

Abstract
Interleukin (IL)-18 is an important regulator of innate and acquired immune responses. Here we show that both the IL-18 and IL-18 receptor (IL-18R), which are induced in spinal dorsal horn, are crucial for tactile allodynia after nerve injury. Nerve injury induced a striking increase in IL-18 and IL-18R expression in the dorsal horn, and IL-18 and IL-18R were upregulated in hyperactive microglia and astrocytes, respectively. The functional inhibition of IL-18 signaling pathways suppressed injury-induced tactile allodynia and decreased the phosphorylation of nuclear factor κB in spinal astrocytes and the induction of astroglial markers. Conversely, intrathecal injection of IL-18 induced behavioral, morphological, and biochemical changes similar to those observed after nerve injury. Our results indicate that IL-18-mediated microglia/astrocyte interactions in the spinal cord have a substantial role in the generation of tactile allodynia. Thus, blocking IL-18 signaling in glial cells might provide a fruitful strategy for treating neuropathic pain.
Keywords: microglia, astrocytes, neuropathic pain, IL-18, MAPK, NFκB
Introduction
Neuropathic pain is often caused by nerve injury or diseases such as diabetes, acquired immunodeficiency syndrome, or cancer, which damage peripheral nerves. A common symptom of neuropathic pain is tactile allodynia, which is characterized by painful responses to normally innocuous tactile stimuli. The currently available therapeutics for this type of chronic pain are relatively ineffective, and the underlying molecular mechanisms are primarily unknown. Previous studies have focused considerable attention on the directly damaged primary afferents and their influence on the activity of spinal dorsal horn neurons (Woolf and Salter, 2000; Scholz and Woolf, 2002). However, emerging evidence indicates that glial cells in the spinal cord also have a substantial role in the pathogenesis of neuropathic pain (Watkins et al., 2003; Marchand et al., 2005; Tsuda et al., 2005). Indeed, microglia and astrocytes are activated in the spinal dorsal horn after peripheral nerve injury. This spinal glial activation is likely involved in the production and release of pro-inflammatory cytokines, interleukin (IL)-1β, IL-6, and tumor necrosis factor-α (TNF-α), which may augment the nociceptive signals in the spinal cord (Watkins et al., 2003).
IL-18 is a member of the IL-1 family; IL-1β and IL-18 are related closely, and both require intracellular cysteine protease caspase-1 for biological activity (Okamura et al., 1995; Nakanishi et al., 2001; Boraschi and Dinarello, 2006). IL-18 signal transduction is initiated after the formation of a complex between the ligand binding the IL-18 receptor (IL-18R) α chain and the accessory IL-18R β chain. IL-18 is known to be upregulated in several human autoimmune and inflammatory diseases and therefore might represent a novel therapeutic target (Plater-Zyberk et al., 2001; Ten Hove et al., 2001; Carrascal et al., 2003; Boraschi and Dinarello, 2006). Recently, after nerve injury, matrix metalloproteases, but not caspase-1, were found to induce and maintain neuropathic pain through IL-1β cleavage and spinal glial activation (Kawasaki et al., 2008a). In contrast to IL-1β, there has been no study examining IL-18 signaling pathways in the spinal cord after nerve injury. Accumulating evidence indicates that nerve injury results in mitogen-activated protein kinase (MAPK) activation in spinal glial cells and that MAPK inhibitors alleviate nerve injury-induced pain hypersensitivity (Jin et al., 2003; Zhuang et al., 2005, 2006; Obata et al., 2007). Furthermore, nuclear factor κB (NFκB) pathways in spinal glial cells have been implicated in neuropathic pain (Ledeboer et al., 2005b; Meunier et al., 2007). However, it remains unknown how these transduction-related molecules are activated in microglia and astrocytes and contribute to pain hypersensitivity after nerve injury.
In this work, we set out to investigate whether IL-18 and IL-18Rs are induced in the spinal cord and participate in neuropathic pain using the L5 spinal nerve ligation (SNL) model (Kim and Chung, 1992). We now show that nerve injury induces an increase in IL-18 and IL-18R expression in microglia and astrocytes, respectively, in the dorsal horn and that the IL-18-mediated microglia/astrocyte interaction is crucial for the development and maintenance of tactile allodynia through glial cell-specific signal transduction cascades.
Materials and Methods
Animals.
Male Sprague Dawley rats weighing 200–250 g were used. Specific pathogen-free C57BL/6 mice, purchased from Clea Japan, were also used. IL-18−/− mice were backcrossed with C57BL/6 mice; F8 mice were used (Takeda et al., 1998). All mice weighed 25–30 g at the time of surgery. All procedures were approved by the Hyogo College of Medicine Committee on Animal Research and were performed in accordance with the National Institutes of Health guidelines on animal care.
Surgical procedures.
All procedures were performed with the rats and mice under pentobarbital anesthesia (50 mg/kg, i.p.). Additional doses of the anesthetics were given as needed. In all animals, no surgery was performed on the right side. Special care was taken to prevent infection and to minimize the influence of inflammation. The hair of the animal's lower back was shaved, and the skin was sterilized with 0.5% chlorhexidine and covered with clean paper. Sterile operating instruments were used. To produce an L5 SNL (Kim and Chung, 1992), a skin incision was made in the midline lumbar region (L4–S1). The L6 transverse process was identified, freed of muscular attachments, and partially removed with the help of bone ronguers. This exposed the L5 spinal nerve. The L5 ventral ramus was isolated and freed from the adjacent nerves, and then the L5 spinal nerve was tightly ligated with silk suture and transected distal to the ligature. After surgery, the wound was washed with saline and closed in layers (fascia and skin) with 3-0 silk thread. Animals were allowed to survive for 1, 3, 7, or 14 d after surgery (n = 4 for immunohistochemistry for each time point). In sham-operated animals, the nerve was exposed without ligation.
Drug treatments.
The intrathecal delivery of 10 μl of the Toll-like receptor 4 (TLR4) agonist lipopolysaccharide (LPS; 20 μg; Fluka), the p38 inhibitor SB203580 (1 or 10 μg; Calbiochem), IL-18 (50 or 500 ng, or 5 μg; R & D Systems), IL-1β (50 ng; R & D Systems), or the NFκB inhibitor SN50 (1 or 10 μg; Calbiochem) was performed as described previously (Fukuoka et al., 2001). Briefly, a laminectomy of the L5 vertebra was performed under adequate anesthesia. The dura was cut, a soft tube (Silascon; outer diameter, 0.64 mm; Kaneka Medix) was inserted into the subarachnoid space of the spinal cord, and the tip of the catheter was implanted at the L5 spinal segmental level. Most experiments were conducted at day 1 after LPS or IL-18 injection. PBS was used as the vehicle control. To obtain a sustained drug infusion, an ALZET osmotic pump (7 d pump, 1 μl/h; DURECT) was filled with normal goat IgG (100 ng/μl; R & D Systems), anti-IL-18 antibodies (10 or 100 ng/μl; R & D Systems), anti-IL-18R antibodies (10 or 100 ng/μl; R & D Systems), or IL-18-binding protein (IL-18BP; 5 or 50 ng/μl; R & D Systems) in PBS. The pump was sutured under the skin in the animal's lower back, and the associated catheter was implanted intrathecally 12 h before SNL or 7 d after SNL.
Small interfering RNA (siRNA) that targets TLR4 (TLR4 siRNA; 5′-GGAUCUUUCUUAUAACUAUtt-3′ and 5′-AUAGUUAUAAGAAAGAUCCac-3′) was designed using the target finder and design tool (Ambion). Negative controls were bioinformatically designed with the latest information about micro-RNA seed regions and sequence alignment algorithms to minimize interactions with any transcript in the transcriptomes of human, mouse, and rat. To obtain a sustained drug infusion, an ALZET osmotic pump (3 d pump, 1 μl/h; DURECT) was filled with TLR4 siRNA (10 pmol/μl) or negative siRNA (10 pmol/μl), and the associated catheter was implanted intrathecally 24 h before SNL. TLR4 siRNA or negative siRNA complexes were prepared immediately before administration by mixing the RNA solution (10 μm in nuclease-free water) with a transfection reagent, Lipofectamine RNAiMAX (Invitrogen), in a ratio of 1:4 (w:v).
Behavioral analysis.
All rats and mice were tested for mechanical hypersensitivity of the plantar surface of the hindpaw 1 d before SNL and 1, 3, 5, 7, 10, 12, or 14 d after SNL. Room temperature and humidity remained stable for all experiments. On each testing day, the rats and mice were brought into the behavior room 1 h before the test session to allow them to habituate to the environment. Mechanical hypersensitivity was assessed with a Dynamic Plantar Aesthesiometer (Ugo Basile), which is an automated von Frey-type system (Obata et al., 2005, 2007). To measure animal hindpaw mechanical thresholds, animals were placed in plastic cages with a wire mesh floor and allowed to acclimate for 15 min before each test session. A paw-flick response was elicited by applying an increasing force (measured in grams) focused on the middle of the plantar surface of the hindpaw. The force applied was initially below detection threshold and then increased from 1 to 5 or 50 g in 0.1 or 1 g steps over 20 s, then held at 5 or 50 g for an additional 10 s. The rate of force increase was 0.25 or 2.5 g/s. The threshold was taken as the force applied to elicit a reflex removal of the hindpaw. This was defined as the mean of three measurements at 1 min intervals. The variability between trials was ∼0.2 or 2 g.
Immunohistochemistry.
The rats were deeply anesthetized with sodium pentobarbital and perfused transcardially with 1% paraformaldehyde in 0.1 m phosphate buffer (PB), pH 7.4, followed by 4% paraformaldehyde in 0.1 m PB, 7 d after surgery (n = 4 at each time point). After the perfusion, the L5 spinal cord segments and L4/5 dorsal root ganglia (DRGs) were dissected out and postfixed in the same fixative for 12 h, then replaced with 20% sucrose overnight. Transverse spinal sections (free floating, 20 μm) and L4/5 DRGs (10 μm) were cut and processed for IL-18, IL-18R, neuronal-specific nuclear protein (NeuN), microtubule-associated protein 2 (MAP2), glial fibrillary acidic protein (GFAP), ionized calcium-binding adapter molecule 1 (Iba1), phosphorylated-p38 (p-p38), p-NFκB p65 at Ser-536, and ED-1 immunohistochemistry according to previously described methods (Noguchi et al., 1995). The polyclonal primary antibodies were used in the following dilutions: IL-18 (1:400; R & D Systems), IL-18R (1:100; R & D Systems), GFAP (1:1000; Millipore Bioscience Research Reagents), Iba1 (1:1000; Wako), p-p38 (1:1000; Cell Signaling Technology), and p-NFκB p65 at Ser-536 (1:100; Cell Signaling Technology). The monoclonal primary antibodies were used in the following dilutions: NeuN (1:2000; Millipore Bioscience Research Reagents), MAP2 (1:5000; Sigma), and ED-1 (1:400; Serotec). For the double immunofluorescent staining for IL-18 and NeuN, MAP2, GFAP, Iba1, p-p38, or p-NFκB, the tyramide signal amplification (PerkinElmer Life Sciences) fluorescence procedures (Michael et al., 1997) were used to detect staining for rabbit anti-IL-18 polyclonal antibody (1:10,000; R & D Systems). An image in a square (316 × 236 μm) on the medial two-thirds of the superficial dorsal horn (laminae I–III), as described previously (Molander et al., 1984), was captured under a 20× objective, and six to eight nonadjacent sections per each rat (each time point, n = 4) were randomly selected and measured with a computer-assisted imaging analysis system (NIH Image version 1.61). Quantitative assessment of immunostaining was performed by determining the immunofluorescence intensity within the fixed area of the medial superficial dorsal horn (laminae I–III), where most of the positive cells were found after our treatments. To assess immunofluorescence staining of cells quantitatively, the immunofluorescence intensity of the p-NFκB or GFAP was also determined as the average pixel intensity within each cell.
