

Abstract
Huntingtin-interacting protein 1 (HIP1) was identified through its interaction with htt (huntingtin), the Huntington's disease (HD) protein. HIP1 is an endocytic protein that influences transport and function of AMPA and NMDA receptors in the brain. However, little is known about its contribution to neuronal dysfunction in HD.
We report that the Caenorhabditis elegans HIP1 homolog hipr-1 modulates presynaptic activity and the abundance of synaptobrevin, a protein involved in synaptic vesicle fusion. Presynaptic function was also altered in hippocampal brain slices of HIP1−/− mice demonstrating delayed recovery from synaptic depression and a reduction in paired-pulse facilitation, a form of presynaptic plasticity. Interestingly, neuronal dysfunction in transgenic nematodes expressing mutant N-terminal huntingtin was specifically enhanced by hipr-1 loss of function. A similar effect was observed with several other mutant proteins that are expressed at the synapse and involved in endocytosis, such as unc-11/AP180, unc-26/synaptojanin, and unc-57/endophilin.
Thus, HIP1 is involved in presynaptic nerve terminal activity and modulation of mutant polyglutamine-induced neuronal dysfunction. Moreover, synaptic proteins involved in endocytosis may protect neurons against amino acid homopolymer expansion.
Keywords: HIP1, synapse, synaptic plasticity, polyglutamine, Huntington's disease, neurodegeneration
Introduction
Huntingtin-interacting protein 1 (HIP1) was identified as an interacting partner of huntingtin (htt), the protein whose gene is mutated in Huntington's disease (HD) (Kalchman et al., 1997; Wanker et al., 1997). Mutant htt contains an expanded polyglutamine (polyQ) tract that influences its interaction with HIP1, suggesting a role in HD pathogenesis (Kalchman et al., 1997; Hackam et al., 2000; Gervais et al., 2002).
HIP1 contains several conserved domains that are important for function including the following: an AP180 NH2-terminal homology (ANTH) domain enabling membrane attachment, a central coiled-coil region, and a C-terminal THATCH domain (Metzler et al., 2001; Mishra et al., 2001; Waelter et al., 2001; Senetar et al., 2004). HIP1 has been implicated in numerous cellular processes. First, HIP1 partially colocalizes and copurifies with clathrin-coated vesicles and is involved in clathrin-mediated endocytosis of cell surface receptors (Metzler et al., 2001; Mishra et al., 2001; Waelter et al., 2001). Studies in primary neurons of HIP1−/− mice demonstrate altered clathrin-dependent AMPA receptor internalization, supporting a role of HIP1 in neurotransmitter receptor endocytosis and the modulation of synaptic activity, in agreement with the enrichment of HIP1 in dendrites and at postsynaptic sites (Metzler et al., 2003; Okano et al., 2003; Yao et al., 2006). Second, HIP1 has tumorigenic properties because the overexpression of HIP1 is associated with prostate and colon cancers, perhaps by dysregulation of receptors including the epidermal growth factor receptor (Rao et al., 2002, 2003). Additionally, a nuclear role for HIP1 has been discovered because it can regulate the transcriptional activity of the androgen receptor (Mills et al., 2005). Third, HIP1−/− mice have complex phenotypes demonstrating a role in development. HIP1−/− mice display spinal deformity early in adulthood and muscle wasting in their hindlimbs, show abnormal hematopoieses, and have reproductive defects associated with reduced sperm counts and motility (Metzler et al., 2003; Oravecz-Wilson et al., 2004; Khatchadourian et al., 2007). Finally, the binding of HIP1 to mutant htt is altered by polyQ expansion and coexpression of HIP1 with mutant htt in HEK 293T cells induces caspase-dependent cell death in which free HIP1 binds to the related protein Hippi, recruiting caspase-8 to initiate apoptosis (Kalchman et al., 1997; Hackam et al., 2000; Gervais et al., 2002).
To better understand HIP1 function under normal and pathological conditions, we examined the Caenorhabditis elegans HIP1 homolog Huntingtin-interacting protein related 1 (hipr-1). Here, we provide evidence that hipr-1 influences cholinergic neurotransmission at the neuromuscular junction and alters the abundance of synaptobrevin along the ventral nerve cord. Additionally, we demonstrate a role for HIP1 at CNS presynaptic nerve terminals in mice. Furthermore, neuronal dysfunction caused by mutant htt-exon1 was enhanced by hipr-1 loss of function (LOF), suggesting a protective function of HIP1. Finally, synaptic proteins regulating endocytosis and vesicle recycling also protected against polyQ toxicity indicating that synaptic vesicle transport may be a site of mutant polyQ-induced neuronal dysfunction. Together, our data reveal a novel function for HIP1 at the presynaptic nerve terminal and show HIP1 belongs to a group of synaptic proteins that protect neurons from dysfunction induced by mutant polyQs.
Materials and Methods
C. elegans handling, strain construction, and phenotypic characterization.
All strains were maintained and handled following standard methods. The hipr-1 insertion/deletion allele tm2207 was generated by the National Bioresource Project for the Nematode (Tokyo, Japan). hipr-1(tm2207) consists of a 340 bp deletion corresponding to nucleotides 2928–3269 of cosmid ZK370, along with the addition of 8 nt (ATATTTTC). This insertion/deletion eliminates a portion of exon 3 and all of exon 4 creating a frameshift, leading to a stop codon after a 12 codon missense mutation. hipr-1(tm2207) was outcrossed seven times before any analyses were made. The presence of the tm2207 deletion in all strain constructions was confirmed by PCR. Oligos used to detect tm2207 were as follows: tm2207-f, AGGTCATCGTTCCAGATTCC; tm2207-r, GCATCGTCTGTCTGAGTTCT.
