

Abstract
Amyotrophic lateral sclerosis (ALS) is characterized by motoneuron (MN) degeneration, generalized weakness, and muscle atrophy. The premature death of MNs is thought to be a determinant in the onset of this disease. In a transgenic mouse model of ALS expressing the G86R mutant superoxide dismutase 1 (mSOD1), we demonstrated previously that CREB (cAMP response element-binding protein)-binding protein (CBP) and histone acetylation levels were specifically decreased in nuclei of degenerating MNs. We show here that oxidative stress and mSOD1 overexpression can both impinge on CBP levels by transcriptional repression, in an MN-derived cell line. Histone deacetylase inhibitor (HDACi) treatment was able to reset proper acetylation levels and displayed an efficient neuroprotective capacity against oxidative stress in vitro. Interestingly, HDACi also upregulated CBP transcriptional expression in MNs. Moreover, when injected to G86R mice in vivo, the HDACi sodium valproate (VPA) maintained normal acetylation levels in the spinal cord, efficiently restored CBP levels in MNs, and significantly prevented MN death in these animals. However, despite neuroprotection, mean survival of treated animals was not significantly improved (<5%), and they died presenting the classical ALS symptoms. VPA was not able to prevent disruption of neuromuscular junctions, although it slightly delayed the onset of motor decline and retarded muscular atrophy to some extent. Together, these data show that neuroprotection can improve disease onset, but clearly provide evidence that one can uncouple MN survival from whole-animal survival and point to the neuromuscular junction perturbation as a primary event of ALS onset.
Keywords: sodium valproate, HDAC inhibitor, CREB-binding protein, motor dysfunction, neuroprotection, acetylation
Introduction
Amyotrophic lateral sclerosis (ALS) is a fatal neurodegenerative disease that affects upper and lower motoneurons (MNs), leading to progressive muscle wasting, paralysis, and death within 2–5 years of diagnosis (Pasinelli and Brown, 2006). Although most of ALS cases occur sporadically, ∼10% are inherited. Among these, 10–20% are attributable to mutations in the gene encoding the superoxide dismutase-1 (SOD1), one of the main free-radical scavenging enzymes that protect cells against oxidative stress (Rosen et al., 1993; Andersen et al., 2003). Different transgenic mice bearing mutations on the sod1 gene have been developed as ALS models, among which is the G86R mouse strain. These mice bear the equivalent G85R human mutation on the mouse sod1 gene. They die within ∼120 d, presenting typical ALS symptoms: motoneuronal death, muscular atrophy, and paralysis (Ripps et al., 1995; Dupuis et al., 2000).
Premature death of MNs is believed to be determinant in the onset of ALS. However, the primary event leading to pathogenicity is still a matter of debate. All cell types implicated in the motor unit could be potential targets of toxicity (i.e., MNs, myocytes, astrocytes, microglia, and Schwann cells). Cell-specific knock-in or knock-down studies of the mutant sod1 gene have not so far brought a clear answer to that question (Gong et al., 2000; Boillee et al., 2006; Miller et al., 2006). Nevertheless, retraction of motor axon from synaptic connection to muscle and fragmentation of the neuromuscular junction are detected before any MN death, suggesting that ALS could be seen as a “dying-back” axonopathy (Fischer et al., 2004; Jokic et al., 2006). We have also shown that muscular metabolic defects and mitochondrial dysfunctions were early symptoms of the pathology (Dupuis et al., 2003, 2004). Furthermore, this idea that MN death could be dissociated from motor dysfunction has been recently strengthened by a study in which MN death was genetically prevented by Bax deletion (Gould et al., 2006).
Our recent work demonstrated that the cAMP response element-binding protein (CREB)-binding protein (CBP), a transcriptional coactivator displaying histone acetyltransferase (HAT) activity (Bannister and Kouzarides, 1996), is specifically lost in lumbar spinal cord MNs of G86R mice (Rouaux et al., 2003). Together with histone deacetylases (HDACs), HATs control gene transcription by finely tuning acetylation homeostasis (Verdone et al., 2005). Consistent with this, we also found decreased histone acetylation levels in the degenerating MNs (Rouaux et al., 2003). Based on these findings, we undertook to correct aberrant gene transcription by interfering with the HAT/HDAC balance with HDAC inhibitors (HDACis) to prevent neuronal death in vivo, hypothesizing that maintaining proper acetylation levels would preserve the transcription of neuroprotective genes. Indeed, several studies have reported the beneficial effects of HDACis on different aspects of neurodegeneration (Langley et al., 2005). Here, we tested the effect of a chronic treatment of G86R mice with sodium valproate (VPA). We provide an in vitro and in vivo proof of concept that VPA is a potent neuroprotective molecule but found that the distal pathology progresses despite neuroprotection.
Materials and Methods
HDAC inhibitors and antibodies.
VPA, sodium butyrate (NaBu), trichostatin A (TSA), and α-bungarotoxin (α-BGT) were purchased from Sigma (St. Louis, MO). Anti-CBP, anti-acetylated histone 3 (AcH3), and anti-p75 antibodies were obtained from Millipore (Billerica, MA). Anti-human synaptophysin antibody was obtained from Dako (Trappes, France). HRP-conjugated goat-anti-rabbit antibody was from Jackson ImmunoResearch (West Grove, PA). Alexa Fluor 488 donkey-anti-rabbit antibody was from Invitrogen (Eugene, OR). Goat anti-choline acetyltransferase (ChAT) and biotinylated goat-anti-rabbit antibody were from Millipore.
NSC34 cell culture.
NSC34 cell line was kindly provided by Dr. Cashman (Cashman et al., 1992). Cells were seeded at a density of ∼1 × 106 cells/ml in 96-well plates for cell survival measurements, 24-well plates for transfection experiments, 12-well plates for Western blot assays, and six-well plates for quantitative PCR (Q-PCR) analysis. Cells were grown in DMEM (Invitrogen) supplemented with 10% fetal bovine serum (Invitrogen), 100 U/ml penicillin, and 100 μg/ml streptomycin (Eurobio, Les Ulis, France) at 37°C and 5% CO2. After 24 h, cells were serum starved for 12 h before being exposed or not to a 15 min pulse of hydrogen peroxide (H2O2) at a final concentration in the millimolar range. The HDAC inhibitors tested were added to media during and after the H2O2 pulse. MTT [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-2H-tetrazolium bromide] assays (cell survival) were performed on NSC34 cells 48 h after treatment as described previously (Rouaux et al., 2003).
Promoter activity assay and cotransfection experiments.
The cbp promoter-luciferase reporter pGL3 construct was kindly provided by Dr. E. D. Adamson (Yu et al., 2004). NSC34 cells were transfected as described previously (Rouaux et al., 2003) using polyethylenimine (25 kDa) as DNA carrier. Briefly, cells grown in 24-well plates were transfected with 0.75 μg of reporter plasmid per well, spun for 5 min at 1500 rpm, and incubated at 37°C for 2 h. Fresh medium was then added, and cells were incubated for an additional 3 h at 37°C, treated (H2O2 and HDACi) for 12 h, and lysed in a Promega (Madison, WI) lysis buffer. Luciferase activity was read in a single tube luminometer (Berthold Detection Systems, Pforzheim, Germany). Expression vectors used for cotransfection experiments are pRC-CMV wild-type (WT)-SOD1 and pRC-CMV G86R-SOD1. One microgram of each expression vector and 0.5 μg of reporter vector were used. Luciferase activity was read 20 h after transfection.
Western blot analysis.
Western blots were performed as described previously (Rouaux et al., 2003) with typically 50 μg of total cell extracts run on 7, 10, or 13% SDS-acrylamide gels for CBP, actin, or histone detection, respectively. Specific bands were detected by enhanced chemiluminescence (GE Healthcare, Buckinghamshire, UK). Size and intensity of specific bands were quantified using an NIH Image 1.62 software to allow the representation of the results as histograms.
Quantitative and classical (reverse transcription)-PCR analysis.
Total RNA was extracted from animal tissues or NSC34 cells using Trizol (Invitrogen) according to the manufacturer's instructions. cDNA syntheses were performed using 1 μg of total RNA (iScript cDNA Synthesis kit; Bio-Rad, Hercules, CA) with the following primers: cbp, forward, 5′-tgg gta acc agc cag ctt ta-3′; reverse, 5′-cag gga cat tgg gtt aaa tga-3′; bcl2, forward, 5′-ctg caa atg ctg gac tga aa-3′; reverse, 5′-tct act tcc tcc gca atg ct-3′; smn, forward, 5′-aag gca cag cca gaa gaa aa-3′; reverse, 5′-tca cag gtc ggg gaa agt ag-3′; 18S, forward, 5′-cgt ctg ccc tat caa ctt tcg-3′; reverse, 5′-ttc ctt gga tgt ggt agc cg-3′.
Q-PCR analysis was performed on iCycler System (Bio-Rad). The reactions were performed in 25 μl, in 96-well plates, using iQSYBR Green Supermix (Bio-Rad). A specific standard curve for each gene was performed in parallel to the analysis. Each sample was analyzed in duplicate. PCR conditions were 3 min at 94°C, followed by 40 cycles of 45 s at 94°C and 10 s at 60°C. Results were analyzed by using the iCycler software (Bio-Rad) and normalized for the reference gene encoding the 18S ribosomal subunit. Semiquantitative reverse transcription (RT)-PCR analysis was performed as described previously (Dupuis et al., 2004).