Western blotting.
Tissue samples from the L4–L6 spinal cord segments were lysed by homogenizing in 200 μl of lysis buffer containing 20 mm Tris-HCl, pH 8.0, 150 mm NaCl, 1 mm EDTA, 1% Igepal CA-630, 2 mm Na3VO4, 0.5 mm DTT, 1 mm PMSF, 1 μg/ml pepstatin, 5 μg/ml leupeptin, 9 μg/ml aprotinin, and 10% glycerol. Lysates were centrifuged at 14,400 × g for 60 min, and the concentration of protein in each sample (supernatant) was determined using Bio-Rad dye binding. Samples with equal amounts of protein were then separated by 10–20% PAGE, and the resolved proteins were electrotransferred to Hybond-P Nitrocellulose (GE Healthcare). Membranes were incubated with 5% nonfat milk in Tris buffer containing Tween 20 (TBST; 10 mm Tris-HCl, pH 8.0, 150 mm NaCl, and 0.2% Tween 20) for at least 10 min at room temperature and incubated with the polyclonal primary antibody for IL-18 (1:500; R & D Systems), TLR4 (1:400; Cell Signaling Technology), IL-18R (1:500; R & D Systems), p-NFκB (1:200; Cell Signaling Technology), total NFκB (1:100; Cell Signaling Technology), and the monoclonal primary antibody for β-actin (1:1000; Sigma) at 4°C overnight. Membranes were then washed twice with TBST and probed with rabbit anti-goat or goat anti-rabbit IgG conjugated with horseradish peroxidase (Vector Laboratories) at room temperature for 2 h. Membranes were finally washed several times with TBST to remove unbound secondary antibodies and visualized using enhanced chemiluminescence (Pierce). The density of specific bands was measured with a computer-assisted imaging analysis system (ATTO Densitograph version 4.02) and normalized against a loading control (β-actin or total NFκB).
In situ hybridization histochemistry.
For the in situ hybridization histochemistry (ISHH), the tissue was sectioned with a cryostat, thaw mounted onto Matsunami Adhesive Silan-coated glass slides (Matsunami), and stored at −80°C until ready for use. The procedure for ISHH was performed according to previously described methods (Yamanaka et al., 1999; Kobayashi et al., 2008). Briefly, the rat IL-18 and IL-18BP cRNA probes corresponding to nucleotides 75–526 and 44–557, respectively, were prepared. The sections were treated with 10 μg/ml proteinase K in 50 mm Tris-HCl and 5 mm EDTA for 3 min and acetylated with 0.25% acetic anhydride in 0.1 m triethanolamine; then, 35S-labeled RNA probe (5 × 106 cpm/ml) was placed on these sections overnight at 55°C. Hybridized sections were rinsed in 5× SSC and 5 mm DTT for 30 min at 65°C, washed in high-stringency buffer for 30 min at 65°C, and treated with 2 μg/ml RNase A for 30 min at 37°C. Sections were rinsed, dehydrated in an ascending ethanol series, and air dried. For autoradiography, the sections were coated with NTB-3 emulsion (Carestream Health), diluted 6:4 with distilled water at 45°C, and exposed for 8 weeks in light-tight boxes at 4°C. After development in D19 (Carestream Health) and fixation in 24% sodium thiosulfate, the sections were rinsed in distilled water, stained with hematoxylin–eosin, dehydrated in a graded ethanol series, and cleared in xylene.
Statistical analysis.
Differences in changes of values over time of each group were tested using one-way ANOVA, followed by individual post hoc comparisons (Fisher's exact test). One-way ANOVA, followed by individual post hoc comparisons (Fisher's exact test), was used to assess differences of values between the intrathecal groups. A difference was accepted as significant if p < 0.05.
Results
Peripheral nerve injury induces IL-18 upregulation in spinal microglia
To examine the distribution of IL-18 mRNA, we first performed ISHH on sections of the L5 spinal cord (Fig. 1A,B). IL-18 mRNA was detected sporadically in the spinal cord of naive rats, and these labeled nuclei were small in size and rather densely stained by hematoxylin. IL-18 mRNA signals dramatically increased only in the ipsilateral dorsal horn at day 7 after peripheral nerve injury. Next, the expression of IL-18 protein was examined by immunohistochemistry using anti-IL-18 antibody (Fig. 1C,D). Consistent with the results of the ISHH, very few IL-18-immunoreactive (IR) cells were found in the spinal dorsal horn of naive control rats and sham-operated rats. Three days after nerve injury, many more IL-18-IR cells were found in the ipsilateral than the contralateral dorsal horn, predominantly in the superficial dorsal horn (laminae I–III). The increase was first evident at day 1 after surgery and continued for 14 d. We also found that the level of IL-18 in the contralateral spinal cord was not different from that in naive rats (Fig. 1D). IL-18 upregulation after nerve injury was confirmed by Western blotting at day 7 after SNL (Fig. 1E). There was a significant increase in the level of IL-18 protein expression in the ipsilateral spinal cord from SNL rats compared with the naive control (by 173.7 ± 15.1%; n = 4; p < 0.001). The antibody against IL-18 displayed a distinct band on Western blots with an apparent molecular mass of 24 kDa, which corresponds to the size of pro-IL-18, consistent with a previous report (Hedtjärn et al., 2002). In the spinal cord tissues, the 18 kDa mature band was often too weak to be detected.
Figure 1.
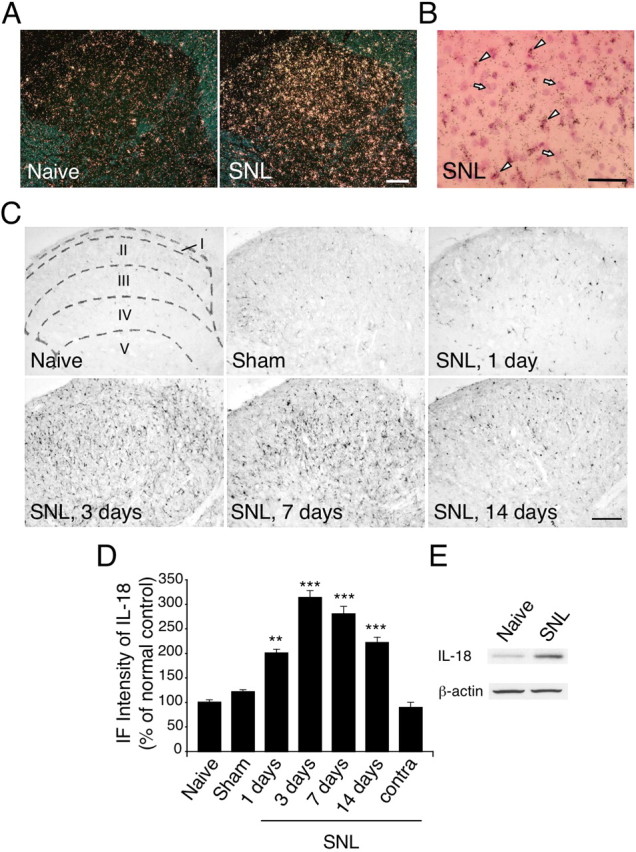
Nerve injury induces the upregulation of IL-18 in the spinal dorsal horn. A, Dark-field images of ISHH show IL-18 mRNA in the spinal dorsal horn of naive control rats and SNL rats. B, Higher-magnification photographs of laminae II–III of the dorsal horn under bright-field illumination in the SNL rats. The sections were stained with hematoxylin–eosin. The arrows and arrowheads indicate samples of the positively labeled glial cells and the negatively labeled neurons, respectively. C, IL-18 immunostaining in the dorsal horn of naive control rats and sham control rats at day 7 after surgery. Immunohistochemistry indicates a substantial increase in the number of IL-18-IR cells in the ipsilateral dorsal horn at days 1, 3, 7, and 14 after SNL. Sketches delineating boundaries of different laminae were superimposed over the spinal sections of naive control rats. D, Immunofluorescence (IF) intensity of IL-18 determined as the average pixel density in the ipsilateral and contralateral (contra) dorsal horn. Results are presented as percentages of the values normalized to the mean values of the naive control. Data represent mean ± SEM; n = 4 per group. **p < 0.01, compared with the naive control; ***p < 0.001, compared with the naive control. E, Western blot analysis reveals persistent IL-18 upregulation in the ipsilateral spinal cord at day 7 after SNL. Scale bars: A, C, 100 μm; B, 50 μm.
To identify the cell types that expressed the IL-18 after nerve injury, we performed double immunostaining of the IL-18 with several cell-specific markers: for neurons, NeuN and MAP2; for astrocytes, GFAP; for microglia, Iba1. We found that IL-18 did not colocalize with NeuN, MAP2, or GFAP in the dorsal horn at day 7 after surgery (Fig. 2A). Instead, the majority of the IL-18-IR cells were double labeled with Iba1 (Fig. 2B), indicating that IL-18 is induced in microglia, but not neurons or astrocytes, at day 7. Double immunofluorescence indicated that IL-18 was colocalized with Iba1, but not with GFAP, at days 1, 3, and 14 (data not shown). Iba1-IR cells displayed a hypertrophic morphology, a sign of microglial activation.
Figure 2.