Additional strains used include CB47 unc-11(e47), CB205 unc-26(e205), CB246 unc-64(e246), CB406 unc-57(e406), CB840 dpy-23(e840), CB1072 unc-29(e1072), DA509 unc-31(e928), DH1033 bIs1[vit-2::GFP] (provided by B. Grant, Rutgers University, Piscataway, NJ), JT73 itr-1(sa73), NL2099 rrf-3(pk1426), NM204 snt-1(md290), and NM664 jsIs37[mec-7::SNB-1::GFP] from the Caenorhabditis Genetics Center, which is funded by the National Institutes of Health National Center for Research Resources.
Defecation cycles were scored essentially as described previously (Dal Santo et al., 1999). For statistical analyses, we first calculated a mean defecation cycle period for each animal, and then a mean and SEM for each strain.
Touch tests were done as described previously by scoring for 10 touches at the tail of the animal for a minimum of 200 animals per genotype (Parker et al., 2001). We used C. elegans transgenics expressing N-ter htt fused to CFP under the control of the mec-3 promoter together with YFP under the control of the mec-7 promoter (Parker et al., 2001, 2005). For the evaluation of the effects of HIP1 on polyQ-dependent neuronal dysfunction, polyQ transgenics were crossed to hipr-1(tm2207), and the presence of the tm2207 deletion in all strain constructions was confirmed by PCR.
RNA interference.
RNA interference (RNAi) experiments were performed as described previously (Kamath et al., 2001). Experiments were performed at least three times. The RNAi clone for hipr-1 was obtained from Geneservice (Cambridge, UK). The control RNAi vector for the progeny and defecation phenotypes was the plasmid L4417, which produces dsRNA against green fluorescent protein (GFP). The control RNAi vector for the touch test experiments was the empty vector L4440 (A. Fire, Stanford University, Stanford, CA). Wild-type N2 was used for the progeny count experiments, and the RNAi-sensitive mutation rrf-3(pk1426) was used for the defecation scoring experiments. For the touch response experiments, we used RNAi-sensitive rrf-3(pk1426) transgenics expressing N-ter htt (19Q or 128Q) fused to GFP under the control of the mec-3 promoter.
Quantitative real-time PCR.
Quantitative real-time PCR was done as described previously (Parker et al., 2005).
Microscopy.
Worms were immobilized with 5 mm levamisole in M9 and mounted on 2% agarose pads. Animals were mounted on an Axioscope (Zeiss, Oberkochen, Germany) and observed with fluorescent filter sets. Images were acquired with a CCD camera and MetaVue software (Molecular Devices, Palo Alto, CA). To assess SNB-1::GFP levels in living animals, images were made from at least 100 animals for each strain over seven different trials using MetaVue software (Molecular Devices). Optimal image acquisition conditions were determined for wild-type controls and identical camera gain, exposure settings, and fluorescence filters were used for all subsequent quantitative studies. Morphometric analysis was used to obtain an average gray value of the GFP signals per animal and normalized to controls. The mean and SEM was calculated for each trial and two-tailed t tests were used for statistical analysis. Final figures for presentation were generated using Adobe Photoshop 7.0 (Adobe Systems, San Jose, CA).
Pharmacological assays.
Cholinergic neurotransmission was tested as described previously (Lackner et al., 1999).
Electrophysiology.
HIP1−/− mice and wild-type littermates were tested in blind manner at 2–4 months of age. Mice were anesthetized and killed by decapitation, and the brain was quickly removed and kept in ice-cold artificial CSF (ACSF) consisting of the following: 124 mm NaCl, 26 mm NaHCO3, 10 mm [scap]d-glucose, 3 mm KCl, 2 mm CaCl2, 2 mm MgSO4, and 1.3 mm NaH2PO4, saturated with 95% O2/5% CO2. In some experiments, a 2.5:1.3 ratio of Ca2+/Mg2+ was used. Hippocampal slices (450 μm) were maintained in ASCF at room temperature and during experiments were perfused with ACSF (30°C). In some experiments, 10 μm bicuculline methiodide (Sigma, St. Louis, MO), 1 μm CGP-55845 [(2S)-3-[[(15)-1-(3,4-dichlorophenyl)ethyl]amino-2-hydroxypropyl](phenylmethyl)phosphinic acid] (Tocris Bioscience, Ellisville, MO), and 50 μm [scap]d-AP5 (Sigma) were added to block GABAA, GABAB, and NMDA receptors, respectively. Glass microelectrodes containing ASCF lacking Ca2+ (3 MΩ resistance) were used to record field EPSPs (fEPSPs). Synaptic responses were amplified using an Axopatch 1D (Molecular Devices, Palo Alto, CA). Signals were filtered at 2 kHz and digitized at 10 kHz using pCLAMP6 software. Whole-cell recordings (EPSCs) were made using patch pipettes with (3–5 MΩ) filled with intracellular solution containing the following: 130 mm Cs gluconate, 30 mm HEPES, 11 mm EGTA, 10 mm KCl, 2 mm Mg-ATP, 2 mm QX-314 (lidocaine N-ethyl bromide) (Sigma), and 1 mm CaCl2, pH 7.25 and 290 mOsmol/L. The pipette was positioned onto CA1 pyramidal cell bodies using a 40× water immersion objective on a Nikon (Tokyo, Japan) E600FN microscope equipped with a C2400 Hamamatsu (Hamamatsu, Japan) infrared camera. Cells that did not have stable baseline synaptic responses or input resistance were not studied. We excluded 5 of 20 neurons that had EPSCs that facilitated <120% on 10 Hz stimulation. No differences in breakthrough cell membrane potential or capacitance were detected between neurons from HIP1−/− and wild-type mice.
