral injection.
These experiments were performed as described previously (Simard et al., 2006). Briefly, 1 μl of saline or synthetic Aβ1–42 (1 mg/ml; Bachem Biosciences) was injected into the hippocampus of CD1 mice using a stereotaxic instrument (David Kofp Instruments). Six hours later, they were perfused and their brains removed for histological analyses.
Production and femoral injection of lentiviral vector.
For lentivirus construction, ViraPower T-Rex Lentiviral Gateway Vector kit was used as described by the manufacturer (Invitrogen). The vector pLenti4/TO/V5–DEST was modified to visualize transduction. The Zeocin resistance cassette was replaced by the enhanced green fluorescent protein (GFP) coding region and its phosphoglycerate kinase promoter. The insert was amplified from pSuperior vector as template (OligoEngine) (forward, 5′-CACAAGTGGCCTCGAGCCTCGCACACATTCCACATCCAC-3′; reverse, 5′-CTTGTTCAATCATGGTACCTCTAGCCTTAAGTTCGAGAC-3′) and was inserted in pLenti4/TO/V5-DEST between XhoI and KpnI restriction sites, forming the pLenti/GFP vector. The TLR2 coding sequence was amplified by PCR from a cDNA brain library and cloned in the pENTR4 vector using the SalI and XhoI restriction sites of the polylinker (forward, 5′-CAGACAAAGCGTCGACTCTCAGAGGATGCTAC-3′; reverse, 5′-GAAGTCAGGAACTCGAGGGAGAACCTAGGAC-3′; GenBank accession number AF124741). The TLR2 coding sequence was transferred onto the pLenti/GFP downstream the cytomegalovirus promoter by homologous recombination, forming the pLenti/GFP/TLR2. To prepare lentivirus, 293T cells (gift from Dr. L. Vallières, Université Laval, Québec, Québec, Canada) were transfected with pLenti/GFP/TLR2, pLPI, pLP2, and pLP–vesicular stomatitis virus protein G (20 μg per plasmid) using the calcium chloride technique. Lentiviruses were concentrated by ultracentrifugation (50,000 × g for 2 h) of the 72 h culture supernatant and resuspended in DMEM (Sigma).
APP–TLR2 mice (2.5–3 months of age) were anesthetized with isoflurane, and the right knee was flexed. The intrafemoral space was reached with a 28 gauge needle by applying gentle twisting and pressure between the condyles at the top of the femur. The needle was replaced by a 30 gauge needle, and 2 × 107 lentiviruses (20 μl) were injected. APP–TLR2 mice treated with control lentivirus (pLenti/GFP) or pLenti/GFP/TLR2 were tested at 6 months of age for spatial learning and memory.
Behavioral analyses.
Mice were housed four or five per cage in a colony maintained in a room under natural lighting conditions with a 12 h light/dark cycle (on at 6:00 A.M.) and tested during the “light on” phase of the day. Food and water were provided ad libitum. Behavioral experimenter was blinded as to the genetic and treatment status of animals. To assess hippocampal-dependent spatial learning and memory, mice were trained in the T-water maze task. In this paradigm, we evaluate the mouse's ability to remember the spatial location of submerged platform. The T-maze apparatus (length of stem, 64 cm; length of arms, 30 cm; width, 12 cm; height of walls, 16 cm) was made of clear fiberglass and filled with water (23 ± 1°C) at a height of 12 cm. A platform (11 × 11 cm) was placed at the end of the target arm and was submerged 1 cm below the surface. The acquisition phase allows to evaluate animals for left–right spatial learning. During the first two trials, platforms were placed on each arms of the maze to test the spontaneous turning preference of the mouse. After these two trials, the least chosen arm was reinforced by the escape platform. The mice were placed in the stem of the T maze and choose to swim either left or right until they found the submerged platform and escape to it, to a maximum of 60 s. After reaching the platform, the mice remained on it for 20 s and then were immediately placed back in the maze. If the animals did not find the platform within this limit, they were gently guided onto it. Repeated trials were presented on the same day up to a maximum of 48 trials. A rest period of at least 10–15 min intervened between each block of 10 trials. A mouse was considered to have learned the task when it made no errors in a block of five consecutive trials. The reversal learning phase was then conducted 48 h later. During this phase, the same protocol was repeated, except that the mice were trained to find the escape platform on the opposite side to that on which they had learned on acquisition phase. The number of trials to reach the criterion (five of five correct choices made on consecutive trials) was measured.
Based on the animal's natural tendency to prefer the dark environment, the animals were also evaluated in retention of nonspatial memory for one-trial passive avoidance task. The passive avoidance apparatus (Ugo Basile) was divided into two sections, one illuminated (the start compartment) and one dark (escape compartment). The floor of each compartment contained a grid, with only the dark compartment being electrified by a generator. On the training day, mice were placed into the lighted compartment for 60 s acclimation period. The guillotine door was then opened, and the latency to enter the dark side was recorded. Immediately after entering the dark compartment, the door was closed and an electric shock (0.5 mA for 2 s) was delivered. The mouse was kept in the dark compartment for 10 s before being returned to its home cage. On the next day, the mice were again placed in the light compartment, and the time, step through latency to enter the dark side, was measured for up to 300 s.
Stereological analysis.
Free-floating sections were treated 30 min with a permeabilization/blocking solution containing 0.4% Triton X-100, 4% goat serum, and 1% bovine serum albumin (Sigma-Aldrich) in potassium PBS (KPBS). Sections were thereafter immunostained 60 min with a polyclonal anti-Aβ (6E10, 1:3000; Millipore Bioscience Research Reagents), washed three times for 5 min in KPBS, and then incubated with a secondary antibody goat anti-mouse cyanine 3 conjugated (1:1000; Jackson ImmunoResearch) for 60 min. Unbiased stereological analysis was performed as described previously (Simard et al., 2006). The contours of the cortex and hippocampus areas were traced as virtual overlay on the steamed images, and areas were calculated. For 3- and 6-month-old APP mice, the area occupied by all Aβ-labeled plaques was determined. For plaque analysis in the 9-month-old APP mice, the Stereo Investigator software (MicroBrightField) sequentially chose counting frames in cortex and hippocampus. Areas of analyzed hippocampal or cortex regions were calculated as well as areas occupied by plaques in those counting frames.
Determination of Aβ1–42 levels by Western blot.
Hemi-forebrains were homogenized in a buffer containing 8 m urea, 0.5% SDS, 2% β-mercaptoethanol, and a protease inhibitor cocktail (1:100; Sigma-Aldrich). Brain homogenates of 6-month-old APP and APP–TLR2 mice were run on a precast 10–20% SDS-polyacrylamide Tris-Tricine gel (Bio-Rad) and transferred onto polyvinylidene fluoride membranes (PerkinElmer Life and Analytical Sciences). Blots were probed with a polyclonal antibody directed against Aβ1–42 (ab10148, 1:1500; Abcam) in 1 m Tris-HCl, pH 8.0, 5 m NaCl, 5% skim milk, and 0.05% Tween 20. Blots were visualized using anti-rabbit secondary antibody tagged with horseradish peroxidase (1:1000; Jackson ImmunoResearch) and enhanced chemiluminescence (PerkinElmer Life and Analytical Sciences) and then analyzed on a gel imaging system (scanner Agfa Arcus II; NIH Image J software version 1.32j). Membranes were stripped in 25 mm glycine-HCl, pH 2.0, containing 1% SDS to allow β-actin revelation using first a mouse β-actin antibody (MAB1501, 1:3500; Millipore Bioscience Research Reagents) and then a goat anti-mouse peroxidase-conjugated secondary antibody (1:1500; Jackson ImmunoResearch).