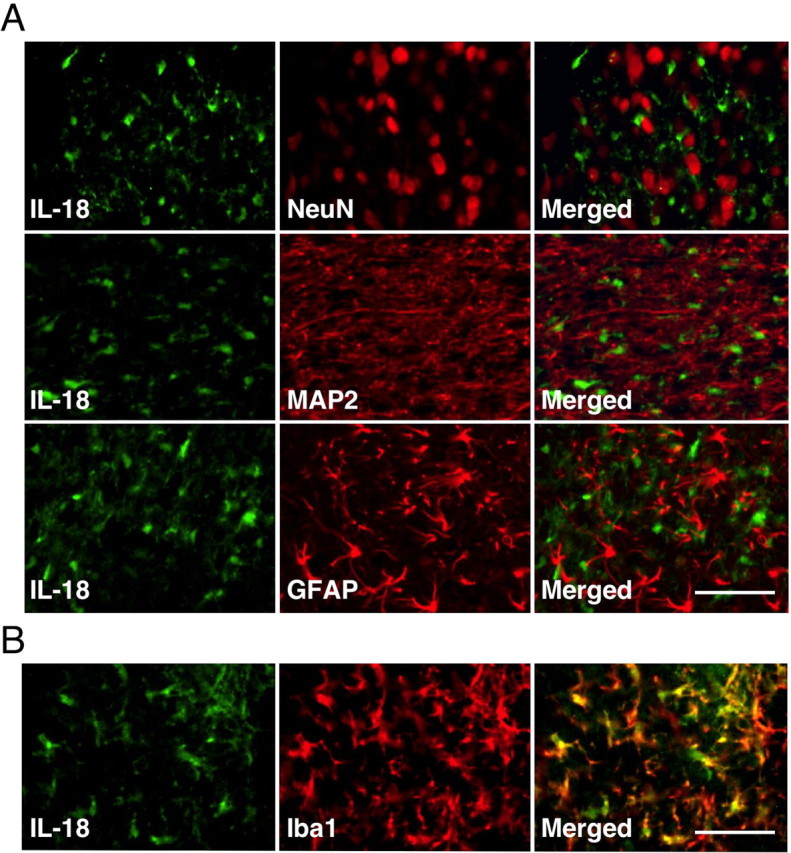
SNL induces IL-18 upregulation in spinal microglia, but not in neurons or astrocytes. A, Double immunostaining of green reaction product for IL-18 and red product for NeuN, MAP2, or GFAP in the ipsilateral dorsal horn at day 7 after surgery. Double staining of IL-18 with NeuN and MAP2, neuronal markers, shows no colocalization. Double labeling of IL-18 with GFAP, an astrocyte marker, indicates that IL-18 is not increased in spinal astrocytes. B, Double immunofluorescence shows that IL-18 (green) is colocalized with Iba1 (red) in the superficial dorsal horn (laminae I–III) at day 7 after SNL. Scale bars, 50 μm.
IL-18 upregulation in spinal microglia occurs through a TLR4/p38 MAPK pathway
TLRs play a key role in host defense during pathogen infection by regulating and linking innate and adaptive immune responses (Akira and Takeda, 2004; Akira et al., 2006; Marshak-Rothstein, 2006). Microglia, the resident myeloid cells that constitute the innate immune system in the CNS, express TLRs (Olson and Miller, 2004; Jack et al., 2005). A recent study has reported that TLR4 upregulation in spinal microglia contributes to nerve injury-induced pain states (Tanga et al., 2005). To investigate whether the expression of IL-18 in spinal microglia is regulated by TLR4, we injected the TLR4 agonist, LPS, intrathecally into naive rats. One day after injection, the number and intensity of Iba1-IR cells in the dorsal horn was significantly higher in the LPS group than in the vehicle control group (Fig. 3A,B). The activation of p38 MAPK can cause microglial activation in the brain (Koistinaho and Koistinaho, 2002), and p38 MAPK activation appears to regulate the expression of pro-inflammatory cytokines (Ji and Strichartz, 2004). We found that the selective p38 MAPK inhibitor SB203580 dose-dependently blocked the LPS-induced microglial activation. Upregulation of IL-18 was also observed after LPS injection, and this increase in IL-18 expression was abolished by SB203580 (Fig. 3C). IL-18 was mainly expressed in p-p38-IR cells at day 7 after nerve injury (Fig. 3D). Thus, these findings suggest that nerve injury increases the expression of IL-18 in activated microglia through a TLR4/p38 MAPK pathway.
Figure 3.
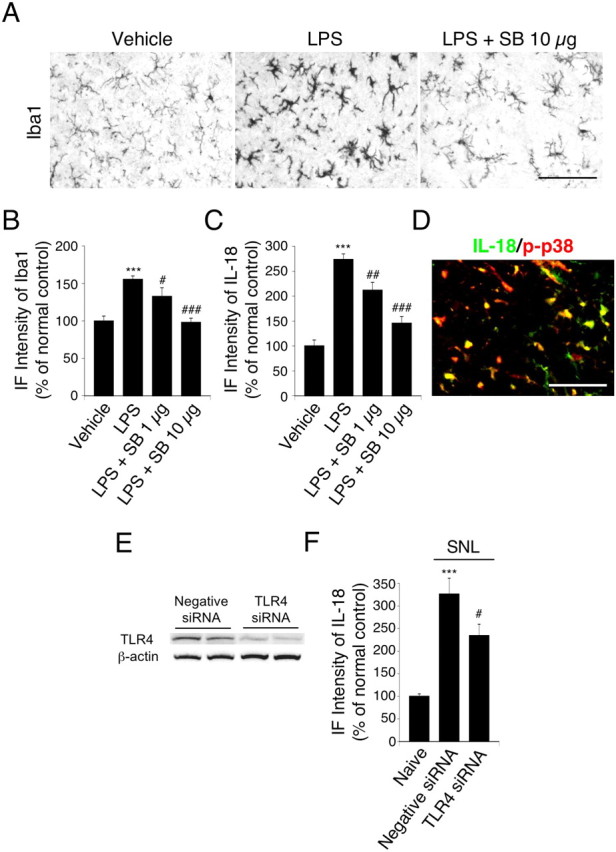
Upregulation of spinal microglial markers and IL-18 by the TLR4 agonist LPS through the p38 MAPK pathway. A, Iba1 immunostaining in the dorsal horn at day 1 after LPS injection. B, C, Immunofluorescence (IF) intensity of Iba1 and IL-18 determined as the average pixel density in the dorsal horn. For control experiments, PBS was used. Data represent mean ± SEM; n = 4 per group. ***p < 0.001, compared with the vehicle control group; #p < 0.05, compared with the LPS group; ##p < 0.01, compared with the LPS group; ###p < 0.001, compared with the LPS group. D, Double immunofluorescence shows that IL-18 (green) is colocalized with p-p38 MAPK (red) in the superficial dorsal horn (laminae I–III) at day 7 after nerve injury. Two single-stained images were merged. Scale bar, 50 μm. E, Protein expression of TLR4 in the ipsilateral spinal cord at day 3 after L5 SNL, as detected by Western blotting. F, IF intensity of IL-18 determined as the average pixel density in the ipsilateral and contralateral dorsal horn. Results are presented as percentages of the values normalized to the mean values of the naive control. Data represent mean ± SEM; n = 4 per group. ***p < 0.001, compared with the naive control; #p < 0.05, compared with the negative siRNA group. SB, SB203580.
To determine whether TLR4 activation is required for IL-18 upregulation in the spinal cord after nerve injury, we assessed the effects of TLR4 RNA interference (RNAi) on expression of IL-18 in the spinal cord. Compared with the rats receiving control siRNAs, a significant reduction of TLR4 proteins was found in the spinal cord at day 3 after the TLR4 siRNA transfer (Fig. 3E). We also found that the level of IL-18 in the ipsilateral spinal cord of the TLR4 siRNA-treated rats was significantly lower than that in the negative siRNA-treated rats (Fig. 3F). Furthermore, knockdown of TLR4 by RNAi in the spinal cord attenuated tactile allodynia after nerve injury (data not shown), consistent with a previous report (Tanga et al., 2005).
IL-18 blockade attenuates nerve injury-induced tactile allodynia
Our results suggest that upregulation of IL-18 in spinal microglia is likely to have an important role in mechanical hypersensitivity after nerve injury. We therefore predicted that suppressing IL-18 in the spinal cord should prevent nerve injury-induced mechanical hypersensitivity. To test this, anti-IL-18 antibodies were delivered intrathecally before surgery and maintained for 7 d via a catheter, the tip of which was implanted at the lumber enlargement. To obtain a sustained drug infusion, the drug was delivered by an osmotic pump connected to a catheter. We found that anti-IL-18 antibody significantly inhibited the injury-induced tactile allodynia at days 1, 3, 5, and 7 after surgery (Fig. 4A). However, intrathecal administration of anti-IL-18 produced no significant changes in the threshold on the contralateral paw (Fig. 4B).
Figure 4.

IL-18 blockade attenuates tactile allodynia induced by peripheral nerve injury. A, Pretreatment with anti-IL-18 antibodies (10 or 100 ng · μl−1 · h−1) attenuated mechanical hypersensitivity induced by nerve injury. Tactile allodynia was determined with a Dynamic Plantar Aesthesiometer at days 1, 3, 5, and 7 after nerve injury. B, Pretreatment with anti-IL-18 antibodies did not change the threshold on the contralateral hindpaw. For control experiments, either PBS or IgG was used. BL, Baseline; Ab, antibody. Data represent mean ± SEM; n = 8 per group. #p < 0.05, compared with the vehicle control group; ##p < 0.01, compared with the vehicle control group; ###p < 0.001, compared with the vehicle control group.
Peripheral nerve injury induces IL-18R upregulation in spinal astrocytes, and IL-18R blockade alleviates nerve injury-induced tactile allodynia
We next investigated the changes in IL-18R expression using an anti-IL-18R antibody. We first performed immunohistochemistry on sections of the L5 spinal cord (Fig. 5A). Very few IL-18R-IR cells were found in the spinal dorsal horn of naive control rats and sham-operated rats. IL-18R levels started to increase in the ipsilateral dorsal horn at day 3 after SNL (Fig. 5B). Many more IL-18R-IR cells were found in the ipsilateral rather than the contralateral dorsal horn, predominantly in the superficial dorsal horn at day 7 after nerve injury. We also found that the level of IL-18R in the contralateral spinal cord was not different from that in naive rats (Fig. 5B). IL-18R upregulation after nerve injury was confirmed by Western blot analysis (Fig. 5C). Western blot analysis revealed the temporal pattern of IL-18R expression based on density changes of the single band at 63 kDa. Consistent with the results obtained by immunohistochemistry, nerve injury induced a 100.2 ± 4.3% increase in IL-18R levels in the ipsilateral spinal cord at day 7, compared with the naive control (n = 4; p < 0.001). Furthermore, double immunostaining showed that IL-18R heavily colocalized with GFAP at day 7 after nerve injury (Fig. 5D). These IL-18R-IR cells in the dorsal horn were predominantly astrocytes, because they coexpressed GFAP but not NeuN, MAP2, or Iba1 (Fig. 5E), throughout the 2 week time course of the experiment (data not shown). Indeed, IL-18-IR and IL-18R-IR cells were clearly distinguishable at day 7 after SNL (Fig. 5E). GFAP-IR cells displayed a hypertrophic morphology, a sign of astroglial activation. We also found the anatomical proximity of IL-18 and IL-18R proteins, indicating probable communication between activated microglia and astrocytes (Fig. 5E).