CA1 synaptic responses were evoked with tungsten electrode stimulation of afferent fibers in the stratum radiatum. Stimulation intensity for experiments was adjusted to yield evoked synaptic responses that were 40% of the maximal fEPSP size (spike-free) or EPSC size in patch-clamped neurons held at −70 mV holding potential. Statistical differences were determined by Student's t test.
Statistical analysis.
One-way ANOVA was used for statistics, and corrected for multiple testing with Tukey's multiple comparison test unless otherwise noted.
Results
Characterization of a hipr-1 deletion mutant
hipr-1, the C. elegans homolog of mammalian HIP1 and yeast SLA2p (Holtzman et al., 1993), is located on chromosome III and maps to the ZK370.3 locus. The protein contains the ANTH, coiled-coil, and THATCH domains present in all HIP1 proteins. The coiled-coil domain in Sla2p and HIP1 enables binding to clathrin-light chain, although the exact mode of interaction differs from yeast to mammals, suggesting differences in the regulation of intracellular transport among different species (Legendre-Guillemin et al., 2005; Newpher et al., 2006; Ybe et al., 2007).
To investigate the function of hipr-1, we used a LOF insertion/deletion allele, designated hipr-1(tm2207) (Fig. 1) and examined basal levels of receptor-mediated endocytosis using vit-2::GFP, the C. elegans vitellogenin homolog YP170 (a yolk protein) fused to GFP (Grant and Hirsh, 1999). Although hipr-1 is expressed in the intestine, normal constitutive endocytosis of YP170-GFP from the intestine into oocytes in hipr-1(tm2207) mutants and controls was observed. This is in agreement with the observed lack of defects in transferrin uptake in HIP1−/− mice (Metzler et al., 2003).
Figure 1.
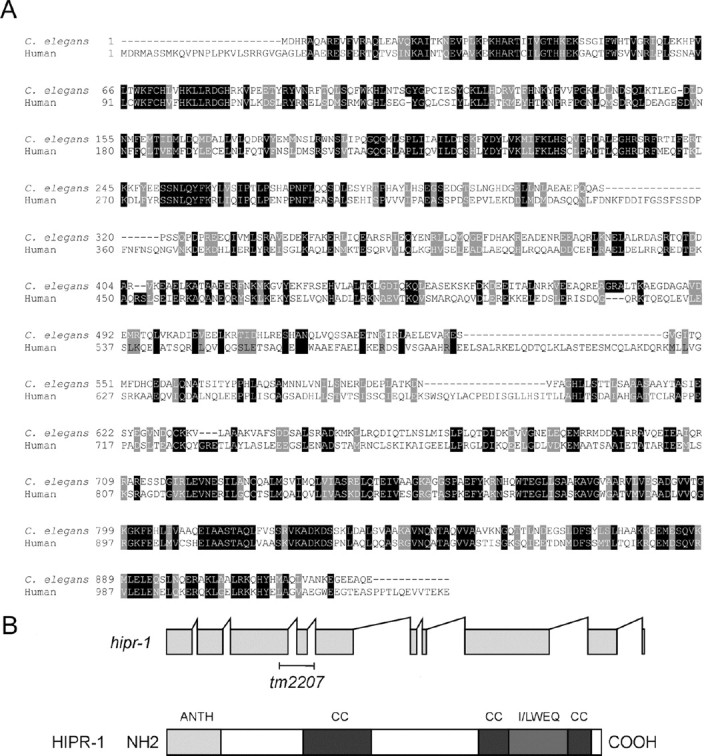
Molecular characterization of hipr-1. A, Sequence alignment (using ClustalW) of human HIP1 and the C. elegans homolog HIPR-1. Identical amino acids are highlighted in black, with similar amino acids in gray. Overall, there is 32% identity and 49% similarity between C. elegans HIPR-1 and human HIP1. B, The hipr-1 insertion/deletion allele tm2207 was obtained by PCR screening of a UV-TMP mutagenesis library. The Coils program was used to predict the probability of coiled-coil domains (Lupas et al., 1991).
hipr-1(tm2207) animals developed slower than wild type, typically reaching adulthood several hours later than wild-type N2 at 20°C, and had several phenotypes. We observed that hipr-1(tm2207) animals were defective in defecation. In C. elegans, defecation is an ultradian rhythmical behavior under the control of the inositol 1,4,5-trisphosphate (IP3) receptor homolog itr-1, and consists of a repeat period of ∼45 s with three motor steps: posterior body wall muscle contraction, anterior body wall muscle contraction, and expulsion (Thomas, 1990). Strong itr-1 mutations greatly extend the cycle time but the cycle remains periodic (Dal Santo et al., 1999). We examined the role hipr-1 in defecation with the tm2207 mutation and by RNA interference. The defecation motor program in hipr-1 mutants and hipr-1(RNAi) animals was intact and maintained periodicity, but the mean defecation cycle was lengthened and was significantly different from controls (Fig. 2A). Furthermore, hipr-1 mutants had reduced brood sizes (Fig. 2B). RNAi experiments to knockdown hipr-1 levels similarly reduced progeny number, worms from control RNAi experiments had normal progeny numbers, whereas itr-1(sa73) animals had reduced progeny numbers in agreement with previous reports (Dal Santo et al., 1999). Thus, hipr-1 has important roles in brood size and the maintenance of the defecation rhythmic behavior.