Animals.
Transgenic male mice with the G86R murine SOD1 mutation (Ripps et al., 1995) were obtained from the animal facility at the Université Louis Pasteur. Mice were bred with female littermates, and transgenic animals were genotyped by PCR amplification of DNA extracted from tail tissue. Nontransgenic age-matched male littermates served as control. Mice received water and regular rodent chow ad libitum. HDAC inhibitors tested were diluted in 0.09% NaCl [vehicle (VEH)], and administration was performed intraperitoneally. The VPA dose to use was evaluated on 16 adult WT mice. Chronic injections were performed daily, from 60 to 105 d of age. Three doses of VPA were tested: 150, 250, and 400 mg · kg−1 · d−1. Acute injections of TSA (2 mg/kg), VPA (250 mg/kg), NaBu (640 mg/kg), or VEH were performed on symptomatic G86R (n = 6 in each group). Six WT littermates were used as control. All animals were killed by decapitation. Animal manipulations followed current European Union regulations.
Survival and clinical assessment.
Clinical assessment was estimated daily, according to a clinical rating scale going from score 4 to 0. Score 4 is attributed to asymptomatic G86R mice, relative to their WT littermates. Score 3 corresponds to an alteration in hindlimb extension when the animal is hung by the tail. Appearance of score 3 is considered the disease onset. Score 2 is attributed when any slight alteration in the locomotion is observed. Score 1 represents an asymmetrical or symmetrical paralysis of the limbs. Score 0 corresponds to the stage at which animals are unable to roll over within 10 s after being pushed on their back. Mice were killed when they reached score 0.
Weight assessment and motor function testing.
Mice were weighed twice a week. Grip strength was assessed on forelimbs, twice a week, using a grip strength meter (Bioseb, Chaville, France). Measurements were performed from 60 d of age to the end of mouse life. Animals were placed over a metallic grid that they instinctively grabbed to try to stop the involuntary backward movement performed by the manipulator until the pulling force overcame their grip strength. The strength meter scored the peak pull force. Three measurements were collected at each time, and the best one was retained.
Tissue preparation.
Lumbar spinal cords, cerebellum, and soleus and gastrocnemius muscles were carefully dissected from G86R or WT mice and immediately frozen in liquid nitrogen for additional biochemical analysis or postfixed for 24 h in 4% paraformaldehyde (PFA) and cryoprotected in 20% sucrose during 24 h at 4°C for immunohistochemistry.
Histone acetylation measurement.
Tissue lysates from spinal cord or cerebellum were obtained by homogenizing each sample in 300 μl of ice-cold lysis buffer for 30 s at 30 Hz in a Qiagen (Haan, Germany) TissueLyser, and histones were acid extracted as described by Rouaux et al. (2003). Five micrograms of purified histones were separated on 13% polyacrylamide gels and further processed for Western blot analysis.
Immunohistochemistry.
Serial lumbar spinal cord tissue sections from L3–L5 spinal cord segments were used for motoneuronal analysis. The sections were mounted onto gelatin-coated slides and kept at −80°C. Briefly, slides were dried for 45 min on a hot plate (70°C), rehydrated for 10 min in PBS, and fixed for 10 min with 4% PFA. For CBP and AcH3 immunostainings, endogenous peroxidases were inactivated by 10 min of incubation in 1% H2O2/PBS. Antigenic sites were then uncovered in a citrate buffer (1.8 mm citric acid and 8 mm sodium citrate), warmed for 10 min in a microwave oven, and cooled down on ice. Sections were permeabilized for 10 min in 1% Triton X-100/PBS before being blocked for 30 min in 5% goat serum/0.1% Triton X-100/PBS. Primary antibodies were applied overnight, and secondary antibodies were applied for 1 h. Revelation was performed using the Vectastain ABC kit (Vector Laboratories, Burlingame, CA). For ChAT immunostaining, the method was adapted to a FITC-conjugated secondary antibody. Pictures were taken with a Nikon (Tokyo, Japan) digital camera DXM 1200. ChAT-positive cells were counted in the ventral horn from six spinal cord sections per animal as described previously (Dupuis et al., 2004). For counting innervated synapses, 40-μm-thick longitudinal sections of PFA-fixed, sucrose-embedded, and frozen soleus muscles were cut and stained with antibodies against synaptophysin and α-BGT. The same protocol was used for p75 immunostaining. Usually 5–6 sections were analyzed per animal, which represents ∼100–150 nicotinic acetylcholine receptor (nAChR) clusters. Immunostaining was analyzed confocally (Laser Scanning System 510; Zeiss, Oberkochen, Germany).
Myelinated axon area measurements.
Lumbar spinal cords were dissected from animals and postfixed in 4% PFA for 48 h. The fourth lumbar ventral roots (L4 VRs) were dissected and stained by 1% osmium tetroxide, dehydrated, and embedded in Araldite. The area of myelinated axons was measured on 1.5-μm-thick sections using the LuciaG software. Nerve fiber occupancy in L4 VRs was expressed as the percentage of the nerve fascicle size occupied by myelinated nerve fibers. Two square fields (150 μm section) were analyzed for each ventral root. Approximately 100–150 fibers were analyzed in each field.
Muscle fiber area measurements.
Gastrocnemius muscles were transversally cryosectioned (10 μm) and stained with 0.1% toluidine blue, and fiber areas were measured with the LuciaG software in two square fields. At least 40 fibers were analyzed in each field.
Carnitine assay.
Total plasma l-carnitine concentrations were determined by a spectrophotometric enzymatic assay using carnitine acetyltransferase (CAT; EC 2.3.1.7) and 5,5-dithiobis (2-nitrobenzoic acid) (DTNB) as thiol group color reagent. Total carnitine was quantified after deesterification by alkaline hydrolysis as described previously (Wan and Hubbard, 1998). Diluted sample (50 μl) was mixed with 200 μl of the primary reagent (200 μm DTNB and 600 μm acetyl-CoA in 200 μm phosphate buffer, pH 7.5), and the reaction was started with 25 μl of diluted start reagent (CAT from pigeon muscle, 123 U/mg protein) to give a final activity of 1.53 U/sample. All specific reagents used were from Sigma. The reaction mixture was incubated for 15 min at 37°C, and absorbance was read at 415 nm. Calibrators containing 10–100 μm l-carnitine were prepared by dilution of the l-carnitine stock solution. The assay was linear for carnitine concentrations up to 200 μm. All specimens were assayed in replicate in the same analytical run.
Electrophysiological recording of spontaneous activity.
All recordings were made with a standard EMG apparatus (Dantec, Les Ulis, France) in accordance with the guidelines of the American Association of Electrodiagnostic Medicine. Mice were anesthetized with 1 mg/kg ketamine chlorhydrate and 0.5 mg/kg xylazine. A monopolar needle electrode (diameter, 0.3 mm; 9013R0312; Medtronic, Minneapolis, MN) was inserted into the tail of the mouse to ground the system. Recordings were made with a concentric needle electrode (diameter, 0.3 mm; 9013S0011; Medtronic). Gastrocnemius muscles were monitored on both sides (right and left) for at least 2 min. Only spontaneous activity with a peak-to-peak amplitude of at least 50 μV was considered to be significant. Fibrillations were graded from 0 to 4: no fibrillations (0), persistent single strains of potentials (longer than 2–3 s) in at least two areas of the muscle (1), moderate number of potentials in three or more areas (2), many potentials in all areas (3), or full interference pattern of potentials (4).
Statistical analysis.
Data are expressed as the mean ± SEM. All statistical analysis were performed on PRISM version 4.0b software (GraphPad, San Diego), using an ANOVA followed by the post hoc Newman–Keuls multiple-comparisons test for classical analysis, a two-way ANOVA followed by a Bonferroni post-test for the grip strength analysis or by means of Kaplan–Meier curves for survival, score onsets, and weigh loss onset.
Results
HDACi protects from oxidative stress-induced MN death
Oxidative stress is described as one of the possible major causes of motoneuronal death in ALS (Liu et al., 1998). To test whether HDACi treatment could be used as a therapeutic approach to prevent MN death in ALS mice, we first used a simplified cellular model, the NSC34 cell line, a hybridoma of spinal cord motoneurons and neuroblastoma (Cashman et al., 1992), in which cell death can be induced by oxidative stress (Cookson et al., 1998). Treated with a pulse of H2O2, NSC34 cells died in a dose-dependent manner (Fig. 1A). Interestingly, death was accompanied by a progressive decrease of both CBP protein and histone H3 acetylation levels (Fig. 1B), two features that we already observed in MNs of symptomatic SOD1(G86R) mice (Rouaux et al., 2003). Several HDACis have been reported to promote cell survival both in vitro and in vivo, and we tested three HDACis (VPA, TSA, and NaBu) for their ability to counteract H2O2-induced NSC34 cell death. All of the three HDACis were able to reverse oxidative stress-induced histone deacetylation in a dose-dependent manner (Fig. 1C). In addition, HDACi treatment could also rescue NSC34 cells from death, with different efficiencies (Fig. 1D). Altogether, these results show that HDACis efficiently restored histone acetylation and counteracted cell death in a cellular model that mimics oxidative stress-induced motoneuronal death.
Figure 1.