In situ hybridization and immunohistochemistry.
Every 12th section of brain slices was mounted on Colorfrost/Plus microscope slides (Fisher Scientific), starting from the end of the olfactory bulb to the end of the cortex. In situ hybridization histochemical localization of TLR2, NMDA receptors R1 and R2A and transforming growth factor β1 (TGFβ1) was performed using 35S-labeled cRNA probes. Riboprobes synthesis and preparation and in situ hybridization were performed according to a protocol described previously (Laflamme et al., 1999). Plasmids were linearized, and sense and antisense cRNA probes were synthesized with appropriate RNA polymerase, as described in supplemental Table 2 (available at www.jneurosci.org as supplemental material). All plasmids were analyzed for sequence confirmation an orientation. Slides were exposed at 4°C to x-ray films (Biomax; Eastman Kodak), and semiquantitative analysis of hybridization signals were performed under a Northern Light Desktop Illuminator (Imaging Research) using a Sony Camera Video System and the NIH Image J software version 1.32j. The expression levels were quantified for three (NMDA-R1 and NMDA-R2A) and 12 (TLR2) sections per brain, and means were used for statistical analyses.
Dual labeling combining immunocytochemistry and in situ hybridization was performed as described previously (Laflamme et al., 2001). Images were captured using a Nikon Eclipse 80i microscope equipped with a digital camera (QImaging), processed to enhance contrast and sharpness using Adobe Photoshop 7 (Adobe Systems), and then assembled using Adobe Illustrator (Adobe Systems).
Results
TLR2 expression in response to Aβ
To assess whether Aβ protein alone can activate TLR2 gene expression in the brain, we injected Aβ1–42 in the hippocampus of wild-type (WT) CD1 mice. Six hours after the injection, we studied the expression level of the TLR2 gene by in situ hybridization. As shown in Figure 1, the expression of TLR2 gene was greatly increased after Aβ1–42 infusion (Fig. 1b) compared with their saline-treated littermates (Fig. 1a). We thus quantitatively measured the optical density of hybridization signal for TLR2 transcript on the vicinity of the injection sites. The values obtained for saline- and Aβ1–42-injected groups were 0.018 ± 0.002 and 0.033 ± 0.004, and a Student's t test analysis revealed a statistical difference between the groups (p < 0.05) (Fig. 1c). The injection of other APP-derived peptides, C31 and CTF-57, did not induce similar effects as depicted by supplemental Figure 1 (available at www.jneurosci.org as supplemental material). These data demonstrate that TLR2 transcriptional activation is specific to the injection of Aβ1–42 peptide.
Figure 1.
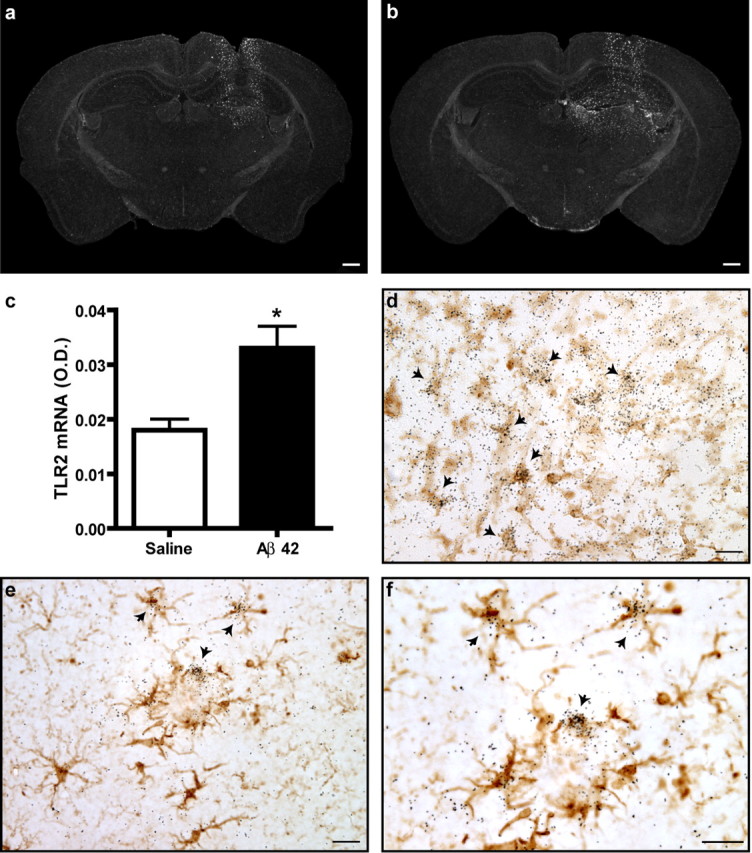
TLR2 gene expression in response to exogenous and endogenous Aβ. A solution containing either sterile saline (a) or synthetic Aβ1–42 peptide (b) was injected into the hippocampus of CD1 mice. TLR2 mRNA expression was determined by in situ hybridization (agglomeration of silver grains) 6 h after the intracerebral infusion. c, TLR2 mRNA expression levels (as determined by optical density quantification) were significantly increased in areas distal to the injection site of Aβ1–42-treated animals. *p < 0.05 (Student's t test; saline, n = 3; Aβ1–42, n = 5). d, Tissue sections were stained with an antibody directed against iba1 to reveal microglia (brown cells) and hybridized with TLR2 mRNA probe. TLR2 mRNA signal always colocalized with iba1-immunoreactive cells surrounding the injection site (indicated with arrows). e, f, Activated microglia had also a positive signal for TLR2 transcript in the brain APP mice (iba1+/TLR2+). Scale bars: a, b, 500 μm; d–f, 20 μm. Error bars indicate SEM.
Microglia are the immune cells of the brain and are known to express inflammatory molecules. To determine whether these cells were responsible for the TLR2 induction in the presence of exogenous Aβ1–42, we performed a colocalization study (Fig. 1d). We found that TLR2-expressing cells surrounding the injection site were all immunoreactive for the microglial marker ionized calcium-binding adapter molecule 1 (iba1). This is also the case in the natural course of AD, because several double-labeled cells were found in the brain of APP mice that did not receive exogenous Aβ1–42 (Fig. 1e,f). These data demonstrate that microglial TLR2 is induced in response to exogenous Aβ1–42 administration and endogenous Aβ deposits. Numerous reports have shown that microglia have the ability to originate from the bone marrow in the brain of mouse models of AD. To determine whether TLR2 transcript was expressed in resident and/or infiltrating microglia, we created APP chimeric mice that were transplanted with bone marrow cells expressing GFP. We labeled GFP-infiltrating cells by immunohistochemistry (brown cells) and found positive TLR2-expressing cells in both resident and bone-marrow-derived microglia (BMDM) surrounding Aβ deposits (supplemental Fig. 2, available at www.jneurosci.org as supplemental material). However, it was not possible to quantify the exact proportion of each type of TLR2-expressing cells, because the hybridization signal decreased considerably attributable to the immunohistochemical procedure using a primary antibody directed against GFP.