Figure 2.

Phenotypic characterization of hipr-1 mutants. A, Defecation cycle times were lengthened in hipr-1(tm2207) mutants. The mean time interval between consecutive posterior body muscle contractions was scored. *p < 0.01 versus wild type; **p < 0.001 versus RNAi controls; ***p < 0.001 versus wild type. Error bars indicate SEM (n > 20). B, Average brood size was reduced by mutation or RNAi of hipr-1 (n > 30). *p < 0.001 compared with wild type; **p < 0.001 versus RNAi controls; ***p < 0.001 versus wild type. Error bars indicate SEM.
hipr-1 modulates presynaptic function
Next, we investigated neuronal function in hipr-1 mutants. To this end, we used pharmacological indicators of neuronal transmission including aldicarb, an acetylcholinesterase inhibitor, or levamisole, a nicotinic acetylcholine receptor agonist. Both compounds result in hyperactive cholinergic synapses, muscle hypercontraction, and paralysis (Nonet et al., 1993). Worms defective in presynaptic calcium-dependent vesicle release were resistant to aldicarb, whereas worms with postsynaptic defects were also resistant to levamisole. Similar to other worm mutations in genes encoding synaptic proteins, we observed that hipr-1(tm2207) mutants were resistant to aldicarb, but sensitive to levamisole (Fig. 3A,B). These data suggest a role of hipr-1 in presynaptic function and perhaps the regulation of neurotransmission and synaptic vesicle cycling.
Figure 3.

hipr-1 regulates synaptic activity and localization of synaptobrevin. A, Cholinergic neuronal transmission was measured by determining the onset of paralysis induced by the cholinesterase inhibitor aldicarb. hipr-1(tm2207) mutants were resistant to the paralytic effects of aldicarb. Strains unc-31, unc-64, and unc-29 are deficient for CAPS, syntaxin, and a non-α-subunit of the nicotinic acetylcholine receptor, respectively. Data points are the mean and SEM for at least five independent experiments. *Significantly different from wild type (p < 0.001). B, hipr-1 mutants were sensitive to paralysis by the nicotinic acetylcholine agonist levamisole. unc-29 mutants were resistant to the paralytic effects levamisole, because it acts on receptors in body wall muscle. *p < 0.01, **p < 0.001 versus wild type. C, SNB-1::GFP expression in the ventral cord for jsIs37 and hipr-1(tm2207);jsIs37 animals. Scale bar, 5 μm.
hipr-1 modifies the accumulation of synaptobrevin in the ventral nerve cord
To directly visualize effects of hipr-1 on synaptic morphology and function, we examined the distribution of the synaptic protein synaptobrevin fused to GFP in living hipr-1 mutants. Synaptobrevin is associated with synaptic vesicles and plays a role in regulating synaptic vesicle fusion at the synaptic terminal. The abundance of synaptobrevin at the plasma membrane is determined by relative rates of exocytosis and endocytosis (Dittman and Kaplan, 2006). Defects in endocytosis result in the accumulation of synaptobrevin at the plasma membrane, whereas dysfunction in exocytosis results in the accumulation of vesicles at nerve terminals.
We examined the expression of a synaptobrevin GFP fusion protein (SNB-1::GFP) that is expressed in mechanosensory neurons by the jsIs37 transgene and is typically observed as one or two large patches along the ventral nerve cord just posterior to the animals' vulva (Koushika et al., 2004). To assess the effect of hipr-1 on SNB-1::GFP expression, we constructed hipr-1(tm2207);SNB-1::GFP animals and quantified the size and intensity of the GFP signals from these patches as average gray values using morphometric analysis.
We observed a significant increase in the average gray value of the GFP signals in hipr-1 mutants (1238 ± 60) versus the control jsIs37 strain (986 ± 27; t test, p < 0.01) (Fig. 3C). The accumulation of synaptobrevin at the synapse suggests dysregulation of the synaptic vesicle cycle perhaps because of the proposed function of HIP1 in intracellular transport. This is consistent with the observation that other endocytic proteins, such as AP180 and endophilin A, have roles in the recycling of surface synaptobrevin and increase the surface fraction of synaptobrevin in C. elegans (Sieburth et al., 2005; Dittman and Kaplan, 2006). These data suggest that hipr-1 functions in mechanosensory neurons and regulates the distribution and function of presynaptic proteins.