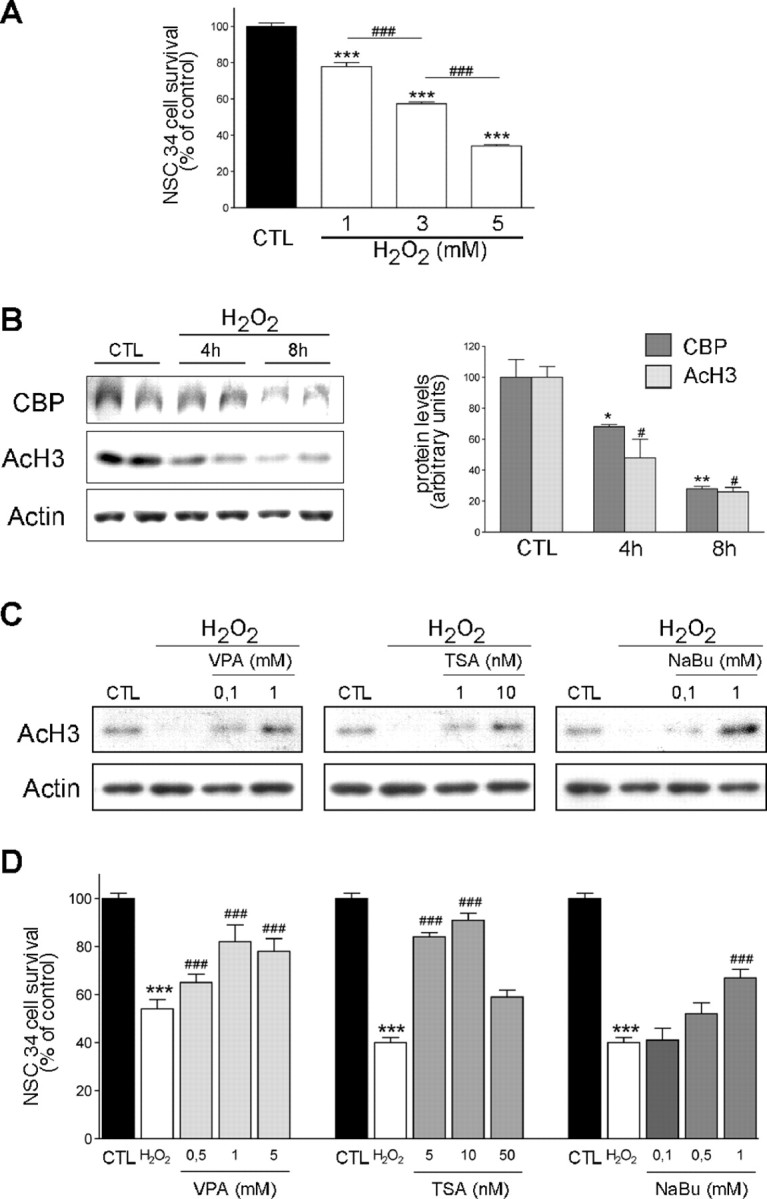
Oxidative stress-induced NSC34 death is accompanied by CBP and histone acetylation loss and is reversed by HDACis. A, NSC34 survival measurements performed in conditions of increasing concentrations of H2O2. ***p < 0.001 compared with control condition; ###p < 0.001 compared with precedent H2O2 concentration. B, Left, Western blot analysis of CBP and AcH3 protein levels in NSC34 cells treated or not with a pulse of 3 mm H2O2. Right, Actin was used as control for loading and served as reference for CBP and AcH3 band quantification. *p < 0.05 and **p < 0.01 compared with CBP levels in control condition; #p < 0.05 compared with AcH3 levels in control condition. C, Western blot analysis of AcH3 levels in NSC34 cells treated or not with a pulse of 3 mm H2O2, in presence or absence of increasing concentrations of HDACis (VPA, TSA, or NaBu). Actin served as gel loading control. D, NSC34 survival measurements in presence or absence of increasing concentrations of HDACis (VPA, TSA, or NaBu). ***p < 0.001 compared with control condition; ###p < 0.001 compared with H2O2 condition. CTL, Control.
Oxidative stress and mutant SOD1 induce transcriptional repression of the cbp gene, which can be upregulated by HDACi treatment
Taking advantage of this homogenous cellular model of MNs (in contrast to the whole spinal cord), we further investigated which mechanism could underlie CBP downregulation under oxidative stress conditions. Figure 2A shows that a 15 min pulse of H2O2 induced a transcriptional repression of the cbp gene within 4 h. The same result was observed with a reporter luciferase gene driven by the cbp promoter (Fig. 2B). Interestingly, overexpression of SOD1(G86R) strongly repressed cbp promoter activity, whereas the WT SOD1 had no effect (Fig. 2C). This result suggests that expression of SOD1(G86R) in ALS transgenic animals could intrinsically exert a transcriptional repression of cbp gene in the lumbar spinal cord, thus triggering MN death. By blocking histone deacetylation, HDACis act as transcriptional activators; therefore, we next tested whether they could directly induce cbp gene expression. Figure 2D shows that HDACis were able to increase cbp promoter-driven luciferase activity in a dose-dependent manner, both under control and oxidative stress conditions. Except for NaBu, the efficient doses in reversing oxidative stress-induced cbp transcriptional repression (1 mm VPA and 10 nm TSA) corresponded to those exhibiting neuroprotective effects (Fig. 1D).
Figure 2.

Oxidative stress-induced CBP loss results from transcriptional repression that can be reversed by HDACis. A, cbp expression level was assessed by Q-PCR in NSC34 cells maintained in control conditions (CTL) or treated for 8 h with increasing concentrations of H2O2. B, cbp promoter-driven activity in control or oxidative stress (H2O2) conditions. C, NSC34 cells were transiently cotransfected with cbp promoter-luciferase reporter, together with the empty vector pRC-CMV, or vectors encoding either the WT or the mutated SOD1(G86R) protein. D, Effects of HDACis (VPA, TSA, or NaBu) on cbp promoter-driven activity in NSC34 cells under control (solid bars) or oxidative stress (hatched bars). **/##p < 0.01 and ***/###p < 0.001 when compared with control (*) or H2O2 (#) conditions (A, B, D) or to WT SOD1 condition (C).
Chronic VPA treatment in vivo prevents histone deacetylation in the spinal cord of symptomatic ALS mice
We then undertook to treat ALS mice with an HDACi to see whether protecting MNs could provide beneficial effects in the pathology. We chose to treat mice with VPA, a Food and Drug Administration (FDA)-approved molecule used as an antiepileptic and against bipolar disorders for many years (Perucca, 2002). VPA, as a short fatty acid, also presents the advantages of being soluble in aqueous buffers and of easily crossing the brain–blood barrier, compared with more complex molecules such as TSA or suberoylanilide hydroxamic acid. Preliminary studies were performed to estimate the dose of VPA necessary to increase histone acetylation levels in nervous tissue. VPA was intraperitoneally injected into the mice at 150 and 300 mg/kg, which correspond to the classical range of VPA tested in epileptic rodent models (Tremolizzo et al., 2002). Figure 3A shows that VPA significantly increased histone acetylation levels in cerebellum as well as in thoracic and lumbar spinal cord 24 h after a single injection in a dose-dependent manner, showing that VPA indeed could reach lower MNs. This result was also observed in other non-nervous tissues (data not shown). In spinal cord, the effect of VPA was observed as soon as 2 h after a single injection at a dose of 250 mg/kg and lasted at least 24 h (Fig. 3B). We then wondered whether a chronic injection of VPA could reverse histone deacetylation in lumbar spinal cord MNs from symptomatic G86R mice. Mice were daily intraperitoneally injected with a dose of 250 mg/kg from 60 d of age (asymptomatic) until the end stage, and the spinal cords were processed for acetylation level assessment. As controls, other cohorts of G86R mice and wild-type littermates received chronic injections of VEH and were killed at matching ages. As observed previously (Rouaux et al., 2003), histone acetylation levels were dramatically decreased in MN nuclei from untreated G86R mice, whereas they were maintained in other cell types (Fig. 3C). In contrast, histone acetylation levels in VPA-treated transgenic mice were significantly restored to levels comparable with those of WT littermates (Fig. 3C).
Figure 3.