APP/PS1 mice that bear a mutation in the gene encoding TLR2 exhibit severe memory deficits
We next determined the role of such natural TLR2 induction by Aβ in studying the behavior of a mouse model of AD in a context of TLR2 gene deficiency. For this purpose, APP/PS1 mice were bred with TLR2-deficient mice, which share the same C57BL/6J background. All newborn pups were genotyped and included in the different experimental groups. These mice were subjected to hippocampus-based spatial learning in T-water maze and contextual memory storage in passive avoidance paradigm to evaluate the importance of TLR2 receptor in the course of the disease. In the T-water maze test, mice were trained to find a hidden platform for the acquisition of spatial memory and mean of trials needed to learn the task is presented in Figure 2a. All groups exhibited comparable swimming performance and motivation to escape from the water during initial training session. There were no effects of genotype or age on number of trials to reach criterion. We then experienced hippocampus-dependant cognitive flexibility by allowing the mice to find a platform in the opposite arm of the T-water maze task apparatus (reversal phase) (Fig. 2b). Both wild-type and TLR2 knock-out mice showed equally successful number of trials to criterion during the reversal task. Because of their AD behavioral phenotype, the trials made by APP mice were increased at 6 and 9 months compared with controls. However, the decline in behavioral performance of APP–TLR2 mice began as early as 3 months, as evidenced by a significantly increased in the number of trials made to reach the platform (5.0 ± 0.9 for APP mice vs 12.0 ± 2.8 for APP–TLR2 mice; p < 0.05). At 6 and 9 months of age, the spatial memory decline of the APP–TLR2 mice (22.0 ± 4.0 and 32.7 ± 4.4 trials, respectively) was also significantly higher than the decline observed in APP mice (14.3 ± 2.0 and 18.9 ± 2.7 trials, respectively).
Figure 2.

TLR2 deficiency accelerates the onset and severity of spatial memory impairment in APP/PS1 mice. a, Animals were trained to reach the hidden platform on the T-water maze apparatus, and mean of trials to accomplish the task was presented. All the different mouse groups had similar acquisition phases at 3, 6, and 9 months of age. b, The reversal learning phase was conducted 48 h later. APP–TLR2 transgenic mice exhibited a greater decline of their spatial memory capacity than their control and APP mice. *p < 0.05, **p < 0.01 (two-way ANOVA was performed revealing a significant interaction between factors age and genotype, and multiple comparisons of genotype simple effects in each level of age was performed by Bonferroni's post hoc test). Error bars indicate SEM.
We also determined the contextual, nonspatial memory of each group with the passive avoidance paradigm. Passive avoidance training was performed by introducing the mice to the light compartment with the dark compartment closed. In this task, animals were trained to avoid the dark compartment by receiving an electrical shock. The latency for entering the dark side of the apparatus was measured 1 d after the training session. There were no significant differences for initial latencies to enter the dark compartment between the groups at all ages (Fig. 3a). However, when tested 1 d later (retention session), APP–TLR2 mice spent less time to reenter the dark compartment compared with the other control groups, revealing an impairment of the memory based on punishment (Fig. 3b). The values obtained for the latencies were 209.2 ± 27.0 versus 99.5 ± 23.6, 154.3 ± 23.3 versus 83.4 ± 13.3, and 124.7 ± 20.6 versus 41.6 ± 7.8 for 3-, 6-, and 9-month-old APP and APP–TLR2 animals, respectively. A two-way ANOVA revealed a significant effect of groups (p < 0.05) but no effect of age, suggesting that the behavior of APP–TLR2 is significantly different than the other strains (p < 0.001). Because electric foot shocks were used in passive avoidance task, we verified whether all animals displayed similar nociceptive responses and found that no difference in term of foot-shock sensitivity threshold was present between groups (data not shown). These results demonstrate a greater cognitive deficit in APP–TLR2 mice compared with their APP littermates. The lack of TLR2 in our model of AD results in an accelerated spatial and contextual memory deficits. These data suggest that TLR2 play an important role in memory formation.
Figure 3.

Behavioral analysis of APP–TLR2 transgenic mice unravels a contextual memory deficit. a, All mouse groups showed similar step-through latency on the single training trial of the passive avoidance test. b, Twenty-four hours after the conditioning test, APP–TLR2 mice showed significantly lower latency to enter the dark compartment of the passive avoidance apparatus. Mean latencies are depicted for each mouse groups at 3, 6, and 9 months of age. Pairwise group comparison revealed a significant main effect between APP–TLR2 and other groups of mice. ***p < 0.001; n = 9–12 (two-way ANOVA, Tamhane post hoc test). No specific pairwise comparisons could be done at each age because of lack of a group × age interaction. Error bars indicate SEM.
Amyloid-β analysis in the brain of APP mice in a context of TLR2 deficiency
In addition to behavioral changes, AD is associated with accumulation of Aβ. We thus determined whether the lack of TLR2 expression in microglia affected the accumulation and production of Aβ in the mouse brains. We first measured Aβ immunoreactivity in coronal sections of APP and APP–TLR2 with the 6E10 antibody specific for the 1–17 fragment of the Aβ human protein. Area of Aβ deposits was measured for two sections identical for each animals, and the percentage of area occupied by plaques was determined in the hippocampus and cortex. At 3 and 6 months of age, area covered by immunoreactive Aβ deposits appeared significantly lower (p < 0.01 and p < 0.05, respectively) in APP–TLR2 brains (0.0037 ± 0.0007 and 0.41 ± 0.09%) compared with their APP littermates (0.020 ± 0.005 and 0.83 ± 0.16%) (Fig. 4a–f). In contrast, 9-month-old brains exhibited comparable amount of Aβ deposits (3.11 ± 0.58% for APP and 2.94 ± 0.50% for APP–TLR2) (Fig. 4g–i). These results revealed a lower rate of Aβ deposition in APP–TLR2 mice on early phases of the disease.
Figure 4.

Amyloid plaque formation is delayed in the brain of APP mice that bear a mutation in the gene encoding TLR2. a–i, Anti-Aβ immunoreactivity in the brains of APP (a, d, g) and APP–TLR2 (b, e, h) mice. APP mice exhibited higher amyloid loads than APP–TLR2 mice at 3 (a, b) and 6 (d, e) months of age. g, h, Such a difference in the plaque formation was attenuated at later time points, because the surface occupied by the positive Aβ signal was similar in both groups of APP mice at 9 months of age. c, f, i, Percentage of area covered by senile plaques in the brain of mice killed at 3, 6, and 9 months, respectively. *p < 0.05, **p < 0.01 (Student's t test; APP, n = 6–11; APP–TLR2, n = 8–11). Error bars indicate SEM. Scale bars, 250 μm.