Recovery after synaptic depression is reduced in HIP1−/− mice
Synaptobrevin is a component of the vesicle fusion machinery and influences synaptic vesicle endocytosis, recycling, and neurotransmitter release (Deak et al., 2004). Thus, HIP1 may be linked to neurotransmitter release by its regulation of synaptobrevin. To determine whether HIP1 has a role in presynaptic function in mammals, we examined neuronal activity in HIP1−/− mice (Metzler et al., 2003). The functional role for HIP1 in clathrin-mediated endocytosis in vitro suggested that certain forms of synaptic vesicle recycling could be altered in HIP1−/− mice (Metzler et al., 2001). Synaptic transmission at CNS presynaptic nerve terminals is normally maintained by a small pool of releasable and recycling synaptic vesicles [∼25 vesicles (Sudhof, 2000)]. This pool can be depleted by extensive stimulation and synaptic vesicle release becomes rate limiting and dependent on the regeneration of vesicles by clathrin-mediated endocytosis. To determine whether recovery from synaptic depression is altered in HIP1−/− mice, a prolonged stimulation paradigm (10 Hz; 200 s) was applied to deplete the releasable synaptic vesicle pool, and subsequent recovery was monitored by stimulating at a lower frequency (0.33 Hz) (Fig. 4). The NMDA receptor blocker AP5 was included in the ACSF to prevent any tetanus-induced changes in synaptic transmission. Evoked synaptic responses (EPSC, EPSP) initially facilitated and subsequently depressed to a constant low level (n = 9) (Fig. 4A). The expanded time base in Figure 4B shows that there was a trend toward a larger initial potentiation during the first 4 s of the tetanus in HIP1−/− mice compared with controls (p > 0.06). In addition, the rate of recovery from synaptic depression was significantly impaired in HIP1−/− mice (Fig. 4C) (significance indicated by *p < 0.05). After 3 min of recovery, the average EPSC amplitude amounted to 58% of the baseline response in HIP1−/− mice in contrast to wild-type mice that showed >86% of recovery from depression. These results demonstrated that recovery of synaptic transmission at the presynaptic nerve terminal is impaired in HIP1−/− mice after synaptic depression induced by prolonged nerve stimulation.
Figure 4.

Recovery from synaptic depression is delayed in slices from HIP1−/− mice. A, HIP1−/− neurons require more time to recover from synaptic depression in comparison with wild-type littermate controls. CA1 pyramidal cells were voltage clamped at −60 mV and stimulated every 3 s via Schaffer collaterals to obtain baseline synaptic responses in the presence of AP5. A prolonged tetanus (10 Hz, 200 s; bar) was delivered at time 0 to induce synaptic depression including depletion of the readily releasable synaptic vesicle pool. Recovery from depression was studied by recording EPSCs at the original stimulation rate (data were averaged from n = 9 wild-type and n = 6 HIP1−/− neurons). B, Same data as in A were plotted on an expanded time base to compare the initial potentiation of EPSCs during the tetanus demonstrating enhanced facilitation in HIP1−/− mice (mean ± SEM). C, The recovery after synaptic depression is significantly decreased in HIP1−/− neurons in comparison with wild-type neurons. The same data as in A were replotted to show only the synaptic responses at the end of the tetanus and most of the recovery phase. Each point represents six averaged EPSCs ± SEM (t test; *p < 0.05).
HIP1−/− mice exhibit enhanced paired-pulse facilitation
The larger initial facilitation during the synaptic depression experiments in HIP1−/− neurons suggests that alterations in short-term synaptic plasticity occur in HIP1−/− mice. Most synapses, including hippocampal CA1–CA3, support various forms of plasticity in response to two or more stimuli. We first studied paired-pulse facilitation (PPF), in which delivery of two stimuli within a second of each other produces an increase in the size of the second synaptic response (Magelby, 1987). This increase in neurotransmitter release is thought to result from residual presynaptic Ca2+ that is still present after the first impulse, thereby increasing the probability of neurotransmitter release after invasion of the second action potential (Wu and Saggau, 1994; Kamiya and Ozawa, 1998). PPF was studied at different interpulse intervals between 40 and 600 ms. The sample fEPSP traces shown in Figure 5A were evoked by delivery of paired pulses between 40 and 60 ms. A significant increase in PPF at interpulse intervals between 60 and 600 ms is evident in HIP1−/− mice compared with wild-type littermates (Fig. 6B) (t test, *p < 0.04). Thus, increased facilitation of neurotransmitter release occurs in HIP1−/− mice when presynaptic neurons receive pairs of action potentials within hundreds of milliseconds of each other.
Figure 5.

Short-term plasticity is enhanced in HIP1−/− mice. A, Sample fEPSP traces evoked by delivering paired pulses 40 or 60 ms apart (only the second response is shown for the latter) to CA3–CA1 synapses from wild-type (blue lines; top traces) and HIP1−/− mice (red lines; middle traces). Traces represent average from three hippocampal slices per genotype selected for similar average initial fEPSP slope. Traces have been superimposed in the bottom panel for comparison. ACSF contained 2.0:2.0 mm ratio of Ca2+/Mg2+. B, PPF is significantly increased in HIP1−/− neurons (n = 18) compared with wild-type littermate controls (n = 17). Interpulse intervals between paired pulses were varied between 40 and 600 ms. Each measurement represents the mean ± SEM (t test, *p < 0.04). PPF is elevated but no longer significantly different in HIP1−/− neurons (n = 20) under increased release probability conditions (Ca2+/Mg2+ ratio was increased to 2.5:1.3) in comparison with wild-type neurons (n = 17; mean ± SEM).
Figure 6.

hipr-1 protects against mutant polyQ neuronal dysfunction. A, Mutation or RNAi of hipr-1 decreased touch responsiveness in animals expressing mutant htt. *p < 0.001 compared with 19Q alone; **p < 0.001 versus 128Q alone; ***p < 0.001 versus 128Q RNAi controls (n = 200 for all experiments). B, In vivo analysis revealed no change in the frequency of either the aggregation of huntingtin fusion proteins or dystrophy along axonal processes (n = 200). C, Synaptic genes are required for wild-type touch response. The percentage of wild-type response is given for each LOF synaptic mutant. *p < 0.001 compared with wild type (n = 200). D, Synaptic genes associated with endocytosis protect against mutant polyQ neuronal dysfunction. The percentages of either 128Q or 19Q touch response for each LOF mutant are given. *p < 0.001 versus 128Q animals; **p < 0.001 compared with 19Q controls (n = 200 for all experiments).