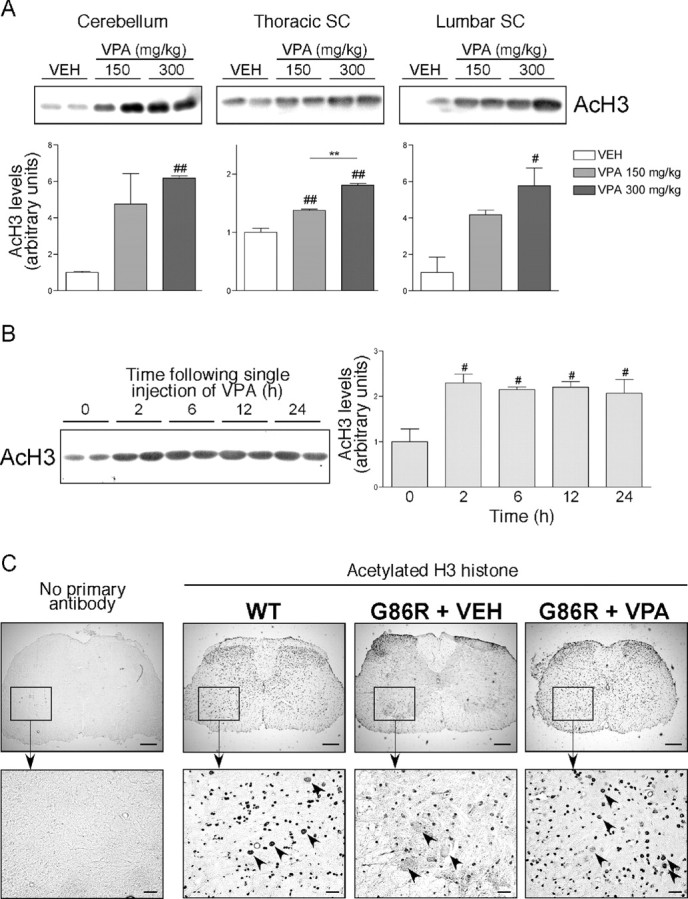
Single and chronic VPA injections increase histone acetylation in nervous tissues in vivo. A, Six 90-d-old WT mice received a single injection of VEH (0.09% NaCl) or 150 or 300 mg/kg VPA. Twenty-four hours later, cerebella and spinal cords (SC) were carefully dissected and processed for acid extraction of histones. Top, Five micrograms of purified histones were then analyzed by Western blot to detect specific AcH3. Bottom, Bands were quantified. #p < 0.05 and ##p < 0.01 compared with VEH condition; **p < 0.01. B, WT mice were injected once with a dose of 250 mg/kg VPA and killed 0, 2, 6, 12, or 24 h later. Left, Spinal cords were dissected and processed for Western blot analysis. Right, Bands were quantified. #p < 0.05 compared with time 0. C, Spinal cord sections from WT (n = 5) or end-stage G86R mice daily injected with VEH (n = 5) or 250 mg/kg VPA (n = 5) were taken, and sections were immunostained using an anti-AcH3 antibody followed by a peroxidase revelation. Representative pictures are shown. Arrowheads depict lumbar motor neurons. Scale bars: top, 200 μm; bottom, 40 μm.
Chronic VPA treatment ensures neuroprotection and restores normal CBP levels in vivo
We then assessed the effects of VPA on survival of lower MNs in these mice. MNs were counted in the ventral horns of lumbar spinal cords from 105-d-old (score 2) (Fig. 4A, left) and end-stage (score 0) mice (right) (for clinical score assessment, see Material and Methods and Fig. 5A). We found that at 105 d of age, when MN loss was ∼30% in G86R mice compared with age-matched WT mice, chronic VPA treatment completely prevented neuronal death. In the end-stage mice, MN loss was much more important (77.7%), but VPA effect was still significant, and the number of MNs was twofold higher in treated than in untreated transgenic mice (Fig. 4A). These results clearly indicate that VPA treatment efficiently delayed motoneuronal death in vivo. To get more convincing information on the extent of neuroprotection by VPA, we verified the occupancy of myelinated axons in L4 VRs from the spinal cords. Typical sections of WT and G86R mice at the onset of locomotor symptoms (score 2) are shown in Figure 4B, and focus images made on the different groups of mice (treated or not) are shown in Figure 4C. We found a reduction of ∼20% of occupancy of myelinated L4 VR axons in score 2 G86R mice [day 105 (D105)] compared with WT (49.9% vs 71.5%) (Fig. 4D). Here again, the chronic VPA treatment helped in protecting from the loss of myelinated fibers at that age (15% improvement). At the end stage of the disease, the myelinated axons occupancy in VPA-treated animals was still larger than in untreated animals (∼10%) (Fig. 4D), a stage at which we previously counted twice as many large lumbar MN cellular bodies (Fig. 4A). These data further support the neuroprotective role of VPA.
Figure 4.

Chronic VPA administration delays motoneuronal death, maintains high levels of CBP protein, and upregulates CREB/CBP-dependent transcription in vivo. WT and G86R mice were chronically injected with VEH (0.09% NaCl) or VPA (250 mg · kg−1 · d−1) from 60 d of age until score 2 (105 d; G86R-D105) or score 0 (end stage; G86R-ES). A, Nonadjacent spinal cord sections from WT (n = 5) or transgenic mice were immunostained using an anti-ChAT antibody followed by a FITC-conjugated antibody. ChAT-positive MNs with an area >600 μm2 are represented as means ± SEM per section. The counting was performed on G86R-D105 and G86R-ES; white and gray histograms correspond to untreated and VPA-treated animals, respectively. *p < 0.05 and ***p < 0.0001 compared with WT conditions; #p < 0.05 compared with G86R plus VEH condition. B, Representative semithin section of an L4 ventral root from a WT mouse or a G86R-D105. Scale bars, 50 μm. C, D, Representative pictures (C) and quantification (D) of myelinated fibers occupancy in the different groups of mice (a, WT; b, G86R-D105 plus VEH; c, G86R-D105 plus VPA; d, G86R-ES plus VEH; e, G86R-ES plus VPA). Scale bar, 20 μm. D, The percentage of occupancy of myelinated axons within the L4 VR is represented for each group. The data represent means ± SEM. The number of each animal tested is noted within the histogram. Statistical analysis were performed with ANOVA followed by the post hoc Newman–Keuls multiple-comparisons test (Prism). All situations are compared with WT, and in each group, VPA treatment is compared with VEH. *p < 0.05; **p < 0.001. E, Representative pictures of CBP protein immunostaining in lumbar spinal cord from WT and G86R mice (score 0) chronically injected with VEH or VPA (250 mg · kg−1 · d−1). Arrowheads depict lumbar motor neurons. Scale bars, 40 μm. F, Expression of bcl-2 and smn were analyzed by semiquantitative RT-PCR 24 h after injection in spinal cord extracts from 105-d-old WT or G86R (score 2) that received or not an acute injection of VEH, VPA (250 mg/kg), TSA (2 mg/kg), or NaBu (640 mg/kg). Agarose gels were scanned, and specific bands were quantified. The results are represented relative to the expression levels of the house-keeping 18S gene mRNA levels. #p < 0.05 and ##p < 0.001 compared with the untreated G86R condition.
Figure 5.

Chronic VPA delays ALS onset but does not improve mouse survival. A, Time course of clinical rating scale established on a cohort of G86R mice and representing the mean age of appearance of symptoms (see Material and Methods). B–E, G86R mice were chronically injected with VEH (0.09% NaCl; n = 21) or VPA (250 mg · kg−1 · d−1; n = 15) from 60 d of age (score 4) until the end of their life (score 0). B–D, Kaplan–Meier curves representing the proportion of G86R mice with a score >3 (B), >2 (C), and >1 (D). E, Kaplan–Meier curve representing the survival of G86R mice relative to time and treatment.
We also verified that chronic VPA administration had no effect on the expression of the sod1(G86R) gene by Q-PCR experiments performed on lumbar spinal cord extracts (data not shown), suggesting that the neuroprotective effect of VPA does not result from a repression of the mutated sod1 transgene.
Most importantly, we found that CBP levels were maintained in MN nuclei from VPA-treated mice (Fig. 4E), suggesting that, as seen in NSC34 cells, VPA could directly upregulate CBP at the transcriptional level. Together, these results suggest that chronic VPA administration could reverse not only histone deacetylation in MNs but also CBP protein loss, two events that could account for the neuroprotective effect mediated by VPA in vivo. Additionally, we checked by Q-PCR performed on whole lumbar spinal cord extracts whether CBP loss in G86R mice, and its upregulation in VPA-treated mice, could result from transcriptional regulations as observed in the NSC34 cells. However, we failed to detect any significant regulation of CBP mRNA levels in any conditions (untreated and VPA-treated end-stage G86R mice and their age-matched WT littermates; data not shown). It is possible that the heterogeneity of the cellular spinal cord population might confound the transcriptional regulations operated in MNs; this point remains to be established. Nevertheless, we found that treatment with HDACis could improve CREB-dependent transcription in vivo, in G86R mouse lumbar spinal cords. Indeed, expression of the antiapoptotic bcl-2 and the survival of motoneuron smn genes, potentially implicated in neuronal survival and identified as CREB-dependent targets (Impey et al., 2004), was upregulated in response to an acute injection of VPA or two other HDACis (Fig. 4F). Altogether, our results suggest that VPA-mediated neuroprotection could be correlated with maintenance of neuroprotective amounts of CBP protein and upregulation of CREB/CBP-dependent neuroprotective genes.