Because the percentage of area occupied by plaques did not correlate with the behavioral results, we next identified the Aβ species that cause memory deficits. Aβ1–42 has been shown to be the most toxic Aβ isoform that elicits immunological response (Simard et al., 2006) and cognitive deficit (Lue et al., 1999; McLean et al., 1999; Mucke et al., 2000; Naslund et al., 2000; Koistinaho et al., 2001). We thus quantified the amount of Aβ1–42 in the brain of 6-month-old APP and APP–TLR2 mice by Western blots. Relative level results, standardized by the amount of the β-actin protein, demonstrated that APP–TLR2 brains contained a significant (p < 0.05, Student's t test; n = 4) higher Aβ1–42 protein load (0.88 ± 0.14 for APP–TLR2 vs 0.38 ± 0.05 for APP mice) (Fig. 5). These data demonstrate that APP–TLR2 mice have a lower Aβ deposition in the early phase but a higher production of the toxic Aβ1–42 species in the same phase of the disease.
Figure 5.

Enhanced production of Aβ1–42 in the brains of APP–TLR2 mice. Proteins from hemi-forebrains of 6-month-old APP and APP–TLR2 mice were immunoblotted with an anti-Aβ1–42 antibody. β-Actin levels were used for equal protein loading of samples.
Defect in synaptic plasticity of APP–TLR2 mice
Previous studies have documented that soluble forms of Aβ1–42 inhibit one form of plastic synaptic function, the long-term potentiation (LTP), that is closely associated with learning and memory (Lambert et al., 1998; Selkoe, 2002; Rowan et al., 2003; Chang et al., 2006). Induction and expression of LTP depend among others on the regulated activity of NMDA receptors, which are affected in subjects with AD (Ulas and Cotman, 1997; Bi and Sze, 2002). To determine whether synaptic plasticity is defective in our model of APP mice lacking of TLR2 receptor, we examined the mRNA expression of the NMDA receptor subunits NR1 and NR2A. Results of densitometric analysis of hybridization signal revealed a significant decrease (p < 0.05) of the NMDA-R1 expression in the hippocampus of 6-month-old APP–TLR2 mice (0.125 ± 0.011) compared with WT (0.168 ± 0.011) and APP (0.167 ± 0.011) mice (Fig. 6a,c). It is interesting to note that NMDA-R1 mRNA levels were higher in 3-month-old APP mice (0.168 ± 0.012) compared with WT controls (0.111 ± 0.008). Such an increase was not detected in the hippocampus of APP–TLR2 mice. NMDA-R2 mRNA levels were similar in the hippocampus of all groups of mice at 3 months of age (Fig. 6b,d). However, 6-month-old APP–TLR2 mice exhibited a significant decrease (0.157 ± 0.014) compared with WT (0.265 ± 0.033) and APP (0.257 ± 0.017) mice (Fig. 6d). These data are consistent with the severe cognitive impairment and increased levels of Aβ1–42 of APP–TLR2 mice.
Figure 6.

Differential expression levels for the genes encoding NMDA receptors in the hippocampus of APP and APP/TLR2 mice. Relative NMDA-R1 (a) and NMDA-R2A (b) mRNA levels were quantified in the dentate gyrus and the pyramidal cell layer of the hippocampus of WT (n = 8), TLR2 knock-out (n = 6–7), APP (n = 6–11), and APP–TLR2 (n = 9–10) mice as described in Materials and Methods. c, d, Optical densities (O.D.) normalized to the background values. Please note the significantly (*p < 0.05) lower mRNA levels for both receptors in the hippocampus of APP/TLR2 mice compared with the other mouse groups at 6 months of age. This is suggestive of potential synaptic defects of learning and memory, which was evaluated by the different behavioral tests. NMDA-R1 expression levels were also higher in the hippocampus of 3-month-old APP mice when compared with those of WT control mice (*p < 0.05). Two-way ANOVA was performed and revealed a significant interaction between factors age and genotype. Bonferroni's post hoc test was then used for multiple comparisons to determine significance levels between mouse genotypes at either 3 or 6 months of age. Error bars indicate SEM. Scale bars, 500 μm.
Enhanced TGFβ1 expression in the brain of APP–TLR2 mice
TGFβ1 is a potent anti-inflammatory cytokine, which has been found in the brain of AD patients and promotes Aβ deposits in transgenic mouse model of AD (Wyss-Coray et al., 1997). A robust hybridization signal was detected in the brain of 9-month-old APP–TLR2 mice (Fig. 7). The signal was mainly associated with the senile plaques in the cortex as well as in the hippocampus (Fig. 7d,f). The number of positive cells and signal intensity were clearly lower in the same regions of age-matched APP mice (Fig. 7c,e). These data indicate that TGFβ1 is upregulated in the brain of APP mice in a context of TLR2 deficiency.
Figure 7.
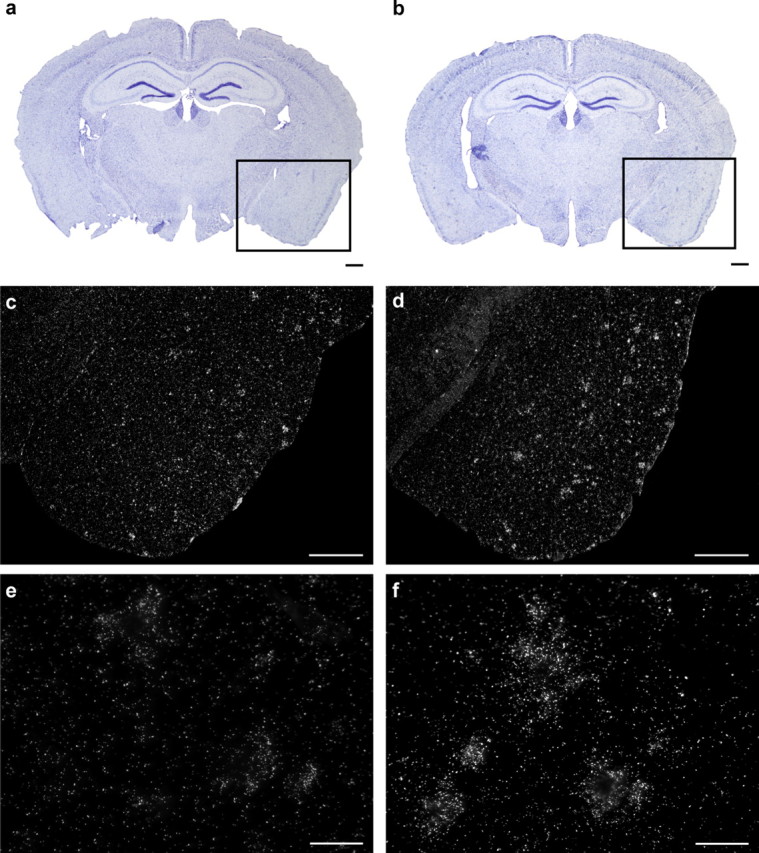
TGFβ1 mRNA expression is increased in regions surrounding Aβ plaques in the brain of TLR2-deficient APP mice. Representative dark-field photomicrograph of in situ hybridization signals showing the expression of TGFβ1 mRNA in brains of 9-month-old APP (c, e) and APP–TLR2 (d, f) mice and the corresponding thionin staining sections (a, b). Scale bars: c, d, 500 μm; e, f, 50 μm.