To determine whether enhanced facilitation persists under conditions favoring greater neurotransmitter release, synaptic responses were recorded under elevated Ca2+ concentrations. Raising the extracellular Ca2+ concentration is known to increase the probability of neurotransmitter release and leads to a decrease in PPF, because the initially high release probability acts as a ceiling and limits additional enhancement (Wu and Saggau, 1994; Kamiya and Ozawa, 1998). Under these conditions, PPF was still enhanced in HIP1−/− mice but the difference was no longer statistically significant in comparison with wild-type littermates (Fig. 5C) (n = 20 and 17, respectively; t test, p > 0.12). Thus, a lack of HIP1 affects the extent of PPF but not the Ca2+ dependency of this form of short-term synaptic plasticity.
hipr-1 protects against mutant polyglutamine-mediated neuronal dysfunction
Changes in synaptic function have been observed in several mouse models of HD (Cepeda et al., 2007). To determine a role of HIP1 in this process, we analyzed whether hipr-1 may act as a modifier of mutant polyQ toxicity in C. elegans neurons. To this end, we used transgenic nematodes showing neuronal cell dysfunction as produced by mutant polyQ expression (Parker et al., 2001). In this system, the expression of mutant N-terminal htt (128Q) fused to a fluorescent protein in the touch receptor neurons causes polyQ-dependent impairment of mechanosensation without cell death, an effect which can be rescued by mutation in genetic pathways associated with longevity and stress response (Parker et al., 2005).
The touch response of hipr-1(tm2207) mutants alone (83 ± 1%) was indistinguishable from wild-type animals (80 ± 1%), suggesting that hipr-1 does not contribute to normal mechanosensation. However, touch response in 128Q animals was impaired by 50% in hipr-1 mutants and by RNAi-knockdown, with no change in 19Q control animals (Fig. 6A). Enhancement of neuronal dysfunction by hipr-1(tm2207) was not attributable to changes in 128Q transgene expression because 128Q mRNA levels were nearly identical in 128Q controls (1.0 ± 0.2) compared with hipr-1(tm2207);128Q animals (1.1 ± 0.2) as determined by quantitative real-time PCR.
The touch receptor neurons at the tail (PLM cells) of 128Q transgenic worms show strong axonal aggregation of htt-fusion proteins and dystrophy of axonal processes, including axonal swelling (Parker et al., 2001). We observed no change in the frequency of aggregates along axonal processes of hipr-1(tm2207);128Q animals compared with 128Q alone (Fig. 6B). Furthermore, we observed no change in the frequency of axonal swelling versus controls (Fig. 6B).
Next, we tested whether the effect of hipr-1 LOF on PLM cell activity in mutant polyQ animals could be attributed to defects in PLM cell differentiation by examining whether neurodevelopmental abnormalities exist. For this purpose, we used two reporters for axonal outgrowth and guidance, including the following: mec-7::YFP that is expressed primarily in the mechanosensory neurons and unc-47::GFP expressed in the GABAergic motor neurons. We observed no difference in neuronal morphology between hipr-1(tm2207) mutants and control strains (data not shown). Together, these data indicated that hipr-1 protects against neuronal dysfunction induced by mutant polyQs, likely acting at the synapse.
Synaptic endocytic proteins protect against mutant polyQ toxicity in nematode neurons
With a putative role for hipr-1 in regulating synaptic function and protection against mutant polyQ toxicity, we examined other known regulators of synaptic function including homologues of syntaxin (unc-64), synaptotagmin (snt-1), the clathrin adaptin protein AP180 (unc-11), synaptojanin (unc-26), endophilin A (unc-57), and the mu2 subunit of adaptor protein complex 2 (dpy-23). The contribution of each mutation to wild-type touch was assessed, and each mutant examined had increased touch insensitivity (Fig. 6C). These data demonstrate that these genes are required for normal mechanosensation.
To determine whether any particular synaptic protein modulated mutant polyQ neurotoxicity, we constructed double mutants expressing either 19Q or 128Q transgenes for each LOF mutant. Mutations that impaired the exocytosis portion of the synaptic cycle, including unc-64/syntaxin, enhanced neuronal dysfunction for both control 19Q and mutant 128Q animals (Fig. 6D). This is likely a non-polyQ-related effect of transgene expression. snt-1/synaptotagmin enhanced 128Q toxicity, but the effects on 19Q could not be examined because we consistently failed to isolate snt-1;19Q animals. We cannot rule out the possibility of genetic linkage between the 19Q transgene and snt-1. However, mutants that affected the endocytosis portion of the cycle enhanced mutant 128Q toxicity with no significant effects compared with 19Q control strains. Mutation in unc-11/AP180 and unc-26/synaptojanin enhanced 128Q toxicity to a level comparable with hipr-1, mutation in unc-57/endophilin A was more severe, whereas mutation of dpy-23/mu2 subunit of adaptor protein complex 2 had no effect (Fig. 6D). Thus, genes that function in synaptic endocytosis may protect against mutant polyQ toxicity, indicating that mutant polyQs may alter synaptic endocytosis.