Figure 5.
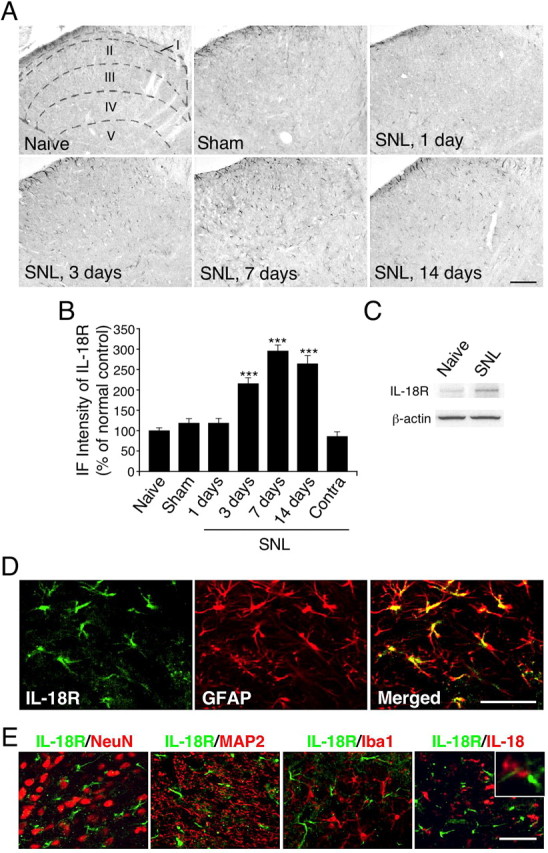
Nerve injury induces the upregulation of the IL-18R in astrocytes in the spinal dorsal horn. A, IL-18R immunostaining in the dorsal horn of naive control rats and sham control rats at day 7. Immunohistochemistry indicates a substantial increase in the number of IL-18R-IR cells in the ipsilateral dorsal horn at days 3, 7, and 14 after SNL. Sketches delineating boundaries of different laminae were superimposed over the spinal sections of naive control rats. B, Immunofluorescence (IF) intensity of IL-18R determined as the average pixel density in the ipsilateral and contralateral (Contra) dorsal horn. Results are presented as percentages of the values normalized to the mean values of the naive control. Data represent mean ± SEM; n = 4 per group. ***p < 0.001, compared with the naive control. C, Western blot analysis reveals persistent IL-18R upregulation in the ipsilateral spinal cord at day 7. D, Double immunofluorescence shows that IL-18R (green) is colocalized with GFAP (red) in the dorsal horn (laminae I–III) at day 7. E, Double immunostaining of green reaction product for IL-18R and red product for NeuN, MAP2, Iba1, or IL-18 in the ipsilateral dorsal horn at day 7. Two single-stained images were merged. The inset shows the anatomical proximity of IL-18 and IL-18R immunoreactivity. Scale bars: A, 100 μm; D, E, 50 μm.
To elucidate whether blocking the IL-18R in the spinal cord inhibits nerve injury-induced mechanical hypersensitivity, rats with nerve injury were intrathecally treated with anti-IL-18R antibodies (10 or 100 ng · μl−1 · h−1). We found that anti-IL-18R treatment diminished SNL-induced mechanical hypersensitivity at days 1, 3, 5, and 7 after nerve injury (Fig. 6A), whereas intrathecal anti-IL-18R injection produced no significant changes in the threshold on the contralateral paw (Fig. 6B).
Figure 6.

IL-18R blockade attenuates tactile allodynia induced by peripheral nerve injury. A, Pretreatment with anti-IL-18R antibodies (10 or 100 ng · μl−1 · h−1) attenuated mechanical hypersensitivity induced by nerve injury. Tactile allodynia was determined with a Dynamic Plantar Aesthesiometer at days 1, 3, 5, and 7 after nerve injury. B, Pretreatment with anti-IL-18R antibodies did not change the threshold on the contralateral hindpaw. For control experiments, either PBS or IgG was used. BL, Baseline; Ab, antibody. Data represent mean ± SEM; n = 8 per group. #p < 0.05, compared with the vehicle control group; ##p < 0.01, compared with the vehicle control group; ###p < 0.001 compared with the vehicle control group.
IL-18 and IL-18R blockade reduces the phosphorylation of NFκB in spinal astrocytes, and the induction of astroglial markers caused by nerve injury
Increasing evidence shows that NFκB activation in spinal glial cells contributes to the generation of neuropathic pain (Ledeboer et al., 2005b; Meunier et al., 2007). Recent studies have shown that posttranslational modification of NFκB subunits, such as p65, contributes significantly to NFκB transactivation potential (Ghosh and Karin, 2002). Phosphorylation of p65 at Ser-536 is proposed to be a key modification that potentiates p65 transactivation function, hence NFκB activation. We therefore performed immunohistochemical staining of NFκB phosphorylation using an anti-p-NFκB p65 at Ser-536 antibody. SNL induced an increase in NFκB phosphorylation in the ipsilateral dorsal horn at day 3 (Fig. 7A,B), consistent with the induction of IL-18R (Fig. 5A,B). These p-NFκB-IR cells were distributed in the superficial dorsal horn throughout the 2 week time course. We also found that the level of p-NFκB in the contralateral spinal cord was not different from that in naive rats (Fig. 7B). The Western blot analysis revealed NFκB activation in the ipsilateral spinal cord at day 7 (Fig. 7C). There was a significant increase in the level of NFκB phosphorylation in the ipsilateral spinal cord from SNL rats compared with the naive control (by 44.4 ± 3.9%; n = 4; p < 0.001). Double labeling showed that most of the p-NFκB-IR signals were colocalized with GFAP, indicating that NFκB is activated in astrocytes (Fig. 7D). Next, to ascertain whether NFκB phosphorylation in spinal astrocytes is regulated by IL-18R, we performed double labeling with p-NFκB and IL-18R. We found that p-NFκB coexpressed with the IL-18R at day 7 after SNL (Fig. 7E).
Figure 7.
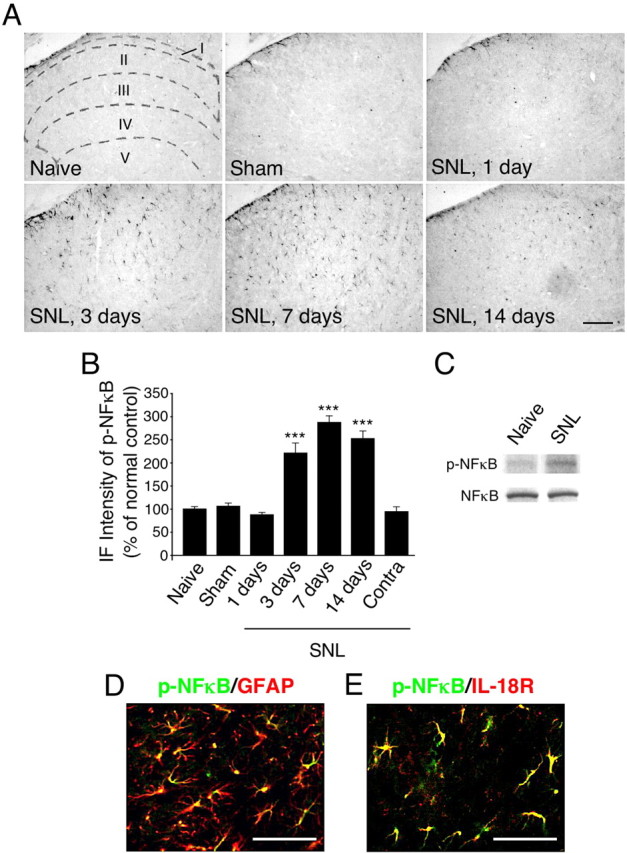
Nerve injury induces the activation of NFκB in astrocytes in the spinal dorsal horn. A, p-NFκB immunostaining in the dorsal horn of naive control rats and sham control rats at day 7. Immunohistochemistry indicates a substantial increase in the number of p-NFκB-IR cells in the ipsilateral dorsal horn at days 3, 7, and 14. Sketches delineating boundaries of different laminae were superimposed over the spinal sections of naive control rats. B, Immunofluorescence (IF) intensity of p-NFκB determined as the average pixel density in the ipsilateral and contralateral (Contra) dorsal horn. Results are presented as percentages of the values normalized to the mean values of the naive control. Data represent mean ± SEM; n = 4 per group. ***p < 0.001, compared with the naive control. C, Western blot analysis reveals persistent NFκB activation in the ipsilateral spinal cord at day 7 after nerve injury. D, E, Double immunofluorescence shows that p-NFκB (green) is colocalized with GFAP or IL-18R (red) in the dorsal horn (laminae I–III) at day 7. Two single-stained images were merged. Scale bars: A, 100 μm; D, E, 50 μm.
To investigate the effect of IL-18 on nerve injury-induced NFκB activation in the spinal cord, the levels of p-NFκB were compared in the vehicle, anti-IL-18, and anti-IL-18R groups (Fig. 8A,B). We found that both anti-IL-18 and anti-IL-18R antibodies inhibited the injury-induced increase in NFκB phosphorylation at day 7 after SNL. These data suggest that activation of NFκB in astrocytes participates in tactile allodynia through IL-18R. We further assessed the effect of anti-IL-18 and anti-IL-18R on nerve injury-induced upregulation of astroglial markers. We found that the intensity of GFAP-IR cells in the ipsilateral dorsal horn increased strikingly at day 7 after surgery, but treatment with both anti-IL-18 and anti-IL-18R blocked this increase (Fig. 8A,C). In addition, the immunofluorescence intensity of p-NFkB and GFAP in individual cells in the dorsal horn of anti-IL-18- and anti-IL-18R-treated rats was significantly lower than in those of vehicle-treated rats (Fig. 8D,E).
Figure 8.