The lentiGFP/TLR2 treatment improves spatial and contextual memory in APP–TLR2 mice
We next attempted to confirm that accelerated evolution of AD in APP–TLR2 mice was caused by the absence of this innate receptor in bone-marrow-derived cells. To achieve this goal, lentiGFP/TLR2 or its control lentiGFP was injected in the bone marrow of APP–TLR2 mice at 2.5–3 months of age, and mice were tested 3 months later. The mean of trials to reach the criterion during the reversal phase in the T-water maze indicates that lentiGFP/TLR2 mice required significantly fewer trials to learn the task than lentiGFP-treated APP–TLR2 transgenic mice (8.6 ± 0.8 vs 18.2 ± 2.1, respectively; p < 0.01) (Fig. 8a). The lentiGFP/TLR2-treated mice performed comparably with the wild-type mice, suggesting a complete rescue of their cognitive deficit. Moreover, the lentiGFP/TLR2 treatment in APP–TLR2 mice resulted in a significant increased of the step through latency in the retention session of the passive avoidance test (71 ± 18 for lentiGFP-treated and 160 ± 30 for lentiGFP/TLR2-treated APP–TLR2 mice; p < 0.05) (Fig. 8b). This suggests a strong improvement of the contextual memory deficit. Therefore, TLR2 expression in bone-marrow-derived cells of APP–TLR2 mice resulted in an enhancement of performance in the passive avoidance and the reversal spatial learning task. LentiGFP/TLR2 treatment had beneficial effects by restoring the memory consolidation process disrupted by TLR2 deficiency in APP mice.
Figure 8.

The lentiGFP/TLR2 treatment in the bone marrow rescues spatial and contextual memory in 6-month-old APP–TLR2 mice. The mean of trials to reach the criterion during the reversal phase in the T-water maze indicates that lentiGFP/TLR2-treated mice required significantly fewer trials to learn the task than APP–TLR2 transgenic animals that were treated with the control lentiGFP virus (a). It is of great interest to note that lentiGFP/TLR2-treated APP–TLR2 transgenic animals performed comparably with wild-type mice. **p < 0.01, *** p < 0.001 (one-way ANOVA; APP–TLR2 reproduced from Fig. 2, n = 9; lentiGFP, n = 11; lentiGFP/TLR2, n = 14). The lentiGFP/TLR2-treated animals also showed a higher step-through latency to enter the dark compartment during the passive avoidance task compared with controls (b). *p < 0.05 (one-way ANOVA; APP–TLR2 reproduced from Fig. 3, n = 9; lentiGFP, n = 5; lentiGFP/TLR2, n = 7). APP–TLR2 mice received the different lentivirus preparations at 2.5–3 months of age and were tested 3 months later. For details, see Materials and Methods. Error bars indicate SEM.
We next determined the nature of the cells that are responsible for these cognitive changes and searched for genetically modified cells in different organs. Although the level of chimera was low in all the infected mice (e.g., <5%), we found positive GFP cells in the spleen, liver, and brain (supplemental Fig. 3, available at www.jneurosci.org as supplemental material). This suggests that the lentiviral constructs successfully modified a proportion of bone marrow cells, which were then able to migrate and help rescuing the cognitive deficit of APP–TLR2 mice.
Discussion
It has been shown that key receptors of innate immune system are involved in Aβ removal. Indeed, the lipopolysaccharide receptor CD14 is upregulated in microglia of AD brains, and polymorphism of this receptor is associated with increased risks of AD (Combarros et al., 2005). CD14 actually interacts with fibrillar Aβ1–42 and facilitates its phagocytosis (Liu et al., 2005). It was also reported that Aβ load in the brain is modulated in part by TLR4 and that activation of TLR2, TLR4, and TLR9 increases Aβ uptake by microglia (Chen et al., 2006; Tahara et al., 2006). These data suggest that expression of these innate immune receptors in microglia act as a natural defense mechanism to prevent Aβ accumulation in the CNS. The critical question then is why do these receptors fail to remove Aβ in the CNS of AD patients and APP mice? It is possible that the phagocytic properties of microglia are decreased with the disease progression or the balance between Aβ production and clearance is deviant in AD subjects. TLR2 may play a critical role in such a balance.
We show in this study that Aβ1–42 triggers TLR2 gene expression in microglia and that deletion of that gene in APP mice results in an accelerated spatial and contextual memory deficits that correlate with increased Aβ1–42 levels in the brain. The accelerated cognitive decline of APP–TLR2 mice was associated with lower NMDA-R1 and NMDA-R2A mRNA levels in the hippocampus and higher levels of the anti-inflammatory cytokine TGFβ1 in the vicinity of Aβ plaques. Finally, APP–TLR2 that received TLR2 gene rescued expression in bone marrow cells no longer exhibited cognitive decline. The presence of TLR2 in bone-marrow-derived microglia is therefore a powerful natural endogenous mechanism to restrict Aβ toxicity and prevent the cognitive decline in this mouse model of AD.
As depicted by the Figure 1, e and f, a small proportion of microglia contained TLR2 hybridization signal around Aβ plaques of APP mice. Although TLR2 was found in resident microglia, a proportion of bone-marrow-derived cells contained mRNA signal for the innate immune receptor (supplemental Fig. 2, available at www.jneurosci.org as supplemental material). Many studies support the idea that bone-marrow-derived cells are able to cross the blood–brain barrier and differentiate into microglia (Hess et al., 2004; Simard and Rivest, 2004; Malm et al., 2005). Moreover, these BMDM are able to phagocyte Aβ, and their selective ablation resulted in an increased of amyloid burden (Simard et al., 2006). Similarly, CC chemokine receptor 2 (CCR2) gene deficiency was found to impair microglia recruitment and increase amyloid deposits in APP mice (El Khoury et al., 2007). CCR2 is the receptor for monocyte chemoattractant protein-1 (CC chemokine ligand 2), which plays a major role for the chemotaxis of monocytes to inflammatory sites. Such a chemoattraction of BMDM is greatly improved with immunization using a wide range of self-antigen (e.g., glatiramer acetate), which is clearly beneficial for APP mice (Butovsky et al., 2006). The present study suggests that TLR2 expression in BMDM plays a major role in preventing cognitive decline because of Aβ1–42 accumulation in the CNS. Upregulating this receptor in monocytoid precursors in the bone marrow may then be a powerful new therapeutic strategy for curing AD.