Discussion
Several studies suggest a function for HIP1 in intracellular transport of cell surface receptors influencing neuronal activity in the CNS (Metzler et al., 2003, 2007). However, the contribution of HIP1 to neuronal pathology in HD is unclear. Here, we provide evidence for a presynaptic function of HIP1. This is based on the resistance of C. elegans hipr-1 mutant motorneurons to aldicarb and changes in presynaptic plasticity and recovery from synaptic depression in hippocampal brain slices from HIP1−/− mice. Moreover, HIP1 and several other presynaptic proteins protect C. elegans neurons from mutant polyQ toxicity.
Previous studies have shown HIP1 localization at primarily postsynaptic sites. This is in agreement with the occurrence of postsynaptic defects in neurotransmitter receptor function and transport in neurons from HIP1−/− mice (Metzler et al., 2003, 2007; Okano et al., 2003; Yao et al., 2006). In this study, we provide the first evidence for a function of HIP1 at the presynaptic nerve terminal. In both nematodes and mice, we observed HIP1 altered neurotransmitter release based on the use of pharmacological and electrophysiological indicators, respectively.
Reduced recovery from synaptic depression in HIP1−/− mice indicates potential defects in endocytosis of synaptic vesicles. This defect could result from lack of HIP1 endocytic function. An alternative explanation is that the subcellular localization of synaptobrevin is altered in HIP1−/− mice, thereby influencing endocytosis of synaptic vesicles. It is known that cultured synapses from synaptobrevin-2−/− mice have slower recovery of synaptic responses subsequent to depletion induced by hypertonic solution (Deak et al., 2004). In addition, neurotransmitter release is greatly reduced in these mice, unlike HIP1 mutants, which do not show any differences in release (data not shown). It is also interesting that synaptobrevin-2−/− mice show enhanced facilitation; we detected a small but significant reduction in paired-pulse facilitation in HIP1 mutants when bathed in saline containing a 2:2 mm ratio of calcium/magnesium saline. However, this difference was restored in saline containing a higher ratio that favors release. These data also raise the question of whether calcium dynamics are altered in HIP1−/− mice, especially because calcium regulates not only exocytosis but also endocytosis (Abenavoli et al., 2001). Future studies are needed to determine whether HIP1 influences vesicle release through altered synaptobrevin distribution in HIP1−/− mice, whether HIP1 has direct effects on the vesicle release machinery independent of synaptobrevin, or whether HIP1 modulates presynaptic calcium and clathrin-mediated endocytosis.
A potential role of HIPR-1 in endocytosis awaits future experimentation. Nevertheless, the ability of HIP1 and Sla2p to bind to clathrin light chain suggests conservation of this function from yeast to mammals. In fact, a clathrin light chain binding site was recently identified in the coiled-coil domain of HIP1 family members, which corresponds to 483ADLLRKN in human HIP1 and KDEQIKN in yeast (Chen and Brodsky, 2005; Legendre-Guillemin et al., 2005; Newpher et al., 2006; Ybe et al., 2007). A similar sequence is present in HIPR-1 and corresponds to 458KDEEITA. This suggests clathrin light chain binding and regulation of clathrin-mediated endocytosis by HIPR-1. Still, the mode of clathrin light chain binding and clathrin assembly may differ from human HIP1 as evidenced by the lack of clathrin heavy chain and AP2 binding sites in HIPR-1.
HIP1 interacts with htt and, therefore, may be a modifier of mutant polyQ cytotoxicity as previously suggested for other htt-interacting proteins (Holbert et al., 2001; Steffan et al., 2001; Goehler et al., 2004; Bae et al., 2005). We found that hipr-1 LOF enhanced touch insensitivity in mutant polyQ transgenic animals independent of aggregation and axonal dystrophy phenotypes, suggesting exacerbation of a defect in synaptic transmission. Our findings that HIP1 modulates presynaptic activity together with the modulation of mutant polyQ cytotoxicity in neurons provide direct evidence for a protective effect of synaptic proteins against mutant polyQ. This hypothesis is supported by the observation that other synaptic proteins involved in endocytosis like unc-11/AP180, unc-26/synaptojanin, and unc-57/endophilin are protective against mutant polyQs. The most severe enhancement of mutant polyQ toxicity was observed with a LOF in unc-57/endophilin A. This is consistent with the previous observation that mutant htt-exon 1 fragments have enhanced binding to endophilin A3 (Sittler et al., 1998).
The role of HIP1 and other synaptic proteins on mutant polyQ cytotoxicity were studied in nematodes expressing N-terminal htt. Additional studies will be needed to determine whether a similar effect occurs in mammalian neurons expressing full-length mutant htt. Nonetheless, it is notable that the generation of an N-terminal htt fragment is a crucial step in the pathogenesis of HD (Crocker et al., 2006; Graham et al., 2006).
An involvement of HIP1 in the pathogenesis of HD is consistent with the observation that mutant htt impairs neurotransmission in mouse models of HD (Usdin et al., 1999; Li et al., 2000; Laforet et al., 2001; Zeron et al., 2002; Vetter et al., 2003). Mutant htt has been shown to directly interact with the cellular machinery required for synaptic transmission and neuronal function including PACSIN 1/syndapin, syntaxin, endophilin A, and N type channels (Sittler et al., 1998; Modregger et al., 2002; Singaraja et al., 2002; Swayne et al., 2005). Our data are consistent with a role for defective endocytosis as an underlying mechanism in HD (Trushina et al., 2006). Importantly, mouse knock-outs of synaptic and/or htt-interacting proteins such as HIP1 (Ferguson et al., 2000; Metzler et al., 2007; Nystuen et al., 2007) often have neurological phenotypes that may be relevant to HD pathogenesis.