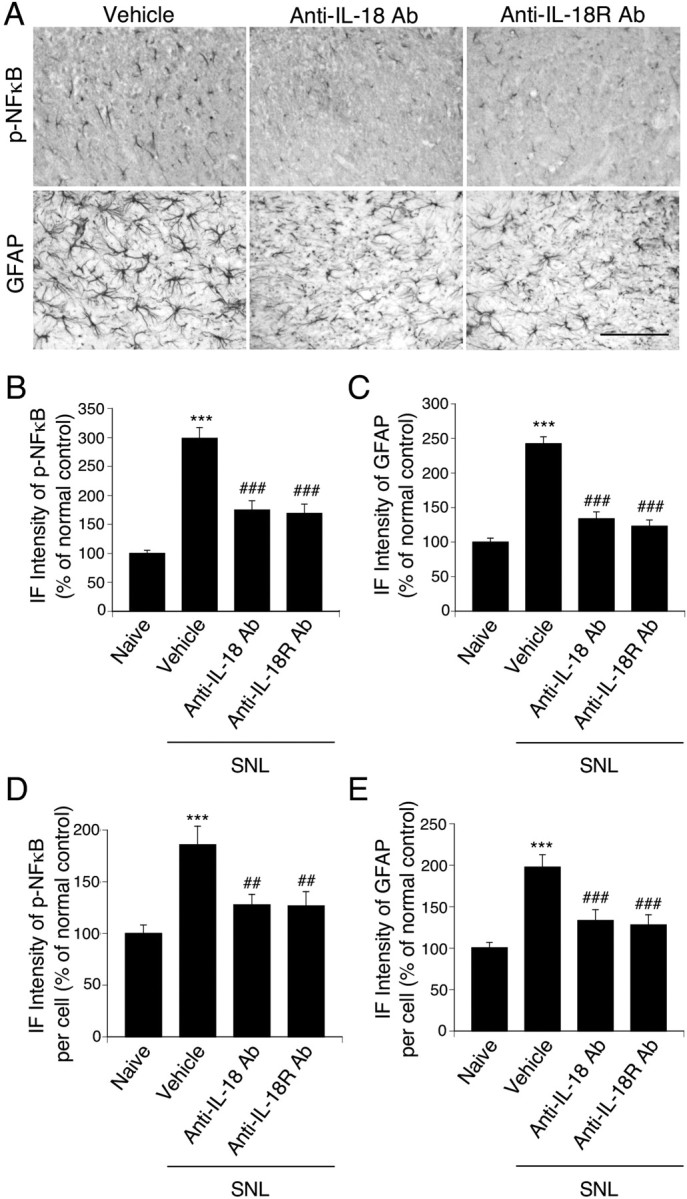
Effects of anti-IL-18 and anti-IL-18R antibodies on the activation of NFκB and the induction of spinal astroglial markers induced by nerve injury. A, p-NFκB and GFAP immunostaining in the superficial dorsal horn at day 7 after surgery. Both anti-IL-18 and anti-IL-18R antibodies suppressed the activation of NFκB and the upregulation of GFAP caused by nerve injury. Scale bar, 100 μm. B, C, Immunofluorescence (IF) intensity of p-NFκB and GFAP determined as the average pixel density in the ipsilateral dorsal horn. D, E, IF intensity of p-NFkB and GFAP in an individual cell determined as the average pixel density in the ipsilateral dorsal horn. For control experiments, PBS was used. Data represent mean ± SEM; n = 4 per group. ***p < 0.001, compared with the naive control; ##p < 0.01, compared with the vehicle control group; ###p < 0.001, compared with the vehicle control group. Ab, Antibody.
Intrathecal IL-18 injection produces tactile allodynia and astroglial activation
To further assess the effects of IL-18 on pain behaviors, we injected IL-18 intrathecally into naive rats. A single intrathecal injection of IL-18 produced tactile allodynia at day 1 in a dose-dependent manner (Fig. 9A), and this allodynia persisted for 2 d (Fig. 9B). The selective NFκB inhibitor, SN50, dose-dependently blocked the IL-18-induced allodynia at day 1 (Fig. 9C).
Figure 9.

Induction of behavioral allodynia and spinal astroglial markers by IL-18 through the NFκB pathway. A, Intrathecal administration of IL-18 induced tactile allodynia at day 1 in a dose-dependent manner. *p < 0.05, compared with the vehicle control group; **p < 0.01, compared with the vehicle control group. B, Time course for IL-18-induced tactile allodynia. **p < 0.01, compared with the pre-operative value; ***p < 0.001, compared with the preoperative value. C, Attenuation of IL-18 action by pretreatment with a NFκB inhibitor, SN50. **p < 0.01, compared with the vehicle control group; ###p < 0.001, compared with the IL-18 group. D, Spinal injection of both IL-18 and IL-1β (50 ng) induced tactile allodynia at 3 and 6 h after injection. *p < 0.05, compared with the preoperative value; ***p < 0.001, compared with the preoperative value. For control experiments, PBS was used. Data represent mean ± SEM; n = 8 per group. BL, Baseline.
To determine whether a low dose of IL-18 (50 ng) produces nociceptive behavior, we examined mechanical sensitivity at earlier time points (e.g., at 3 or 6 h). Injection of IL-18, as well as IL-1β (50 ng), induced marked tactile allodynia at 3 and 6 h after injection (Fig. 9D).
Intrathecal injection of IL-18 (5 μg) caused a 107.1 ± 15.1% increase in the expression of p-NFκB in the spinal dorsal horn at day 1 after injection compared with the vehicle control group (n = 4; p < 0.001). Upregulation of GFAP and Iba1 was also observed after IL-18 injection. There was a significant increase in the expression of GFAP and Iba1 in the spinal dorsal horn from IL-18-treated rats compared with the vehicle control group (by 81.0 ± 6.8 and 28.8 ± 6.7%, respectively; n = 4; p < 0.01). Therefore, these data suggest that IL-18 upregulation in the spinal cord was sufficient to produce tactile allodynia, as well as glial activation, in otherwise naive rats.
Blockade of the IL-18 signaling reverses nerve injury-induced tactile allodynia
IL-18BP is a naturally occurring inhibitor of IL-18 activity (Novick et al., 1999; Kim et al., 2000). IL-18 has a greater affinity for IL-18BP than for IL-18R. Neutralizing IL-18 with IL-18BP reduces inflammation, including hepatic metastasis and systemic or local inflammation (Plater-Zyberk et al., 2001; Ten Hove et al., 2001; Carrascal et al., 2003; Boraschi and Dinarello, 2006). To examine the distribution of IL-18BP mRNA, we first performed ISHH on sections of the L5 spinal cord (Fig. 10A,B). IL-18BP mRNA was detected sporadically in the spinal cord of naive rats. IL-18BP mRNA signals did not change in the ipsilateral dorsal horn 7 d after nerve injury. We then administrated IL-18BP (5 or 50 ng · μl−1 · h−1) intrathecally to SNL rats (Fig. 10C). We found that IL-18BP dose-dependently inhibited nerve injury-induced tactile allodynia at days 1, 3, 5, and 7 after SNL. The threshold on the contralateral paw did not vary significantly from baseline values throughout the period we studied (Fig. 10D).
Figure 10.
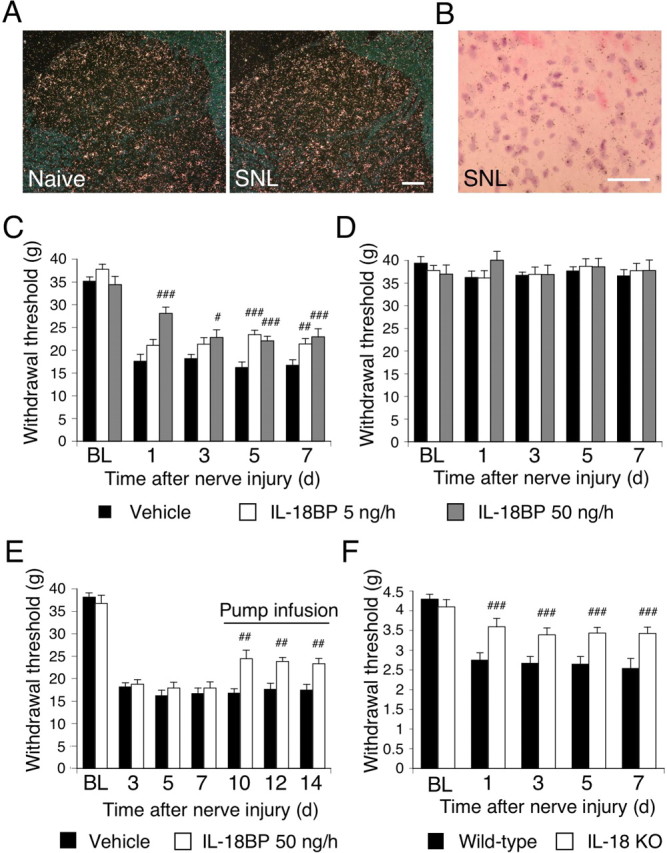
IL-18 signaling contributes to the development and maintenance of tactile allodynia after nerve injury. A, Dark-field images of ISHH show IL-18BP mRNA in the spinal dorsal horn of naive control rats and SNL rats. Scale bar, 100 μm. B, Higher-magnification photographs of laminae II–III of the dorsal horn under bright-field illumination in the SNL rats. Scale bar, 50 μm. C, Pretreatment with IL-18BP (5 or 50 ng · μl−1 · h−1) attenuated mechanical hypersensitivity induced by nerve injury. Tactile allodynia was determined with a Dynamic Plantar Aesthesiometer at days 1, 3, 5, and 7. #p < 0.05, compared with the vehicle control group; ##p < 0.01, compared with the vehicle control group; ###p < 0.001, compared with the vehicle control group. D, Pretreatment with IL-18BP did not change the threshold on the contralateral hindpaw. E, Posttreatment with IL-18BP reversed nerve injury-induced tactile allodynia. IL-18BP was delivered intrathecally via an osmotic pump starting on post-SNL day 7. ##p < 0.01, compared with the vehicle control group. F, Mechanical hypersensitivity was significantly attenuated at days 1, 3, 5, and 7 after nerve injury in IL-18 knock-out (KO) mice relative to wild-type mice. ###p < 0.001, compared with the wild-type control. For control experiments, PBS was used. Data represent mean ± SEM; n = 8 per group. BL, Baseline.
Several reports have shown that inhibition of spinal glial activation attenuates the existing hypersensitivity in a rat model of neuropathy (Katsura et al., 2006, 2008; Zhuang et al., 2006). To investigate whether inhibition of IL-18 would reverse established mechanical hypersensitivity, a treatment mode more relevant to a clinical situation, we infused IL-18BP intrathecally via an osmotic pump at day 7 after establishment of SNL-induced mechanical hypersensitivity (Fig. 10E). This treatment effectively reversed the nerve injury-induced pain hypersensitivity at days 10, 12, and 14 after surgery. We also observed that IL-18-deficient mice displayed significantly attenuated tactile allodynia at days 1, 3, 5, and 7 after surgery compared with the wild-type controls (Fig. 10F). In addition, we found that the injury-induced increase in GFAP and Iba1 expression in the ipsilateral dorsal horn was reduced in IL-18-deficient mice compared with the wild-type controls (by 34.8 ± 4.9 and 37.4 ± 7.0%, respectively; n = 4; p < 0.001). These results suggest that the IL-18 signaling cascade in spinal glial cells might have a role in the development and maintenance of tactile allodynia caused by nerve injury.