Motoneuronal protection by chronic VPA treatment delays disease onset but does not increase the lifespan of transgenic ALS mice
We then tested the effects of chronic VPA treatment (250 mg · kg−1 · d−1) on disease onset and survival. To this aim, we first established a rating scale representing disease progression and ranging from score 4 (no visible symptoms) to score 0 (dying mouse) (see Material and Methods). Figure 5A shows the mean ages of appearance and duration of each score, as assessed on a cohort of untreated G86R mice. Treated and untreated mice were then evaluated for their onset in each of these scores (Fig. 5B–E). Statistical analysis of Kaplan–Meier curves showed that onset of score 3 (i.e., first ALS symptoms) was significantly delayed in VPA-treated G86R mice by 10% (Fig. 5B) (p = 0.027). Onsets of scores 2 and 1 were also significantly delayed but to a lesser extent (Fig. 5C,D). Overall and despite the neuroprotective effect observed on lumbar MNs, we found no significant effect of chronic VPA treatment on the lifespan of G86R mice [110.5 ± 1.46 d for untreated animals (n = 21) vs 114.3 ± 2.34 d for VPA-treated animals (n = 15) (Fig. 5E)]. A lower dose of VPA (150 mg · kg−1 · d−1) did not modify the mean lifespan of animals [108.9 ± 1.96 d for untreated animals (n = 12) vs 108.6 ± 4.48 d for VPA-treated animals (n = 9)], whereas increasing VPA concentrations to 400 mg · kg−1 · d−1 was toxic to G86R mice (data not shown). We also conducted a survival study with another HDACi, sodium butyrate, at a dose that has previously been shown to display in vivo neuroprotective effect on a Huntington's disease mouse model (Ferrante et al., 2003), but we found no beneficial effect on G86R mice (supplemental material 1, available at www.jneurosci.org). Together, these data show that protecting against MN death could only provide mild beneficial effects at the onset of the disease and did not prevent mice from dying with the same clinical symptoms typical of ALS. We thus evaluated more carefully different criteria of motor dysfunction in treated and untreated mice, to further analyze the lack of effect of VPA on mouse survival (more precisely, the muscular and the neuromuscular junction aspects).
Chronic VPA administration prevents muscular alteration in mouse only at the first stages of ALS
The muscle functions were then further evaluated, first by measuring muscle strength with the grip test. Two-way ANOVA analysis of the results showed that muscular strength of G86R mice was gradually reduced as the disease progressed (p < 0.0001) and that VPA-treated transgenic mice presented a significant increase in motor performances (p < 0.0001) compared with untreated mice (Fig. 6A). This difference was observed very early, after 2 weeks of treatment, at ∼11 weeks of age, when mice did not present any motor symptoms (score 4). Altogether, these results show that VPA treatment induced a rapid and sustained increase in skeletal muscle strength. Second, we measured muscle fiber area in our group of experimental mice and found that, if the size of fibers was diminished as expected because of muscular atrophy during the course of the disease, VPA did prevent muscular atrophy at the early stage of the disease (score 2) (Fig. 6B). Nevertheless, muscle fiber area was not significantly different at the end stage. These results are in line with the previous observation, in that mice with muscle fibers that are preserved from atrophy by VPA also present a better score at the grip test, and underline that muscle function is protected only at the early stage of the disease.
Figure 6.

Chronic VPA administration delays but does not prevent loss of muscle function and does not improve body mass. WT and G86R mice were chronically injected with VEH (0.09% NaCl) or VPA (250 mg · kg−1 · d−1) from 60 d of age until score 2 (105 d; D105) or score 0 (end stage). A, Evolution of muscle strength assessed by grip test twice a week. ▿, G86R (n = 21); ♦, G86R plus VPA (n = 15). B, Muscle fiber area from WT or G86R at D105 and end stage was measured (in square micrometers) on transversal cross sections of gastrocnemius muscle (10 μm thick). C, Evolution of body weight during the survival experiment. □, WT (n = 8); ■, WT plus VPA (n = 10); ▵, G86R (n = 21); ▴, G86R plus VPA (n = 15). D, Concentration of total plasmatic l-carnitine was measured in the different groups of animals at D105 and end stage. Results are given in micromoles per liter. B, D, White and gray histograms correspond to untreated and VPA-treated animals, respectively. The data represent means ± SEM. The number of each animal tested is noted within the histogram. Statistical analysis was performed with ANOVA followed by the post hoc Newman–Keuls multiple-comparisons test (Prism). All situations are compared with WT, and in each group, VPA treatment (gray histograms) is compared with VEH (white histograms). *p < 0.05; **p < 0.01; ***p < 0.001.
We wanted to further check whether VPA could have some toxic side effects on mice at the dose tested in vivo. It is known that VPA decreases the body mass in rodents (Wolden-Hanson et al., 1998), and we then wondered whether the lack of a significant effect on mice survival could result from a deleterious effect of the molecule on body mass. This question is of particular importance because it is now well established that a dramatic loss of body mass is one of the symptoms of the disease in G86R mice and is related to major metabolic dysfunction (Dupuis et al., 2004). Chronic VPA treatment indeed reduced the gain of body mass of WT animals (Fig. 6C). This effect was observed as soon as 1 week after the beginning of injections and was maintained during all of the time of treatment (p < 0.0001). Surprisingly, VPA had no significant effect on the body mass of transgenic animals. Indeed, although G86R mice presented a characteristic lighter body mass than their WT littermates (p < 0.0001), no difference was found between untreated and VPA-treated G86R mice (Fig. 6C). VPA treatment had no effect on onset of body weight loss either (data not shown), suggesting that variation of mouse body mass cannot account for the lack of beneficial VPA effect on G86R mouse survival. It might be noteworthy that prevention of muscular atrophy (Fig. 6B) may have accounted for the retardation of the appearance of symptoms, at least at the early stage of the disease.
The most frequent alterations reported for VPA, in cases of acute intoxication, are hepatotoxicity and alteration of carnitine metabolism (for review, see Lheureux et al., 2005). Moreover, because a recent study showed that l-carnitine treatment of SOD1(G93A) mice significantly delayed the onset of signs of disease (including deterioration of motor activity) and extended the lifespan of the mice (Kira et al., 2006), a drop in l-carnitine levels induced by VPA might be deleterious in the course of the pathology. We measured l-carnitine levels in the plasma of our experimental groups of mice under chronic treatment of VPA (Fig. 6D). We found that VPA did slightly but not significantly decrease plasmatic levels of carnitine in the WT animals. Of note, plasmatic l-carnitine concentrations increased during the course of the disease, and VPA lowered them back to the normal levels found in WT animals at D105. VPA did not modify increased levels found in G86R mice at the end stage of disease. It is thus unlikely that, at the dose tested, VPA could trigger toxic side effects on animals through modulation of plasmatic carnitine levels.
Chronic VPA treatment delays but does not prevent neuromuscular denervation
Progressive paralysis observed in ALS results from progressive denervation of skeletal muscles (Bjornskov et al., 1984). The delay of symptom onset and the increase in muscle strength seen under VPA treatment suggest that this molecule could retard skeletal muscle denervation. To test this hypothesis, we performed electromyographic analysis on score 3 transgenic mice and their WT age-matched littermates (100 d of age). Recording of the resting activity in the gastrocnemius muscle of untreated G86R mice (Fig. 7A, middle) consistently revealed abnormal spontaneous activity with fibrillation potentials graded from 3 to 4 (see Material and Methods). Numerous fasciculations were also observed. In contrast, recording the resting activity in the gastrocnemius muscle of chronically VPA-treated mice showed strongly reduced abnormal spontaneous activity with fibrillation potentials graded from 1 to 2 and without any fasciculation (right). As expected, neither fibrillation nor fasciculation were observed in WT animals (left).
Figure 7.
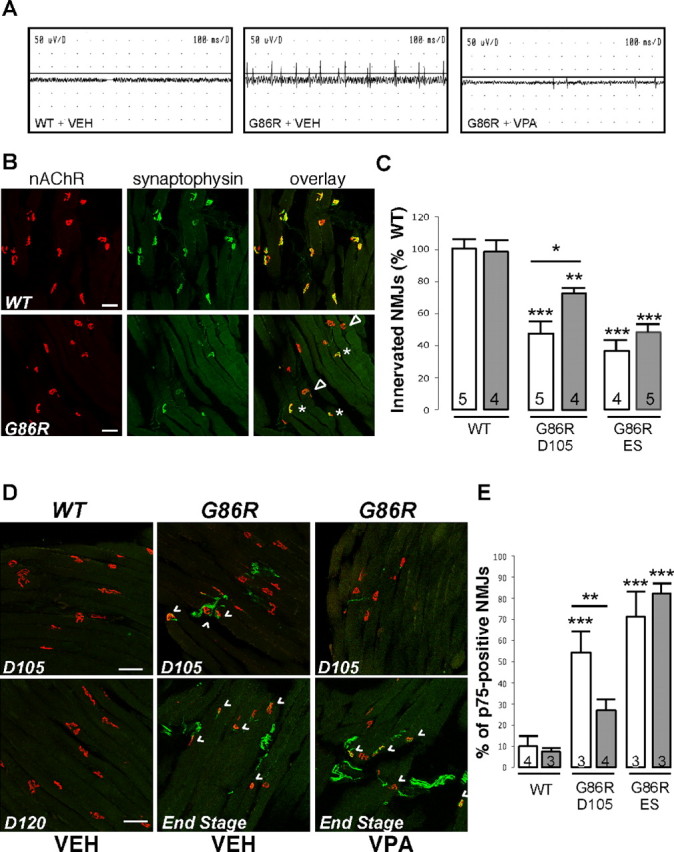
Chronic VPA delays but does not prevent skeletal muscle denervation. A, Representative electromyography recordings performed on 100-d-old WT mice (n = 4) or G86R mice (score 3) daily injected with VEH (n = 3) or VPA (n = 3). B, Typical confocal photographs representing innervated NMJs in the gastrocnemius muscle of a WT and a G86R (end-stage) mouse. nAChRs are labeled with fluorescent α-BGT binding (red), and nerve terminals are labeled by immunochemistry of synaptophysin (green). Fully and partially innervated nAChR clusters are denoted by asterisks and arrowheads, respectively, Scale bars, 25 μm. C, Count of innervated NMJs in each experimental group. D, Visualization of nAChR (α-BGT; red) and p75 immunoreactivity (green) in WT and mutant G86R mice chronically treated with VPA or not as noted. A representative overlay (confocal analysis) is shown for each animal group. NMJs counted as positive are shown with arrowheads. WT mice corresponding to each disease stage tested were killed as noted (D105 and D120). Scale bars, 25 μm. E, Counts of p75-positive NMJs in the gastrocnemius muscle of WT and G86R chronically treated with vehicle or VPA. C, E, The data represent means ± SEM. The number of each animal tested is noted within the histogram. Statistical analysis were performed with ANOVA followed by the post hoc Newman–Keuls multiple-comparisons test (Prism) All situations are compared with WT, and in each group, VPA treatment (gray histograms) is compared with VEH (white histograms). *p < 0.05; **p < 0.01; ***p < 0.001.