Studies using passive immunization against Aβ have highlighted the possibility that brain Aβ burden can be reduced by a peripheral sink mechanism (DeMattos et al., 2001). This theory actually proposes an efflux of soluble Aβ from the brain, across the blood–brain barrier, into the plasma and that this protein can be degraded in peripheral organs. Transporter and carrier proteins have been found in the blood–brain barrier to control the exchange of smaller peptides, including Aβ, from the brain to the periphery (Mackic et al., 1998; Shibata et al., 2000; Sagare et al., 2007). Therefore, it is possible that peripheral immunocompetent cells, expressing TLR2 after the lentiviral injection, contribute to the Aβ clearance. In such a case, these cells would not necessary need to enter the CNS to remove Aβ from the circulation.
It is interesting to note that the deficit of memory capacities was not associated with plaque formation in the brain APP–TLR2 mice; actually, they had less plaques at both 3 and 6 months of age than APP mice. Accumulation of Aβ plaques commonly indicates AD progression in these mouse models. However, many reports have suggested that soluble Aβ, more specifically the Aβ1–42 oligomers, may better correlate with both neurodegeneration and memory impairment (Lue et al., 1999; McLean et al., 1999; Mucke et al., 2000; Naslund et al., 2000; Koistinaho et al., 2001). Soluble forms of Aβ are known to accumulate in the brain of AD patients and APP mice (Gong et al., 2003; Lesne et al., 2006). It is also demonstrated that Aβ1–42 induced deficits in synaptic plasticity, a critical component of the mechanisms involved in learning and memory (Selkoe, 2002). Moreover, two studies have shown that passive immunization using anti-Aβ antibodies resulted in the reversal of memory impairment without reduction in amyloid plaque burden (Dodart et al., 2002; Kotilinek et al., 2002). According to these studies, Aβ1–42 accumulation in the CNS of APP–TLR2 mice is more indicating of AD progression than the area occupied by plaques. Moreover, we found the same quantity of total Aβ in brain homogenates of APP and APP–TLR2 mice (Western blot; data not shown), which supports the ratio of soluble Aβ and plaque formation in early phases of the disease. We propose that TLR2 in BMDM is essential to eliminate the most toxic form of Aβ explaining the significant increase in Aβ1–42 levels in the CNS of APP–TLR2 mice.
Expression of NMDA receptors was performed to test whether the rapid cognitive decline and high Aβ1–42 levels were associated with changes in synaptic plasticity. We found lower NMDA-R1 and NMDA-R2A mRNA levels in the hippocampus of APP–TLR2 mice. The hippocampus is known to be vulnerable in AD. Cognitive tests used here explored the hippocampus-based spatial learning and contextual memory storage and can detect alterations in synaptic plasticity, especially LTP (Tsien et al., 1996; Morris et al., 2003). Aβ1–42 is able to inhibit LTP, affects synapse composition and structure, and causes the loss of glutamate receptors (Snyder et al., 2005; Lacor et al., 2007). Moreover, LTP deficit is rescued by inhibiting Aβ production (Postina et al., 2004), and Aβ induces NMDA receptor endocytosis of cultured cortical neurons (Snyder et al., 2005). Both reversal spatial task and contextual memory paradigm appear to be sensitive assays of NMDA receptor functions and hippocampal integrity in mouse models of AD.
Qualitative analysis of brain tissues showed that TLR2 deficiency resulted in TGFβ1 overexpression. This molecule has profound immunosuppressive and anti-inflammatory effects on microglia (Brionne et al., 2003). Chronic TGFβ1 overexpression promotes cerebrovascular fibrosis and amyloidosis (Wyss-Coray et al., 1997; Ueberham et al., 2005). High TGFβ1 mRNA levels are also detected in senile plaques and correlate with the degree of cerebrovascular amyloidosis in AD-affected patients (Wyss-Coray et al., 1997). Although we have to prove this experimentally, it is tempting to propose that TGFβ1 inhibits the immune/phagocytic properties of BMDM in both AD patients and mouse models of AD. These data fit very well with the increased soluble Aβ, decreased NMDA receptors, and greater cognitive impairment of APP–TLR2 mice.
In conclusion, we found a rapid cognitive decline closely related to the prevalence of soluble Aβ1–42 in the brain of APP mice in a context of TLR2 deficiency. The rescue experiment in cells of the bone marrow provides direct evidence that TLR2 competent BMDM act as a natural innate immune mechanism for restricting Aβ toxicity. Such a mechanism may be absent in AD patients, and restoring TLR2 via gene delivery in monocytoid precursors could therefore be envisaged as a powerful new therapy for treating AD.
Footnotes
This work was supported by the Canadian Institutes in Health Research. Karine L. Richard was supported by a Studentship from the Fonds de la Recherche en Santé du Québec. Serge Rivest holds a Canadian Research Chair in Neuroimmunology. We thank Nataly Laflamme, Martine Lessard, and Marie Michèle Plante for their technical assistance and Isaias Glezer for his help with the statistical analyses.
References
- Akira S, Uematsu S, Takeuchi O. Pathogen recognition and innate immunity. Cell. 2006;124:783–801. doi: 10.1016/j.cell.2006.02.015. [DOI] [PubMed] [Google Scholar]
- Akiyama H, Barger S, Barnum S, Bradt B, Bauer J, Cole GM, Cooper NR, Eikelenboom P, Emmerling M, Fiebich BL, Finch CE, Frautschy S, Griffin WS, Hampel H, Hull M, Landreth G, Lue L, Mrak R, Mackenzie IR, McGeer PL, et al. Inflammation and Alzheimer's disease. Neurobiol Aging. 2000;21:383–421. doi: 10.1016/s0197-4580(00)00124-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bi H, Sze CI. N-methyl-d-aspartate receptor subunit NR2A and NR2B messenger RNA levels are altered in the hippocampus and entorhinal cortex in Alzheimer's disease. J Neurol Sci. 2002;200:11–18. doi: 10.1016/s0022-510x(02)00087-4. [DOI] [PubMed] [Google Scholar]
- Borchelt DR, Ratovitski T, van Lare J, Lee MK, Gonzales V, Jenkins NA, Copeland NG, Price DL, Sisodia SS. Accelerated amyloid deposition in the brains of transgenic mice coexpressing mutant presenilin 1 and amyloid precursor proteins. Neuron. 1997;19:939–945. doi: 10.1016/s0896-6273(00)80974-5. [DOI] [PubMed] [Google Scholar]