To further investigate the effects of IL-18 in the spinal cord on pain behaviors, we injected IL-18 (500 ng) intrathecally into IL-18-deficient mice. IL-18-deficient mice treated with IL-18 showed a decrease in their mechanical nociceptive thresholds at day 1 (vehicle-treated mice, 4.25 ± 0.16 g vs IL-18-treated mice, 2.88 ± 0.23 g; n = 8; p < 0.001).
Nerve injury induces IL-18 upregulation in macrophages in the primary afferents
The L5 SNL model, which is one of the most popular models of neuropathic pain, is unique because the L4 DRG neurons are clearly separated from the axotomized L5 neurons (Fukuoka et al., 2001; Obata et al., 2005). Therefore, we first examined IL-18 and IL-18R immunoreactivity in the injured L5 DRG after L5 SNL (Fig. 11). L5 SNL induced an increased number of IL-18-IR cells in the L5 DRG from the first to 14th day, and furthermore, these IL-18-IR cells included macrophages labeled for ED-1. IL-18 and ED-1 immunoreactivity was never detected in the uninjured L4 DRG, and L5 SNL did not induce an increase in IL-18R, neither in the L4 DRG nor the L5 DRG (data not shown).
Figure 11.

Nerve injury induces the upregulation of IL-18 in macrophages in injured L5 DRG. A, IL-18 immunostaining in the naive DRG and the ipsilateral and contralateral (Contra) L5 DRG at days 1, 3, 7, and 14 after L5 SNL. B, Double immunostaining of green reaction product for IL-18 and red product for macrophage marker ED-1 in the L5 DRG at day 7. Scale bars, 100 μm.
Discussion
There is accumulating evidence supporting a role for spinal microglia in the pathogenesis of nerve injury-induced pain hypersensitivity. For example, the ATP receptor P2X4 and the chemokine receptor CX3CR1 are increased in spinal microglia, and blocking these receptors results in decreased neuropathic pain (Tsuda et al., 2003; Verge et al., 2004). Furthermore, the chemokine receptor CCR2 and the cannabinoid receptor CB2 are also expressed in spinal microglia and contribute to tactile allodynia after nerve injury (Abbadie et al., 2003; Zhang et al., 2003). In the present study, we found that the expression of IL-18 increased in the spinal cord microglia after nerve injury. MAPK activation appears to regulate the expression of pro-inflammatory cytokines, including IL-1β, IL-6, and TNF-α, as well as cyclooxygenase-2 (COX-2) and inducible nitric oxide synthase (Koistinaho and Koistinaho, 2002; Ji and Strichartz, 2004). Because the activation of p38 MAPK in spinal microglia has a critical role in the induction of neuropathic pain (Jin et al., 2003; Clark et al., 2007; Kobayashi et al., 2008), these findings suggest that nerve injury induces IL-18 upregulation in spinal cord microglia via p38 activation, and thus increasing pain hypersensitivity. Indeed, we found that p38 MAPK was activated in IL-18-expressing cells after nerve injury.
There is growing appreciation that signaling through TLRs, which is key to generating innate responses to infection, may have pathogen-independent roles, by initiating responses to host-derived, endogenous ligands (Akira and Takeda, 2004; Akira et al., 2006; Marshak-Rothstein, 2006). Intriguingly, the TLR family has been implicated in the microglial response to nerve damage (Block et al., 2007); peripheral nerve injury induces TLR4 upregulation in spinal microglia, and TLR4-deficient mice do not develop tactile allodynia after nerve injury (Tanga et al., 2005). In the present study, we found that injection of the TLR4 agonist LPS increased IL-18 expression in spinal cord microglia. Furthermore, a p38 MAPK inhibitor, SB203580, diminished this LPS-induced upregulation of Iba1 and IL-18 expression. Because knockdown of TLR4 by RNAi reduced IL-18 upregulation induced by nerve injury, our findings suggest that after nerve injury, microglial activation might occur through the TLR4/p38 MAPK signaling pathway and lead to the enhanced expression of IL-18. One specific example of a TLR4 ligand released after nerve injury are saturated fatty acids, which have been shown to induce the upregulation of COX-2 and other inflammatory markers after interacting with membrane-bound TLR4 of monocyte/macrophage such as microglia (Hwang, 2001).
Not only microglia, but also astrocytes, are activated in the spinal cord after nerve injury, and these activated astrocytes participate in the maintenance of neuropathic pain (Watkins and Maier, 2003; Marchand et al., 2005; Tsuda et al., 2005). For example, the serine protease tissue plasminogen activator and the calcium-binding peptide S100β are selectively expressed in activated astrocytes in the spinal dorsal horn and contribute to nerve injury-induced mechanical hypersensitivity (Tanga et al., 2006; Kozai et al., 2007). Furthermore, mice overexpressing CCL2 (formerly monocyte chemoattractant protein-1) in astrocytes display enhanced nociceptive responses (Menetski et al., 2007). In the present study, we found that the IL-18R increased in spinal astrocytes after nerve injury. Furthermore, both anti-IL-18 and anti-IL-18R antibodies diminished the nerve injury-induced tactile allodynia and reduced the phosphorylation of NFκB in spinal astrocytes and the induction of astroglial markers caused by nerve injury. Conversely, intrathecal injection of IL-18 induced both tactile allodynia and astroglial activation. Because the activation of NFκB in spinal astrocytes is involved in the maintenance of mechanical hypersensitivity (Ledeboer et al., 2005b; Meunier et al., 2007), these findings suggest that nerve injury induces NFκB activation in spinal astrocytes via the IL-18R and that the activation of the IL-18R/NFκB signaling cascade in astrocytes contributes to hypersensitivity to innocuous mechanical stimulation. An unexpected finding in the present study was that anti-IL-18R treatment diminished SNL-induced mechanical hypersensitivity at day 1, although IL-18R levels started to increase at day 3. Because upregulation of Iba1 was also observed after IL-18 injection, and Iba1 expression was reduced in injured IL-18-deficient mice, we cannot exclude the possibility that IL-18R is expressed in not only astrocytes but also microglia (Prinz and Hanisch, 1999).
Based on the findings described in the present study, we hypothesize the molecular events leading to neuroinflammation in the spinal dorsal horn to be the following (Fig. 12). First, TLR4 triggers microglial activation to synthesize IL-18 through the activation of p38 MAPK. The IL-18 produced by these activated microglia then stimulates IL-18R on neighboring astrocytes in a paracrine manner and upregulates the expression of IL-18R on these astrocytes. The activation of IL-18R increases the phosphorylation of NFκB in the astrocytes to induce hypertrophy of these cells. The present study also demonstrated that IL-18R was delayed in its enhanced expression but remained elevated after nerve injury. A recent report has demonstrated that both genetically altered mice and rats treated with TLR4 antisense oligodeoxynucleotide displayed significantly decreased expression of not only microglial markers but also astroglial markers in the spinal cord, although TLR4 is expressed predominantly in microglia, not astrocytes (Tanga et al., 2005). Together, these findings suggest that activated microglia in the spinal dorsal horn may be directly responsible for the induction of astroglial activation after nerve injury. Alternatively, IL-18 and other cytokines, produced and released by activated astrocytes, might be involved in microglial activation (Fig. 12).
Figure 12.

Proposed model of IL-18-mediated microglia/astrocyte interaction in the spinal cord. First, TLR4 triggers microglial activation through the p38 MAPK pathway  . IL-18 produced by the TLR4/p38 MAPK signaling cascade in activated microglia stimulates IL-18R on astrocytes in a paracrine manner
. IL-18 produced by the TLR4/p38 MAPK signaling cascade in activated microglia stimulates IL-18R on astrocytes in a paracrine manner  . IL-18 binding to IL-18R increases NFkB phosphorylation in astrocytes and causes GFAP upregulation
. IL-18 binding to IL-18R increases NFkB phosphorylation in astrocytes and causes GFAP upregulation  . IL-18R expression is upregulated by IL-18R stimulation, thus accelerating this signaling pathway in astrocytes
. IL-18R expression is upregulated by IL-18R stimulation, thus accelerating this signaling pathway in astrocytes  . IL-18 or other cytokines might stimulate microglia and astrocytes in an autocrine or paracrine manner and induce production of pro-inflammatory molecules, such as IL-1β, IL-6, and TNF-α, as well as COX-2 and inducible nitric oxide synthase
. IL-18 or other cytokines might stimulate microglia and astrocytes in an autocrine or paracrine manner and induce production of pro-inflammatory molecules, such as IL-1β, IL-6, and TNF-α, as well as COX-2 and inducible nitric oxide synthase  . These pro-inflammatory molecules induced in glial cells sensitize dorsal horn neurons in the spinal cord
. These pro-inflammatory molecules induced in glial cells sensitize dorsal horn neurons in the spinal cord  .
.
Proinflammatory cytokines, such as IL-1β, IL-6, and TNF-α, are released by activated glial cells in the spinal cord and play a major role in pain facilitation (Ledeboer et al., 2005a; Clark et al., 2006; Kawasaki et al., 2008b). These cytokines exert their actions, at least partially, through the activation of the transcription factor NFκB. In turn, NFκB regulates the transcription of many inflammatory mediators, including those for proinflammatory cytokines, chemokines, and adhesion molecules (Ledeboer et al., 2005b; Meunier et al., 2007). We found that NFκB was activated in IL-18R-expressing cells after nerve injury, and furthermore, an NFκB inhibitor, SN50, dose-dependently blocked the IL-18-induced allodynia. Although it is important to establish how glia–glia interactions cause signaling to neurons in the spinal dorsal horn, our results indicate that IL-18 signaling in spinal glial cells has a crucial role in the pathogenesis of tactile allodynia after nerve injury. In fact, IL-18-deficient mice displayed significantly attenuated tactile allodynia compared with the wild-type controls. However, intrathecal injection of anti-IL-18 or anti-IL-18R only partially suppressed SNL-induced mechanical hypersensitivity. Considering that in naive mice the intraplantar injection of IL-18 induced dose- and time-dependent mechanical hypernociception (Verri et al., 2007), we cannot rule out the possibility that IL-18 produced by other cells may also contribute to mechanical hypersensitivity, to some extent, after injury. Indeed, we found that nerve injury also induced an increase in IL-18 expression in ED-1-IR macrophages in the injured DRG. Furthermore, recent reports have shown that induction of type-2 metabotropic glutamate receptors is mediated by acetylation mechanisms that involve transcription factors of the NFκB family in DRG neurons (Chiechio et al., 2006, 2007). Alternatively, other cytokines, chemokines, or neurotrophins produced by glial cells are likely involved in the pathogenesis of neuropathic pain. For example, a recent report showed that ATP-stimulated spinal microglia signal to lamina I neurons, causing a collapse of their transmembrane anion gradient (Coull et al., 2005).