To check whether VPA could have had a direct effect on membrane potential of myocytes (independently of motor innervation), we tested the electromyographic response to treatment of mice with two doses of VPA (250 and 400 mg/kg) after a right sciatic nerve axotomy. All groups of mice, whether treated or not, presented with a full interference pattern of spontaneous potentials in the right gastrocnemius muscle 3 and 8 d after axotomy of the right sciatic nerve (supplemental material 2, available at www.jneurosci.org). These results suggest that treatment with VPA does not interfere with the electrical activity of normal and denervated skeletal muscle.
Thus, it seems that VPA treatment may delay muscle denervation in ALS mice, and we further counted the number of innervated synapses in muscles. To this aim, the presynaptic terminals were identified on longitudinal soleus sections using an anti-synaptophysin antibody, and nAChRs were labeled by fluorescent α-BGT binding. Figure 7B represents typical photographs obtained after confocal analysis of a WT and a score 1 G86R mouse. We found that the number of innervated neuromuscular junctions (NMJs) decreased in nontreated animals and that VPA treatment slightly helped in preventing denervation but only at the early stage of the disease (Fig. 7C).
Finally, we performed an immunohistological labeling against p75, which is highly expressed in both MN presynaptic terminals and terminal Schwann cells after denervation (You et al., 1997). Figure 7D shows representative sections of each type of animal (WT and G86R, treated or not, D105 and end-stage mice) photographed using confocal microscopy. Quantification is presented on Figure 7E. We observed that most of the NMJs were p75 positive in end-stage G86R animals, either treated or not. VPA significantly delayed p75 expression at D105, although p75-positive NMJs tend to be more present in D105 VPA-treated animals than in WT or WT VPA-treated mice (27 vs 8–10%).
Thus, we can conclude that chronic VPA administration modestly delays life extent and motor performance in vivo.
Discussion
Modulation of cbp gene transcription by oxidative stress and HDACis
In a previous report, we evidenced a loss of CBP in MN nuclei from G86R mice spinal cord (Rouaux et al., 2003). Herein, we show that an acute oxidative stress is able to downregulate cbp gene transcription. Loss of CBP is a recurrent phenomenon observed in different neurological disorders. Interestingly, it has been shown to occur through a diversity of mechanisms depending on the pathological context [for review, see Rouaux et al. (2004) and Saha and Pahan (2006)], but so far, cbp transcriptional repression has only been reported in a presenilin knock-out mouse model presenting increased neurodegeneration and memory alterations (Saura et al., 2004). It is thought that MN death in ALS involves oxidative stress (for review, see Simpson et al., 2003). Consistent with this, we found that overexpression of the mutant sod1(G86R) strongly repressed cbp promoter-driven luciferase activity, suggesting that chronic SOD1 dysfunction in transgenic animals and subsequent oxidative stress could be responsible for the drop of CBP levels. Remarkably, cbp was found among the downregulated genes in a gene expression profiling performed on MNs microdissected from spinal cords of ALS patients (Jiang et al., 2005).
In a therapeutic view, if cbp gene transcription were repressed in MNs, then a drug that proves efficient at reactivating its transcription (and subsequent CBP-dependent transcription) might favor neuroprotection. For a few years, because of their capacity to activate transcription by blocking deacetylation, HDACis have been tested as potential neuroprotective agents in several cellular and animal models of neurodegenerative diseases (Rouaux et al., 2004; Langley et al., 2005; Saha and Pahan, 2006). It is believed that global deacetylation will induce transcriptional repressions associated with pathologies. Such genetic alterations are observed in the spinal cord of transgenic mice and ALS patients (Dupuis et al., 2000; Ishigaki et al., 2002; Malaspina and de Belleroche, 2004; Jiang et al., 2005). Thus, inhibiting HDAC activity could remove repressive blocks from promoters, thereby providing neuroprotection. Herein, we present for the first time evidence that HDACis can combat neurodegeneration through modulation of the cbp transcriptional pathway (cbp and its targeted genes), not only in simplified cellular models in response to oxidative stress, but also in vivo in the pathological context of ALS. Interestingly, HDACi treatment did not revert neuronal apoptosis induced by electrical activity depletion (Rouaux et al., 2004), a model in which the CBP protein is degraded by posttranslational caspase-6 dependent mechanisms (Rouaux et al., 2003). Together, these results suggest that the presence of CBP is required, or at least more effective than random reacetylation with HDACi treatment, in counteracting neuronal death, either because its other functions (molecular scaffolding and transcriptional coactivator) are needed or because HDAC inhibition is not specific to CBP-dependent genes.
VPA delays MN death but does not significantly increase the mean survival of ALS mice
The capacity of HDACis to modulate SOD1(G86R) mice lifespan has recently been investigated in several other studies. Sugai et al. (2004) showed that a chronic preonset VPA treatment significantly increased mice survival of SOD1(G93A) mice. Our results thus contrast with this study, although both reports showed a neuroprotective effect of VPA in vivo. However, some differences are found in the experimental design between studies. Whereas we treated G86R mice at 250 mg · kg−1 · d−1 intraperitoneally, Sugai and coworkers administrated VPA in drinking water at 0.26% w/v, which corresponds to approximately twice our dose (530 mg · kg−1 · d−1). It is thus possible that at a higher dose, VPA may have affected other genes and cellular pathways. Indeed, gene-profiling experiments showed that different doses of VPA induced different panels of genes (Massa et al., 2005), so they might also have different therapeutic effects. However, in our hands, a higher dose of VPA (400 mg · kg−1 · d−1) injected intraperitoneally was lethal to the G86R mice population. It is noteworthy that not only the mode of injection is different in the two studies, but also the mouse strains used [i.e., SOD1(G93A) vs SOD1(G86R)], which do not display the same pathological time course and may not have the same sensitivity/metabolism for VPA. Nevertheless, VPA therapy was also established as ineffectual in a retest performed on the G93A mice strain (n = 18 nontreated and n = 19 treated animals), given at preonset of disease (60 d of age) by subcutaneous pump [131.6 d control group vs 125.9 d VPA-treated group (Scott et al., 2007) (see also details on http://www.als.net/)]. Overall, the only currently available FDA-validated drug to treat ALS is riluzole (Bensimon et al., 1994). In the ALS mouse model, this molecule has been found to increase survival by 10% (Gurney et al., 1996, 1998), a threshold that can be considered clinically significant. In this respect, our data are obtained with a sufficient number of treated mice (n = 15) to assess with a power of >80% that we can reach 10% survival improvement. As we only observed 5% (not statistically significant) increase in lifespan, we concluded that VPA, although protecting MNs efficiently, does not display clinically significant improvement in survival.
Nevertheless, it has been shown recently that treatment with another HDACi, phenyl butyrate (PBA), resulted in significant enhancement of survival and improvement in clinical and neuropathological phenotypes of the SOD1(G93A) mice (Ryu et al., 2005; Petri et al., 2006). PBA was shown to increase histone acetylation (Petri et al., 2006), block apoptotic signaling, upregulate bcl-2 gene (Ryu et al., 2005), and increase MN survival, life extent, and motor performances in vivo (Ryu et al., 2005; Petri et al., 2006). Although here again, the strategy was aimed at resetting global histone acetylation levels, the 22% survival improvement under PBA treatment observed by Ryu et al. (2005) was associated with a significant increase in body weight (∼20%), which is a major difference with our study, in which we observed that VPA had no effect on diseased animal body weight. This discrepancy seems of prime interest, because we previously showed that increasing adiposity and body weight of G86R mice by feeding them with a high-energetic diet significantly increased their survival (Dupuis et al., 2004). Thus, the lack of beneficial effect of VPA on energy deficit of G86R mice may be an explanation why it failed to improve mice survival although an efficient neuroprotection was observed.
Overall, these data indicate the poor efficiency of HDACi treatment for ALS therapy despite the neuroprotective efficiency of HDACis (see below). The recent results obtained in other laboratories with HDACis (i.e., PBA) may be reconsidered in light of these studies, as they may have revealed beneficial effects on ALS mice by other means than only a strict neuroprotection.