- Brionne TC, Tesseur I, Masliah E, Wyss-Coray T. Loss of TGF-beta 1 leads to increased neuronal cell death and microgliosis in mouse brain. Neuron. 2003;40:1133–1145. doi: 10.1016/s0896-6273(03)00766-9. [DOI] [PubMed] [Google Scholar]
- Bsibsi M, Ravid R, Gveric D, van Noort JM. Broad expression of Toll-like receptors in the human central nervous system. J Neuropathol Exp Neurol. 2002;61:1013–1021. doi: 10.1093/jnen/61.11.1013. [DOI] [PubMed] [Google Scholar]
- Butovsky O, Koronyo-Hamaoui M, Kunis G, Ophir E, Landa G, Cohen H, Schwartz M. Glatiramer acetate fights against Alzheimer's disease by inducing dendritic-like microglia expressing insulin-like growth factor 1. Proc Natl Acad Sci USA. 2006;103:11784–11789. doi: 10.1073/pnas.0604681103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang EH, Savage MJ, Flood DG, Thomas JM, Levy RB, Mahadomrongkul V, Shirao T, Aoki C, Huerta PT. AMPA receptor downscaling at the onset of Alzheimer's disease pathology in double knockin mice. Proc Natl Acad Sci USA. 2006;103:3410–3415. doi: 10.1073/pnas.0507313103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen K, Iribarren P, Hu J, Chen J, Gong W, Cho EH, Lockett S, Dunlop NM, Wang JM. Activation of Toll-like receptor 2 on microglia promotes cell uptake of Alzheimer disease-associated amyloid beta peptide. J Biol Chem. 2006;281:3651–3659. doi: 10.1074/jbc.M508125200. [DOI] [PubMed] [Google Scholar]
- Combarros O, Infante J, Rodriguez E, Llorca J, Pena N, Fernandez-Viadero C, Berciano J. CD14 receptor polymorphism and Alzheimer's disease risk. Neurosci Lett. 2005;380:193–196. doi: 10.1016/j.neulet.2005.01.082. [DOI] [PubMed] [Google Scholar]
- DeMattos RB, Bales KR, Cummins DJ, Dodart JC, Paul SM, Holtzman DM. Peripheral anti-A beta antibody alters CNS and plasma A beta clearance and decreases brain A beta burden in a mouse model of Alzheimer's disease. Proc Natl Acad Sci USA. 2001;98:8850–8855. doi: 10.1073/pnas.151261398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dodart JC, Bales KR, Gannon KS, Greene SJ, DeMattos RB, Mathis C, DeLong CA, Wu S, Wu X, Holtzman DM, Paul SM. Immunization reverses memory deficits without reducing brain Abeta burden in Alzheimer's disease model. Nat Neurosci. 2002;5:452–457. doi: 10.1038/nn842. [DOI] [PubMed] [Google Scholar]
- El Khoury J, Toft M, Hickman SE, Means TK, Terada K, Geula C, Luster AD. Ccr2 deficiency impairs microglial accumulation and accelerates progression of Alzheimer-like disease. Nat Med. 2007;13:432–438. doi: 10.1038/nm1555. [DOI] [PubMed] [Google Scholar]
- Glezer I, Simard AR, Rivest S. Neuroprotective role of the innate immune system by microglia. Neuroscience. 2007;147:867–883. doi: 10.1016/j.neuroscience.2007.02.055. [DOI] [PubMed] [Google Scholar]
- Gong Y, Chang L, Viola KL, Lacor PN, Lambert MP, Finch CE, Krafft GA, Klein WL. Alzheimer's disease-affected brain: presence of oligomeric A beta ligands (ADDLs) suggests a molecular basis for reversible memory loss. Proc Natl Acad Sci USA. 2003;100:10417–10422. doi: 10.1073/pnas.1834302100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science. 2002;297:353–356. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- Hess DC, Abe T, Hill WD, Studdard AM, Carothers J, Masuya M, Fleming PA, Drake CJ, Ogawa M. Hematopoietic origin of microglial and perivascular cells in brain. Exp Neurol. 2004;186:134–144. doi: 10.1016/j.expneurol.2003.11.005. [DOI] [PubMed] [Google Scholar]
- Hsiao K, Chapman P, Nilsen S, Eckman C, Harigaya Y, Younkin S, Yang F, Cole G. Correlative memory deficits, Abeta elevation, and amyloid plaques in transgenic mice. Science. 1996;274:99–102. doi: 10.1126/science.274.5284.99. [DOI] [PubMed] [Google Scholar]
- Koistinaho M, Ort M, Cimadevilla JM, Vondrous R, Cordell B, Koistinaho J, Bures J, Higgins LS. Specific spatial learning deficits become severe with age in beta-amyloid precursor protein transgenic mice that harbor diffuse beta-amyloid deposits but do not form plaques. Proc Natl Acad Sci USA. 2001;98:14675–14680. doi: 10.1073/pnas.261562998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotilinek LA, Bacskai B, Westerman M, Kawarabayashi T, Younkin L, Hyman BT, Younkin S, Ashe KH. Reversible memory loss in a mouse transgenic model of Alzheimer's disease. J Neurosci. 2002;22:6331–6335. doi: 10.1523/JNEUROSCI.22-15-06331.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacor PN, Buniel MC, Furlow PW, Clemente AS, Velasco PT, Wood M, Viola KL, Klein WL. Aβ oligomer-induced aberrations in synapse composition, shape, and density provide a molecular basis for loss of connectivity in Alzheimer's disease. J Neurosci. 2007;27:796–807. doi: 10.1523/JNEUROSCI.3501-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laflamme N, Lacroix S, Rivest S. An essential role of interleukin-1beta in mediating NF-κB activity and COX-2 transcription in cells of the blood–brain barrier in response to a systemic and localized inflammation but not during endotoxemia. J Neurosci. 1999;19:10923–10930. doi: 10.1523/JNEUROSCI.19-24-10923.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laflamme N, Soucy G, Rivest S. Circulating cell wall components derived from gram-negative, not gram-positive, bacteria cause a profound induction of the gene-encoding Toll-like receptor 2 in the CNS. J Neurochem. 2001;79:648–657. doi: 10.1046/j.1471-4159.2001.00603.x. [DOI] [PubMed] [Google Scholar]
- Lambert MP, Barlow AK, Chromy BA, Edwards C, Freed R, Liosatos M, Morgan TE, Rozovsky I, Trommer B, Viola KL, Wals P, Zhang C, Finch CE, Krafft GA, Klein WL. Diffusible, nonfibrillar ligands derived from Abeta1–42 are potent central nervous system neurotoxins. Proc Natl Acad Sci USA. 1998;95:6448–6453. doi: 10.1073/pnas.95.11.6448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lesne S, Koh MT, Kotilinek L, Kayed R, Glabe CG, Yang A, Gallagher M, Ashe KH. A specific amyloid-beta protein assembly in the brain impairs memory. Nature. 2006;440:352–357. doi: 10.1038/nature04533. [DOI] [PubMed] [Google Scholar]
- Liu Y, Walter S, Stagi M, Cherny D, Letiembre M, Schulz-Schaeffer W, Heine H, Penke B, Neumann H, Fassbender K. LPS receptor (CD14): a receptor for phagocytosis of Alzheimer's amyloid peptide. Brain. 2005;128:1778–1789. doi: 10.1093/brain/awh531. [DOI] [PubMed] [Google Scholar]