Animal models suggest suppression of IL-18 bioactivity as a novel therapeutic concept, specifically for the treatment of chronic inflammatory diseases such as rheumatoid arthritis, Crohn's disease, and psoriasis (Plater-Zyberk et al., 2001; Ten Hove et al., 2001; Carrascal et al., 2003; Boraschi and Dinarello, 2006). IL-18BP, a naturally occurring and specific inhibitor of IL-18, neutralizes IL-18 activity (Novick et al., 1999; Kim et al., 2000) and has been shown to be safe in patients (Plater-Zyberk et al., 2001; Boraschi and Dinarello, 2006). Importantly, we also demonstrate that the expression of IL-18BP mRNA did not change in the ipsilateral dorsal horn after nerve injury, in contrast to marked upregulation of IL-18 and IL-18R. Some reports have shown that a glial inhibitor suppressed the development of neuropathic pain but had no effect on its maintenance (Raghavendra et al., 2003; Kozai et al., 2007). However, we found that not only pretreatment but also posttreatment with IL-18BP reversed nerve injury-induced tactile allodynia. Many patients in pain clinics suffer from neuropathic pain, and one of the most distinct symptoms is tactile allodynia. Furthermore, drug treatment typically is not initiated until well after the onset of allodynia in clinical settings. Thus, the ability of a therapeutic to reverse established mechanical hypersensitivity might translate to increased clinical utility. In conclusion, here we demonstrate that IL-18-mediated microglia/astrocyte interaction in the dorsal horn enhances neuropathic pain processing after nerve injury. Blocking IL-18 signaling cascade in the spinal cord may represent a new approach to effectively treat clinical neuropathic pain.
Footnotes
This work was supported in part by Grant-in-Aid for Young Scientists and Grants-in-Aid for Scientific Research from the Japan Society for the Promotion of Science; an Open Research Center Grant, Hyogo College of Medicine from the Japanese Ministry of Education, Science, and Culture; and Grants-in-Aid for Researchers Hyogo College of Medicine 2008. We thank Y. Wadazumi and N. Kusumoto for technical assistance. We thank D. A. Thomas for correcting the English usage.
References
- Abbadie C, Lindia JA, Cumiskey AM, Peterson LB, Mudgett JS, Bayne EK, DeMartino JA, MacIntyre DE, Forrest MJ. Impaired neuropathic pain responses in mice lacking the chemokine receptor CCR2. Proc Natl Acad Sci U S A. 2003;100:7947–7952. doi: 10.1073/pnas.1331358100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akira S, Takeda K. Toll-like receptor signalling. Nat Rev Immunol. 2004;4:499–511. doi: 10.1038/nri1391. [DOI] [PubMed] [Google Scholar]
- Akira S, Uematsu S, Takeuchi O. Pathogen recognition and innate immunity. Cell. 2006;124:783–801. doi: 10.1016/j.cell.2006.02.015. [DOI] [PubMed] [Google Scholar]
- Block ML, Zecca L, Hong JS. Microglia-mediated neurotoxicity: uncovering the molecular mechanisms. Nat Rev Neurosci. 2007;8:57–69. doi: 10.1038/nrn2038. [DOI] [PubMed] [Google Scholar]
- Boraschi D, Dinarello CA. IL-18 in autoimmunity: review. Eur Cytokine Netw. 2006;17:224–252. [PubMed] [Google Scholar]
- Carrascal MT, Mendoza L, Valcárcel M, Salado C, Egilegor E, Tellería N, Vidal-Vanaclocha F, Dinarello CA. Interleukin-18 binding protein reduces b16 melanoma hepatic metastasis by neutralizing adhesiveness and growth factors of sinusoidal endothelium. Cancer Res. 2003;63:491–497. [PubMed] [Google Scholar]
- Chiechio S, Copani A, De Petris L, Morales ME, Nicoletti F, Gereau RW., 4th Transcriptional regulation of metabotropic glutamate receptor 2/3 expression by the NF-kappaB pathway in primary dorsal root ganglia neurons: a possible mechanism for the analgesic effect of L-acetylcarnitine. Mol Pain. 2006;2:20. doi: 10.1186/1744-8069-2-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiechio S, Copani A, Gereau RW, 4th, Nicoletti F. Acetyl-L-carnitine in neuropathic pain: experimental data. CNS Drugs. 2007;21(Suppl 1):31–38. doi: 10.2165/00023210-200721001-00005. [DOI] [PubMed] [Google Scholar]
- Clark AK, D'Aquisto F, Gentry C, Marchand F, McMahon SB, Malcangio M. Rapid co-release of interleukin 1beta and caspase 1 in spinal cord inflammation. J Neurochem. 2006;99:868–880. doi: 10.1111/j.1471-4159.2006.04126.x. [DOI] [PubMed] [Google Scholar]
- Clark AK, Yip PK, Grist J, Gentry C, Staniland AA, Marchand F, Dehvari M, Wotherspoon G, Winter J, Ullah J, Bean S, Malcangio M. Inhibition of spinal microglial cathepsin S for the reversal of neuropathic pain. Proc Natl Acad Sci U S A. 2007;104:10655–10660. doi: 10.1073/pnas.0610811104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coull JA, Beggs S, Boudreau D, Boivin D, Tsuda M, Inoue K, Gravel C, Salter MW, De Koninck Y. BDNF from microglia causes the shift in neuronal anion gradient underlying neuropathic pain. Nature. 2005;438:1017–1021. doi: 10.1038/nature04223. [DOI] [PubMed] [Google Scholar]
- Fukuoka T, Kondo E, Dai Y, Hashimoto N, Noguchi K. Brain-derived neurotrophic factor increases in the uninjured dorsal root ganglion neurons in selective spinal nerve ligation model. J Neurosci. 2001;21:4891–4900. doi: 10.1523/JNEUROSCI.21-13-04891.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh S, Karin M. Missing pieces in the NF-kappaB puzzle. Cell. 2002;109(Suppl):S81–S96. doi: 10.1016/s0092-8674(02)00703-1. [DOI] [PubMed] [Google Scholar]
- Hedtjärn M, Leverin AL, Eriksson K, Blomgren K, Mallard C, Hagberg H. Interleukin-18 involvement in hypoxic-ischemic brain injury. J Neurosci. 2002;22:5910–5919. doi: 10.1523/JNEUROSCI.22-14-05910.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang D. Modulation of the expression of cyclooxygenase-2 by fatty acids mediated through toll-like receptor 4-derived signaling pathways. FASEB J. 2001;15:2556–2564. doi: 10.1096/fj.01-0432com. [DOI] [PubMed] [Google Scholar]
- Jack CS, Arbour N, Manusow J, Montgrain V, Blain M, McCrea E, Shapiro A, Antel JP. TLR signaling tailors innate immune responses in human microglia and astrocytes. J Immunol. 2005;175:4320–4330. doi: 10.4049/jimmunol.175.7.4320. [DOI] [PubMed] [Google Scholar]
- Ji RR, Strichartz G. Cell signaling and the genesis of neuropathic pain. Sci STKE. 2004;2004:reE14. doi: 10.1126/stke.2522004re14. [DOI] [PubMed] [Google Scholar]
- Jin SX, Zhuang ZY, Woolf CJ, Ji RR. p38 mitogen-activated protein kinase is activated after a spinal nerve ligation in spinal cord microglia and dorsal root ganglion neurons and contributes to the generation of neuropathic pain. J Neurosci. 2003;23:4017–4022. doi: 10.1523/JNEUROSCI.23-10-04017.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katsura H, Obata K, Mizushima T, Sakurai J, Kobayashi K, Yamanaka H, Dai Y, Fukuoka T, Sakagami M, Noguchi K. Activation of Src-family kinases in spinal microglia contributes to mechanical hypersensitivity after nerve injury. J Neurosci. 2006;26:8680–8690. doi: 10.1523/JNEUROSCI.1771-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katsura H, Obata K, Miyoshi K, Kondo T, Yamanaka H, Kobayashi K, Dai Y, Fukuoka T, Sakagami M, Noguchi K. Transforming growth factor-activated kinase 1 induced in spinal astrocytes contributes to mechanical hypersensitivity after nerve injury. Glia. 2008;56:723–733. doi: 10.1002/glia.20648. [DOI] [PubMed] [Google Scholar]
- Kawasaki Y, Xu ZZ, Wang X, Park JY, Zhuang ZY, Tan PH, Gao YJ, Roy K, Corfas G, Lo EH, Ji RR. Distinct roles of matrix metalloproteases in the early- and late-phase development of neuropathic pain. Nat Med. 2008a;14:331–336. doi: 10.1038/nm1723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawasaki Y, Zhang L, Cheng JK, Ji RR. Cytokine mechanisms of central sensitization: distinct and overlapping role of interleukin-1β, interleukin-6, and tumor necrosis factor-α in regulating synaptic and neuronal activity in the superficial spinal cord. J Neurosci. 2008b;28:5189–5194. doi: 10.1523/JNEUROSCI.3338-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SH, Chung JM. An experimental model for peripheral neuropathy produced by segmental spinal nerve ligation in the rat. Pain. 1992;50:355–363. doi: 10.1016/0304-3959(92)90041-9. [DOI] [PubMed] [Google Scholar]
- Kim SH, Eisenstein M, Reznikov L, Fantuzzi G, Novick D, Rubinstein M, Dinarello CA. Structural requirements of six naturally occurring isoforms of the IL-18 binding protein to inhibit IL-18. Proc Natl Acad Sci U S A. 2000;97:1190–1195. doi: 10.1073/pnas.97.3.1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi K, Yamanaka H, Fukuoka T, Dai Y, Obata K, Noguchi K. P2Y12 receptor upregulation in activated microglia is a gateway of p38 signaling and neuropathic pain. J Neurosci. 2008;28:2892–2902. doi: 10.1523/JNEUROSCI.5589-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koistinaho M, Koistinaho J. Role of p38 and p44/42 mitogen-activated protein kinases in microglia. Glia. 2002;40:175–183. doi: 10.1002/glia.10151. [DOI] [PubMed] [Google Scholar]