Motor dysfunctions are not counteracted by VPA treatment in ALS mice
It is noteworthy that, despite neuroprotection, VPA-treated animals died with the neuropathological criteria typical of ALS. Interestingly, several recent studies question the origin of ALS, suggesting that biochemical and molecular events occurring in the muscle fibers (such as hypermetabolism, oxidative stress, and mitochondrial dysfunction) could increase NMJ integrity and spinal MN vulnerability (for review, see Gonzalez de Aguilar et al., 2007). It is thus conceivable that protecting against MN death might not be sufficient to cure ALS. We found that preonset treatment with VPA delayed disease onset in G86R mice and induced a significant amelioration of muscle strength, as well as decreased muscle denervation (EMG studies). This is reminiscent of the fact that the MNs as well as their proximal axons were spared in the treated animals: protecting MN from death may have retarded the disease onset. Recently, in a series of elegant experiments using genetic elimination of programmed cell death in SOD1 mice by crossings with Bax knock-out mutants, Gould et al. (2006) provided evidence that damage to the distal motor axon was an event dissociated from MN death. More recently, Dewil et al. (2007) found that the antiapoptotic effect of inhibition of p38 MAPK (mitogen-activated protein kinase) activation rescued MNs and to a lesser degree the proximal axon but that the distal denervation still progressed. In fact, our experiments aimed at counting the number of innervated motor synapses reached the same conclusions: NMJs were already damaged at D105 in VPA-treated animals, whereas the number of MNs was similar to that of WT animals. Moreover, we found that the low-affinity neurotrophin receptor p75 was highly expressed in NMJs of control and VPA-treated animals at the end stage of the disease. Both of these results suggest that the distal pathology (i.e., denervation) was not prevented by neuroprotection.
Altogether, these data show that at the clinical level, neuroprotection of lower MNs is not sufficient for ALS therapy, because it does not prevent NMJ denervation during the pathology. These data strongly suggest that MN death activation is not the required pathogenic event of ALS and point instead to disruption of MN presynaptic terminals.
Footnotes
C.R. is a recipient of a fellowship from the French Research Ministry. The laboratory was supported by grants from “Association Française contre les Myopathies,” “Association pour la Recherche sur la SLA,” “Association pour la Recherche sur les Maladies Neurodégénératives,” “Association pour la Recherche contre le Cancer,” and “Fédération pour la Recherche sur le Cerveau.” We thank Annie Picchinenna and Marie-Jo Ruivo for their technical assistance. We thank Bastien Fricker and Muriel Hoering for assistance with the p75 immunohistochemistry and muscle fiber counting, respectively.
References
- Andersen PM, Sims KB, Xin WW, Kiely R, O'Neill G, Ravits J, Pioro E, Harati Y, Brower RD, Levine JS, Heinicke HU, Seltzer W, Boss M, Brown RH., Jr Sixteen novel mutations in the Cu/Zn superoxide dismutase gene in amyotrophic lateral sclerosis: a decade of discoveries, defects and disputes. Amyotroph Lateral Scler Other Motor Neuron Disord. 2003;4:62–73. doi: 10.1080/14660820310011700. [DOI] [PubMed] [Google Scholar]
- Bannister AJ, Kouzarides T. The CBP co-activator is a histone acetyltransferase. Nature. 1996;384:641–643. doi: 10.1038/384641a0. [DOI] [PubMed] [Google Scholar]
- Bensimon G, Lacomblez L, Meininger V. A controlled trial of riluzole in amyotrophic lateral sclerosis. ALS/Riluzole Study Group. N Engl J Med. 1994;330:585–591. doi: 10.1056/NEJM199403033300901. [DOI] [PubMed] [Google Scholar]
- Bjornskov EK, Norris FH, Jr, Mower-Kuby J. Quantitative axon terminal and end-plate morphology in amyotrophic lateral sclerosis. Arch Neurol. 1984;41:527–530. doi: 10.1001/archneur.1984.04050170073021. [DOI] [PubMed] [Google Scholar]
- Boillee S, Yamanaka K, Lobsiger CS, Copeland NG, Jenkins NA, Kassiotis G, Kollias G, Cleveland DW. Onset and progression in inherited ALS determined by motor neurons and microglia. Science. 2006;312:1389–1392. doi: 10.1126/science.1123511. [DOI] [PubMed] [Google Scholar]
- Cashman NR, Durham HD, Blusztajn JK, Oda K, Tabira T, Shaw IT, Dahrouge S, Antel JP. Neuroblastoma x spinal cord (NSC) hybrid cell lines resemble developing motor neurons. Dev Dyn. 1992;194:209–221. doi: 10.1002/aja.1001940306. [DOI] [PubMed] [Google Scholar]
- Cookson MR, Ince PG, Shaw PJ. Peroxynitrite and hydrogen peroxide induced cell death in the NSC34 neuroblastoma x spinal cord cell line: role of poly (ADP-ribose) polymerase. J Neurochem. 1998;70:501–508. doi: 10.1046/j.1471-4159.1998.70020501.x. [DOI] [PubMed] [Google Scholar]
- Dewil M, Dela Cruz VF, Van Den Bosch L, Robberecht W. Inhibition of p38 mitogen activated protein kinase activation and mutant SOD1(G93A)-induced motor neuron death. Neurobiol Dis. 2007;26:332–341. doi: 10.1016/j.nbd.2006.12.023. [DOI] [PubMed] [Google Scholar]
- Dupuis L, de Tapia M, Rene F, Lutz-Bucher B, Gordon JW, Mercken L, Pradier L, Loeffler JP. Differential screening of mutated SOD1 transgenic mice reveals early up-regulation of a fast axonal transport component in spinal cord motor neurons. Neurobiol Dis. 2000;7:274–285. doi: 10.1006/nbdi.2000.0292. [DOI] [PubMed] [Google Scholar]
- Dupuis L, di Scala F, Rene F, de Tapia M, Oudart H, Pradat PF, Meininger V, Loeffler JP. Up-regulation of mitochondrial uncoupling protein 3 reveals an early muscular metabolic defect in amyotrophic lateral sclerosis. FASEB J. 2003;17:2091–2093. doi: 10.1096/fj.02-1182fje. [DOI] [PubMed] [Google Scholar]
- Dupuis L, Oudart H, Rene F, Gonzalez de Aguilar JL, Loeffler JP. Evidence for defective energy homeostasis in amyotrophic lateral sclerosis: benefit of a high-energy diet in a transgenic mouse model. Proc Natl Acad Sci USA. 2004;101:11159–11164. doi: 10.1073/pnas.0402026101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrante RJ, Kubilus JK, Lee J, Ryu H, Beesen A, Zucker B, Smith K, Kowall NW, Ratan RR, Luthi-Carter R, Hersch SM. Histone deacetylase inhibition by sodium butyrate chemotherapy ameliorates the neurodegenerative phenotype in Huntington's disease mice. J Neurosci. 2003;23:9418–9427. doi: 10.1523/JNEUROSCI.23-28-09418.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer LR, Culver DG, Tennant P, Davis AA, Wang M, Castellano-Sanchez A, Khan J, Polak MA, Glass JD. Amyotrophic lateral sclerosis is a distal axonopathy: evidence in mice and man. Exp Neurol. 2004;185:232–240. doi: 10.1016/j.expneurol.2003.10.004. [DOI] [PubMed] [Google Scholar]
- Gong YH, Parsadanian AS, Andreeva A, Snider WD, Elliott JL. Restricted expression of G86R Cu/Zn superoxide dismutase in astrocytes results in astrocytosis but does not cause motoneuron degeneration. J Neurosci. 2000;20:660–665. doi: 10.1523/JNEUROSCI.20-02-00660.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez de Aguilar JL, Echaniz-Laguna A, Fergani A, Rene F, Meininger V, Loeffler JP, Dupuis L. Amyotrophic lateral sclerosis: all roads lead to Rome. J Neurochem. 2007 doi: 10.1111/j.1471-4159.2006.04408.x. in press. [DOI] [PubMed] [Google Scholar]
- Gould TW, Buss RR, Vinsant S, Prevette D, Sun W, Knudson CM, Milligan CE, Oppenheim RW. Complete dissociation of motor neuron death from motor dysfunction by Bax deletion in a mouse model of ALS. J Neurosci. 2006;26:8774–8786. doi: 10.1523/JNEUROSCI.2315-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurney ME, Cutting FB, Zhai P, Doble A, Taylor CP, Andrus PK, Hall ED. Benefit of vitamin E, riluzole, and gabapentin in a transgenic model of familial amyotrophic lateral sclerosis. Ann Neurol. 1996;39:147–157. doi: 10.1002/ana.410390203. [DOI] [PubMed] [Google Scholar]
- Gurney ME, Fleck TJ, Himes CS, Hall ED. Riluzole preserves motor function in a transgenic model of familial amyotrophic lateral sclerosis. Neurology. 1998;50:62–66. doi: 10.1212/wnl.50.1.62. [DOI] [PubMed] [Google Scholar]
- Impey S, McCorkle SR, Cha-Molstad H, Dwyer JM, Yochum GS, Boss JM, McWeeney S, Dunn JJ, Mandel G, Goodman RH. Defining the CREB regulon: a genome-wide analysis of transcription factor regulatory regions. Cell. 2004;119:1041–1054. doi: 10.1016/j.cell.2004.10.032. [DOI] [PubMed] [Google Scholar]