- Lue LF, Kuo YM, Roher AE, Brachova L, Shen Y, Sue L, Beach T, Kurth JH, Rydel RE, Rogers J. Soluble amyloid beta peptide concentration as a predictor of synaptic change in Alzheimer's disease. Am J Pathol. 1999;155:853–862. doi: 10.1016/s0002-9440(10)65184-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackic JB, Stins M, McComb JG, Calero M, Ghiso J, Kim KS, Yan SD, Stern D, Schmidt AM, Frangione B, Zlokovic BV. Human blood-brain barrier receptors for Alzheimer's amyloid-beta 1–40. Asymmetrical binding, endocytosis, and transcytosis at the apical side of brain microvascular endothelial cell monolayer. J Clin Invest. 1998;102:734–743. doi: 10.1172/JCI2029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malm TM, Koistinaho M, Parepalo M, Vatanen T, Ooka A, Karlsson S, Koistinaho J. Bone-marrow-derived cells contribute to the recruitment of microglial cells in response to beta-amyloid deposition in APP/PS1 double transgenic Alzheimer mice. Neurobiol Dis. 2005;18:134–142. doi: 10.1016/j.nbd.2004.09.009. [DOI] [PubMed] [Google Scholar]
- Massengale M, Wagers AJ, Vogel H, Weissman IL. Hematopoietic cells maintain hematopoietic fates upon entering the brain. J Exp Med. 2005;201:1579–1589. doi: 10.1084/jem.20050030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLean CA, Cherny RA, Fraser FW, Fuller SJ, Smith MJ, Beyreuther K, Bush AI, Masters CL. Soluble pool of Abeta amyloid as a determinant of severity of neurodegeneration in Alzheimer's disease. Ann Neurol. 1999;46:860–866. doi: 10.1002/1531-8249(199912)46:6<860::aid-ana8>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- Morris RG, Moser EI, Riedel G, Martin SJ, Sandin J, Day M, O'Carroll C. Elements of a neurobiological theory of the hippocampus: the role of activity-dependent synaptic plasticity in memory. Philos Trans R Soc Lond B Biol Sci. 2003;358:773–786. doi: 10.1098/rstb.2002.1264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mucke L, Masliah E, Yu GQ, Mallory M, Rockenstein EM, Tatsuno G, Hu K, Kholodenko D, Johnson-Wood K, McConlogue L. High-level neuronal expression of Aβ1–42 in wild-type human amyloid protein precursor transgenic mice: synaptotoxicity without plaque formation. J Neurosci. 2000;20:4050–4058. doi: 10.1523/JNEUROSCI.20-11-04050.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naslund J, Haroutunian V, Mohs R, Davis KL, Davies P, Greengard P, Buxbaum JD. Correlation between elevated levels of amyloid beta-peptide in the brain and cognitive decline. JAMA. 2000;283:1571–1577. doi: 10.1001/jama.283.12.1571. [DOI] [PubMed] [Google Scholar]
- Postina R, Schroeder A, Dewachter I, Bohl J, Schmitt U, Kojro E, Prinzen C, Endres K, Hiemke C, Blessing M, Flamez P, Dequenne A, Godaux E, van Leuven F, Fahrenholz F. A disintegrin-metalloproteinase prevents amyloid plaque formation and hippocampal defects in an Alzheimer disease mouse model. J Clin Invest. 2004;113:1456–1464. doi: 10.1172/JCI20864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowan MJ, Klyubin I, Cullen WK, Anwyl R. Synaptic plasticity in animal models of early Alzheimer's disease. Philos Trans R Soc Lond B Biol Sci. 2003;358:821–828. doi: 10.1098/rstb.2002.1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sagare A, Deane R, Bell RD, Johnson B, Hamm K, Pendu R, Marky A, Lenting PJ, Wu Z, Zarcone T, Goate A, Mayo K, Perlmutter D, Coma M, Zhong Z, Zlokovic BV. Clearance of amyloid-beta by circulating lipoprotein receptors. Nat Med. 2007;13:1029–1031. doi: 10.1038/nm1635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selkoe DJ. Alzheimer's disease is a synaptic failure. Science. 2002;298:789–791. doi: 10.1126/science.1074069. [DOI] [PubMed] [Google Scholar]
- Shibata M, Yamada S, Kumar SR, Calero M, Bading J, Frangione B, Holtzman DM, Miller CA, Strickland DK, Ghiso J, Zlokovic BV. Clearance of Alzheimer's amyloid-ss(1–40) peptide from brain by LDL receptor-related protein-1 at the blood-brain barrier. J Clin Invest. 2000;106:1489–1499. doi: 10.1172/JCI10498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simard AR, Rivest S. Bone marrow stem cells have the ability to populate the entire central nervous system into fully differentiated parenchymal microglia. FASEB J. 2004;18:998–1000. doi: 10.1096/fj.04-1517fje. [DOI] [PubMed] [Google Scholar]
- Simard AR, Soulet D, Gowing G, Julien JP, Rivest S. Bone marrow-derived microglia play a critical role in restricting senile plaque formation in Alzheimer's disease. Neuron. 2006;49:489–502. doi: 10.1016/j.neuron.2006.01.022. [DOI] [PubMed] [Google Scholar]
- Snyder EM, Nong Y, Almeida CG, Paul S, Moran T, Choi EY, Nairn AC, Salter MW, Lombroso PJ, Gouras GK, Greengard P. Regulation of NMDA receptor trafficking by amyloid-beta. Nat Neurosci. 2005;8:1051–1058. doi: 10.1038/nn1503. [DOI] [PubMed] [Google Scholar]
- Tahara K, Kim HD, Jin JJ, Maxwell JA, Li L, Fukuchi K. Role of toll-like receptor signalling in Abeta uptake and clearance. Brain. 2006;129:3006–3019. doi: 10.1093/brain/awl249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsien JZ, Huerta PT, Tonegawa S. The essential role of hippocampal CA1 NMDA receptor-dependent synaptic plasticity in spatial memory. Cell. 1996;87:1327–1338. doi: 10.1016/s0092-8674(00)81827-9. [DOI] [PubMed] [Google Scholar]
- Ueberham U, Ueberham E, Bruckner MK, Seeger G, Gartner U, Gruschka H, Gebhardt R, Arendt T. Inducible neuronal expression of transgenic TGF-beta1 in vivo: dissection of short-term and long-term effects. Eur J Neurosci. 2005;22:50–64. doi: 10.1111/j.1460-9568.2005.04189.x. [DOI] [PubMed] [Google Scholar]
- Ulas J, Cotman CW. Decreased expression of N-methyl-d-aspartate receptor 1 messenger RNA in select regions of Alzheimer brain. Neuroscience. 1997;79:973–982. doi: 10.1016/s0306-4522(97)00023-7. [DOI] [PubMed] [Google Scholar]
- Verdier Y, Zarandi M, Penke B. Amyloid beta-peptide interactions with neuronal and glial cell plasma membrane: binding sites and implications for Alzheimer's disease. J Pept Sci. 2004;10:229–248. doi: 10.1002/psc.573. [DOI] [PubMed] [Google Scholar]
- Wyss-Coray T, Masliah E, Mallory M, McConlogue L, Johnson-Wood K, Lin C, Mucke L. Amyloidogenic role of cytokine TGF-beta1 in transgenic mice and in Alzheimer's disease. Nature. 1997;389:603–606. doi: 10.1038/39321. [DOI] [PubMed] [Google Scholar]