- Kozai T, Yamanaka H, Dai Y, Obata K, Kobayashi K, Mashimo T, Noguchi K. Tissue type plasminogen activator induced in rat dorsal horn astrocytes contributes to mechanical hypersensitivity following dorsal root injury. Glia. 2007;55:595–603. doi: 10.1002/glia.20483. [DOI] [PubMed] [Google Scholar]
- Ledeboer A, Sloane EM, Milligan ED, Frank MG, Mahony JH, Maier SF, Watkins LR. Minocycline attenuates mechanical allodynia and proinflammatory cytokine expression in rat models of pain facilitation. Pain. 2005a;115:71–83. doi: 10.1016/j.pain.2005.02.009. [DOI] [PubMed] [Google Scholar]
- Ledeboer A, Gamanos M, Lai W, Martin D, Maier SF, Watkins LR, Quan N. Involvement of spinal cord nuclear factor kappaB activation in rat models of proinflammatory cytokine-mediated pain facilitation. Eur J Neurosci. 2005b;22:1977–1986. doi: 10.1111/j.1460-9568.2005.04379.x. [DOI] [PubMed] [Google Scholar]
- Marchand F, Perretti M, McMahon SB. Role of the immune system in chronic pain. Nat Rev Neurosci. 2005;6:521–532. doi: 10.1038/nrn1700. [DOI] [PubMed] [Google Scholar]
- Marshak-Rothstein A. Toll-like receptors in systemic autoimmune disease. Nat Rev Immunol. 2006;6:823–835. doi: 10.1038/nri1957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menetski J, Mistry S, Lu M, Mudgett JS, Ransohoff RM, Demartino JA, Macintyre DE, Abbadie C. Mice overexpressing chemokine ligand 2 (CCL2) in astrocytes display enhanced nociceptive responses. Neuroscience. 2007;149:706–714. doi: 10.1016/j.neuroscience.2007.08.014. [DOI] [PubMed] [Google Scholar]
- Meunier A, Latrémolière A, Dominguez E, Mauborgne A, Philippe S, Hamon M, Mallet J, Benoliel JJ, Pohl M. Lentiviral-mediated targeted NF-kappaB blockade in dorsal spinal cord glia attenuates sciatic nerve injury-induced neuropathic pain in the rat. Mol Ther. 2007;15:687–697. doi: 10.1038/sj.mt.6300107. [DOI] [PubMed] [Google Scholar]
- Michael GJ, Averill S, Nitkunan A, Rattray M, Bennett DL, Yan Q, Priestley JV. Nerve growth factor treatment increases brain-derived neurotrophic factor selectively in TrkA-expressing dorsal root ganglion cells and in their central terminations within the spinal cord. J Neurosci. 1997;17:8476–8490. doi: 10.1523/JNEUROSCI.17-21-08476.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molander C, Xu Q, Grant G. The cytoarchitectonic organization of the spinal cord in the rat. I. The lower thoracic and lumbosacral cord. J Comp Neurol. 1984;230:133–141. doi: 10.1002/cne.902300112. [DOI] [PubMed] [Google Scholar]
- Nakanishi K, Yoshimoto T, Tsutsui H, Okamura H. Interleukin-18 regulates both Th1 and Th2 responses. Annu Rev Immunol. 2001;19:423–474. doi: 10.1146/annurev.immunol.19.1.423. [DOI] [PubMed] [Google Scholar]
- Noguchi K, Kawai Y, Fukuoka T, Senba E, Miki K. Substance P induced by peripheral nerve injury in primary afferent sensory neurons and its effect on dorsal column nucleus neurons. J Neurosci. 1995;15:7633–7643. doi: 10.1523/JNEUROSCI.15-11-07633.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novick D, Kim SH, Fantuzzi G, Reznikov LL, Dinarello CA, Rubinstein M. Interleukin-18 binding protein: a novel modulator of the Th1 cytokine response. Immunity. 1999;10:127–136. doi: 10.1016/s1074-7613(00)80013-8. [DOI] [PubMed] [Google Scholar]
- Obata K, Katsura H, Mizushima T, Yamanaka H, Kobayashi K, Dai Y, Fukuoka T, Tokunaga A, Tominaga M, Noguchi K. TRPA1 induced in sensory neurons contributes to cold hyperalgesia after inflammation and nerve injury. J Clin Invest. 2005;115:2393–2401. doi: 10.1172/JCI25437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obata K, Katsura H, Mizushima T, Sakurai J, Kobayashi K, Yamanaka H, Dai Y, Fukuoka T, Noguchi K. Roles of extracellular signal-regulated protein kinases 5 in spinal microglia and primary sensory neurons for neuropathic pain. J Neurochem. 2007;102:1569–1584. doi: 10.1111/j.1471-4159.2007.04656.x. [DOI] [PubMed] [Google Scholar]
- Okamura H, Tsutsui H, Komatsu T, Yutsudo M, Hakura A, Tanimoto T, Torigoe K, Okura T, Nukada Y, Hattori K, Akita K, Namba M, Tanabe F, Konishi K, Fukuda S, Kurimoto M. Cloning of a new cytokine that induces IFN-gamma production by T cells. Nature. 1995;378:88–91. doi: 10.1038/378088a0. [DOI] [PubMed] [Google Scholar]
- Olson JK, Miller SD. Microglia initiate central nervous system innate and adaptive immune responses through multiple TLRs. J Immunol. 2004;173:3916–3924. doi: 10.4049/jimmunol.173.6.3916. [DOI] [PubMed] [Google Scholar]
- Plater-Zyberk C, Joosten LA, Helsen MM, Sattonnet-Roche P, Siegfried C, Alouani S, van De Loo FA, Graber P, Aloni S, Cirillo R, Lubberts E, Dinarello CA, van Den Berg WB, Chvatchko Y. Therapeutic effect of neutralizing endogenous IL-18 activity in the collagen-induced model of arthritis. J Clin Invest. 2001;108:1825–1832. doi: 10.1172/JCI12097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prinz M, Hanisch UK. Murine microglial cells produce and respond to interleukin-18. J Neurochem. 1999;72:2215–2218. doi: 10.1046/j.1471-4159.1999.0722215.x. [DOI] [PubMed] [Google Scholar]
- Raghavendra V, Tanga F, DeLeo JA. Inhibition of microglial activation attenuates the development but not existing hypersensitivity in a rat model of neuropathy. J Pharmacol Exp Ther. 2003;306:624–630. doi: 10.1124/jpet.103.052407. [DOI] [PubMed] [Google Scholar]
- Scholz J, Woolf CJ. Can we conquer pain? Nat Neurosci. 2002;5(Suppl):1062–1067. doi: 10.1038/nn942. [DOI] [PubMed] [Google Scholar]
- Takeda K, Tsutsui H, Yoshimoto T, Adachi O, Yoshida N, Kishimoto T, Okamura H, Nakanishi K, Akira S. Defective NK cell activity and Th1 response in IL-18-deficient mice. Immunity. 1998;8:383–390. doi: 10.1016/s1074-7613(00)80543-9. [DOI] [PubMed] [Google Scholar]
- Tanga FY, Nutile-McMenemy N, DeLeo JA. The CNS role of Toll-like receptor 4 in innate neuroimmunity and painful neuropathy. Proc Natl Acad Sci U S A. 2005;102:5856–5861. doi: 10.1073/pnas.0501634102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanga FY, Raghavendra V, Nutile-McMenemy N, Marks A, Deleo JA. Role of astrocytic S100beta in behavioral hypersensitivity in rodent models of neuropathic pain. Neuroscience. 2006;140:1003–1010. doi: 10.1016/j.neuroscience.2006.02.070. [DOI] [PubMed] [Google Scholar]
- Ten Hove T, Corbaz A, Amitai H, Aloni S, Belzer I, Graber P, Drillenburg P, van Deventer SJ, Chvatchko Y, Te Velde AA. Blockade of endogenous IL-18 ameliorates TNBS-induced colitis by decreasing local TNF-alpha production in mice. Gastroenterology. 2001;121:1372–1379. doi: 10.1053/gast.2001.29579. [DOI] [PubMed] [Google Scholar]
- Tsuda M, Shigemoto-Mogami Y, Koizumi S, Mizokoshi A, Kohsaka S, Salter MW, Inoue K. P2X4 receptors induced in spinal microglia gate tactile allodynia after nerve injury. Nature. 2003;424:778–783. doi: 10.1038/nature01786. [DOI] [PubMed] [Google Scholar]
- Tsuda M, Inoue K, Salter MW. Neuropathic pain and spinal microglia: a big problem from molecules in “small” glia. Trends Neurosci. 2005;28:101–107. doi: 10.1016/j.tins.2004.12.002. [DOI] [PubMed] [Google Scholar]
- Verge GM, Milligan ED, Maier SF, Watkins LR, Naeve GS, Foster AC. Fractalkine (CX3CL1) and fractalkine receptor (CX3CR1) distribution in spinal cord and dorsal root ganglia under basal and neuropathic pain conditions. Eur J Neurosci. 2004;20:1150–1160. doi: 10.1111/j.1460-9568.2004.03593.x. [DOI] [PubMed] [Google Scholar]
- Verri WA, Jr, Cunha TM, Parada CA, Poole S, Liew FY, Ferreira SH, Cunha FQ. Antigen-induced inflammatory mechanical hypernociception in mice is mediated by IL-18. Brain Behav Immun. 2007;21:535–543. doi: 10.1016/j.bbi.2006.11.005. [DOI] [PubMed] [Google Scholar]
- Watkins LR, Maier SF. Glia: a novel drug discovery target for clinical pain. Nat Rev Drug Discov. 2003;2:973–985. doi: 10.1038/nrd1251. [DOI] [PubMed] [Google Scholar]
- Woolf CJ, Salter MW. Neuronal plasticity: increasing the gain in pain. Science. 2000;288:1765–1769. doi: 10.1126/science.288.5472.1765. [DOI] [PubMed] [Google Scholar]
- Yamanaka H, He X, Matsumoto K, Shiosaka S, Yoshida S. Protease M/neurosin mRNA is expressed in mature oligodendrocytes. Brain Res Mol Brain Res. 1999;71:217–224. doi: 10.1016/s0169-328x(99)00187-4. [DOI] [PubMed] [Google Scholar]
- Zhang J, Hoffert C, Vu HK, Groblewski T, Ahmad S, O'Donnell D. Induction of CB2 receptor expression in the rat spinal cord of neuropathic but not inflammatory chronic pain models. Eur J Neurosci. 2003;17:2750–2754. doi: 10.1046/j.1460-9568.2003.02704.x. [DOI] [PubMed] [Google Scholar]
- Zhuang ZY, Gerner P, Woolf CJ, Ji RR. ERK is sequentially activated in neurons, microglia, and astrocytes by spinal nerve ligation and contributes to mechanical allodynia in this neuropathic pain model. Pain. 2005;114:149–159. doi: 10.1016/j.pain.2004.12.022. [DOI] [PubMed] [Google Scholar]
- Zhuang ZY, Wen YR, Zhang DR, Borsello T, Bonny C, Strichartz GR, Decosterd I, Ji RR. A peptide c-Jun N-terminal kinase (JNK) inhibitor blocks mechanical allodynia after spinal nerve ligation: respective roles of JNK activation in primary sensory neurons and spinal astrocytes for neuropathic pain development and maintenance. J Neurosci. 2006;26:3551–3560. doi: 10.1523/JNEUROSCI.5290-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]