- Ishigaki S, Niwa J, Ando Y, Yoshihara T, Sawada K, Doyu M, Yamamoto M, Kato K, Yotsumoto Y, Sobue G. Differentially expressed genes in sporadic amyotrophic lateral sclerosis spinal cords—screening by molecular indexing and subsequent cDNA microarray analysis. FEBS Lett. 2002;531:354–358. doi: 10.1016/s0014-5793(02)03546-9. [DOI] [PubMed] [Google Scholar]
- Jiang YM, Yamamoto M, Kobayashi Y, Yoshihara T, Liang Y, Terao S, Takeuchi H, Ishigaki S, Katsuno M, Adachi H, Niwa J, Tanaka F, Doyu M, Yoshida M, Hashizume Y, Sobue G. Gene expression profile of spinal motor neurons in sporadic amyotrophic lateral sclerosis. Ann Neurol. 2005;57:236–251. doi: 10.1002/ana.20379. [DOI] [PubMed] [Google Scholar]
- Jokic N, Gonzalez de Aguilar JL, Dimou L, Lin S, Fergani A, Ruegg MA, Schwab ME, Dupuis L, Loeffler JP. The neurite outgrowth inhibitor Nogo-A promotes denervation in an amyotrophic lateral sclerosis model. EMBO Rep. 2006;7:1162–1167. doi: 10.1038/sj.embor.7400826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kira Y, Nishikawa M, Ochi A, Sato E, Inoue M. L-carnitine suppresses the onset of neuromuscular degeneration and increases the life span of mice with familial amyotrophic lateral sclerosis. Brain Res. 2006;1070:206–214. doi: 10.1016/j.brainres.2005.11.052. [DOI] [PubMed] [Google Scholar]
- Langley B, Gensert JM, Beal MF, Ratan RR. Remodeling chromatin and stress resistance in the central nervous system: histone deacetylase inhibitors as novel and broadly effective neuroprotective agents. Curr Drug Targets CNS Neurol Disord. 2005;4:41–50. doi: 10.2174/1568007053005091. [DOI] [PubMed] [Google Scholar]
- Lheureux PE, Penaloza A, Zahir S, Gris M. Science review: carnitine in the treatment of valproic acid-induced toxicity—what is the evidence? Crit Care. 2005;9:431–440. doi: 10.1186/cc3742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu R, Althaus JS, Ellerbrock BR, Becker DA, Gurney ME. Enhanced oxygen radical production in a transgenic mouse model of familial amyotrophic lateral sclerosis. Ann Neurol. 1998;44:763–770. doi: 10.1002/ana.410440510. [DOI] [PubMed] [Google Scholar]
- Malaspina A, de Belleroche J. Spinal cord molecular profiling provides a better understanding of amyotrophic lateral sclerosis pathogenesis. Brain Res Brain Res Rev. 2004;45:213–229. doi: 10.1016/j.brainresrev.2004.04.002. [DOI] [PubMed] [Google Scholar]
- Massa V, Cabrera RM, Menegola E, Giavini E, Finnell RH. Valproic acid-induced skeletal malformations: associated gene expression cascades. Pharmacogenet Genomics. 2005;15:787–800. doi: 10.1097/01.fpc.0000170914.11898.3a. [DOI] [PubMed] [Google Scholar]
- Miller TM, Kim SH, Yamanaka K, Hester M, Umapathi P, Arnson H, Rizo L, Mendell JR, Gage FH, Cleveland DW, Kaspar BK. Gene transfer demonstrates that muscle is not a primary target for non-cell-autonomous toxicity in familial amyotrophic lateral sclerosis. Proc Natl Acad Sci USA. 2006;103:19546–19551. doi: 10.1073/pnas.0609411103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasinelli P, Brown RH. Molecular biology of amyotrophic lateral sclerosis: insights from genetics. Nat Rev Neurosci. 2006;7:710–723. doi: 10.1038/nrn1971. [DOI] [PubMed] [Google Scholar]
- Perucca E. Pharmacological and therapeutic properties of valproate: a summary after 35 years of clinical experience. CNS Drugs. 2002;16:695–714. doi: 10.2165/00023210-200216100-00004. [DOI] [PubMed] [Google Scholar]
- Petri S, Kiaei M, Kipiani K, Chen J, Calingasan NY, Crow JP, Beal MF. Additive neuroprotective effects of a histone deacetylase inhibitor and a catalytic antioxidant in a transgenic mouse model of amyotrophic lateral sclerosis. Neurobiol Dis. 2006;22:40–49. doi: 10.1016/j.nbd.2005.09.013. [DOI] [PubMed] [Google Scholar]
- Ripps ME, Huntley GW, Hof PR, Morrison JH, Gordon JW. Transgenic mice expressing an altered murine superoxide dismutase gene provide an animal model of amyotrophic lateral sclerosis. Proc Natl Acad Sci USA. 1995;92:689–693. doi: 10.1073/pnas.92.3.689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosen DR, Siddique T, Patterson D, Figlewicz DA, Sapp P, Hentati A, Donaldson D, Goto J, O'Regan JP, Deng HX, Rahmani Z, Krizus A, McKenna-Yasek D, Cayabyab A, Gaston SM, Berger R, Tanzi RE, Halperin JJ, Herzfeldt B, Van den Bergh R, et al. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature. 1993;362:59–62. doi: 10.1038/362059a0. [DOI] [PubMed] [Google Scholar]
- Rouaux C, Jokic N, Mbebi C, Boutillier S, Loeffler JP, Boutillier AL. Critical loss of CBP/p300 histone acetylase activity by caspase-6 during neurodegeneration. EMBO J. 2003;22:6537–6549. doi: 10.1093/emboj/cdg615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rouaux C, Loeffler JP, Boutillier AL. Targeting CREB-binding protein (CBP) loss of function as a therapeutic strategy in neurological disorders. Biochem Pharmacol. 2004;68:1157–1164. doi: 10.1016/j.bcp.2004.05.035. [DOI] [PubMed] [Google Scholar]
- Ryu H, Smith K, Camelo SI, Carreras I, Lee J, Iglesias AH, Dangond F, Cormier KA, Cudkowicz ME, Brown RH, Jr, Ferrante RJ. Sodium phenylbutyrate prolongs survival and regulates expression of anti-apoptotic genes in transgenic amyotrophic lateral sclerosis mice. J Neurochem. 2005;93:1087–1098. doi: 10.1111/j.1471-4159.2005.03077.x. [DOI] [PubMed] [Google Scholar]
- Saha RN, Pahan K. HATs and HDACs in neurodegeneration: a tale of disconcerted acetylation homeostasis. Cell Death Differ. 2006;13:539–550. doi: 10.1038/sj.cdd.4401769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saura CA, Choi SY, Beglopoulos V, Malkani S, Zhang D, Shankaranarayana Rao BS, Chattarji S, Kelleher RJ, III, Kandel ER, Duff K, Kirkwood A, Shen J. Loss of presenilin function causes impairments of memory and synaptic plasticity followed by age-dependent neurodegeneration. Neuron. 2004;42:23–36. doi: 10.1016/s0896-6273(04)00182-5. [DOI] [PubMed] [Google Scholar]
- Scott S, Kranz JE, Cole J, Lincecum JM, Thompson K, Kelly N, Bostrom A, Theodoss J, Al-Nakhala BM, Vieira FG, Ramesh T, Ramasubbu J, Heywood JA. Design, power, and interpretation of studies in the standard murine model of ALS. PLoS Med. 2007 doi: 10.1080/17482960701856300. in press. [DOI] [PubMed] [Google Scholar]
- Simpson EP, Yen AA, Appel SH. Oxidative stress: a common denominator in the pathogenesis of amyotrophic lateral sclerosis. Curr Opin Rheumatol. 2003;15:730–736. doi: 10.1097/00002281-200311000-00008. [DOI] [PubMed] [Google Scholar]
- Sugai F, Yamamoto Y, Miyaguchi K, Zhou Z, Sumi H, Hamasaki T, Goto M, Sakoda S. Benefit of valproic acid in suppressing disease progression of ALS model mice. Eur J Neurosci. 2004;20:3179–3183. doi: 10.1111/j.1460-9568.2004.03765.x. [DOI] [PubMed] [Google Scholar]
- Tremolizzo L, Carboni G, Ruzicka WB, Mitchell CP, Sugaya I, Tueting P, Sharma R, Grayson DR, Costa E, Guidotti A. An epigenetic mouse model for molecular and behavioral neuropathologies related to schizophrenia vulnerability. Proc Natl Acad Sci USA. 2002;99:17095–17100. doi: 10.1073/pnas.262658999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verdone L, Caserta M, Di Mauro E. Role of histone acetylation in the control of gene expression. Biochem Cell Biol. 2005;83:344–353. doi: 10.1139/o05-041. [DOI] [PubMed] [Google Scholar]
- Wan L, Hubbard RW. Determination of free and total carnitine with a random-access chemistry analyzer. Clin Chem. 1998;44:810–816. [PubMed] [Google Scholar]
- Wolden-Hanson T, Gidal BE, Atkinson RL. Evaluation of a rat model of valproate-induced obesity. Pharmacotherapy. 1998;18:1075–1081. [PubMed] [Google Scholar]
- You S, Petrov T, Chung PH, Gordon T. The expression of the low affinity nerve growth factor receptor in long-term denervated Schwann cells. Glia. 1997;20:87–100. doi: 10.1002/(sici)1098-1136(199706)20:2<87::aid-glia1>3.0.co;2-1. [DOI] [PubMed] [Google Scholar]
- Yu J, de Belle I, Liang H, Adamson ED. Coactivating factors p300 and CBP are transcriptionally crossregulated by Egr1 in prostate cells, leading to divergent responses. Mol Cell. 2004;15:83–94. doi: 10.1016/j.molcel.2004.06.030. [DOI] [PubMed] [Google Scholar]