In conclusion, we identified an important conserved role for HIP1 in the regulation of presynaptic function and the modulation of polyQ cytotoxicity. Exacerbation of polyQ cytotoxicity was also observed with LOF mutants for other endocytic proteins influencing transport and neurotransmitter release. This is consistent with the notion that htt partner proteins may modify the cytotoxicity of mutant htt, which may potentially influence HD symptoms (Holbert et al., 2001). Thus, together with our previous study of longevity modulators (Parker et al., 2005), our data indicate that C. elegans transgenics expressing mutant polyQs in neurons are useful to search for modifier genes and active compounds that may restore a normal synapse before evaluation in mammalian models of HD pathogenesis and samples from HD patients.
Footnotes
This work was supported by Inserm and University of Paris René Descartes (Paris, France) (C.N.), Natural Sciences and Engineering Research Council of Canada (A.M.R.), and Canadian Institutes of Health Research (CIHR) Grant MT-9133 (M.R.H.). Additional support was provided by Merck Frosst Canada (M.R.H., M. Metzler). M.R.H is a holder of a Canada Research Chair in Human Genetics. J.G. was supported by a CIHR fellowship at the start of this study and is currently a National Alliance for Research on Schizophrenia and Depression Young Investigator Award recipient. J.C.R. holds a CIHR Chair in Learning and Memory. J.A.P was the recipient of an Inserm Young Researcher award. We thank Jean-Louis Bessereau (École Normale Supérieure, Paris, France) for advice on synaptic experiments.
References
- Abenavoli A, Montagna M, Malgaroli A. Calcium: the common theme in vesicular cycling. Nat Neurosci. 2001;4:117–118. doi: 10.1038/83924. [DOI] [PubMed] [Google Scholar]
- Bae BI, Xu H, Igarashi S, Fujimuro M, Agrawal N, Taya Y, Hayward SD, Moran TH, Montell C, Ross CA, Snyder SH, Sawa A. p53 mediates cellular dysfunction and behavioral abnormalities in Huntington's disease. Neuron. 2005;47:29–41. doi: 10.1016/j.neuron.2005.06.005. [DOI] [PubMed] [Google Scholar]
- Cepeda C, Wu N, Andre VM, Cummings DM, Levine MS. The corticostriatal pathway in Huntington's disease. Prog Neurobiol. 2007;81:253–271. doi: 10.1016/j.pneurobio.2006.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crocker SF, Costain WJ, Robertson HA. DNA microarray analysis of striatal gene expression in symptomatic transgenic Huntington's mice (R6/2) reveals neuroinflammation and insulin associations. Brain Res. 2006;1088:176–186. doi: 10.1016/j.brainres.2006.02.102. [DOI] [PubMed] [Google Scholar]
- Dal Santo P, Logan MA, Chisholm AD, Jorgensen EM. The inositol trisphosphate receptor regulates a 50-second behavioral rhythm in C. elegans. Cell. 1999;98:757–767. doi: 10.1016/s0092-8674(00)81510-x. [DOI] [PubMed] [Google Scholar]
- Deak F, Schoch S, Liu X, Sudhof TC, Kavalali ET. Synaptobrevin is essential for fast synaptic-vesicle endocytosis. Nat Cell Biol. 2004;6:1102–1108. doi: 10.1038/ncb1185. [DOI] [PubMed] [Google Scholar]
- Dittman JS, Kaplan JM. Factors regulating the abundance and localization of synaptobrevin in the plasma membrane. Proc Natl Acad Sci USA. 2006;103:11399–11404. doi: 10.1073/pnas.0600784103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferguson GD, Anagnostaras SG, Silva AJ, Herschman HR. Deficits in memory and motor performance in synaptotagmin IV mutant mice. Proc Natl Acad Sci USA. 2000;97:5598–5603. doi: 10.1073/pnas.100104597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gervais FG, Singaraja R, Xanthoudakis S, Gutekunst CA, Leavitt BR, Metzler M, Hackam AS, Tam J, Vaillancourt JP, Houtzager V, Rasper DM, Roy S, Hayden MR, Nicholson DW. Recruitment and activation of caspase-8 by the Huntingtin-interacting protein Hip-1 and a novel partner Hippi. Nat Cell Biol. 2002;4:95–105. doi: 10.1038/ncb735. [DOI] [PubMed] [Google Scholar]
- Goehler H, Lalowski M, Stelzl U, Waelter S, Stroedicke M, Worm U, Droege A, Lindenberg KS, Knoblich M, Haenig C, Herbst M, Suopanki J, Scherzinger E, Abraham C, Bauer B, Hasenbank R, Fritzsche A, Ludewig AH, Bussow K, Coleman SH. A protein interaction network links GIT1, an enhancer of huntingtin aggregation, to Huntington's disease. Mol Cell. 2004;15:853–865. doi: 10.1016/j.molcel.2004.09.016. [DOI] [PubMed] [Google Scholar]
- Graham RK, Deng Y, Slow EJ, Haigh B, Bissada N, Lu G, Pearson J, Shehadeh J, Bertram L, Murphy Z, Warby SC, Doty CN, Roy S, Wellington CL, Leavitt BR, Raymond LA, Nicholson DW, Hayden MR. Cleavage at the caspase-6 site is required for neuronal dysfunction and degeneration due to mutant huntingtin. Cell. 2006;125:1179–1191. doi: 10.1016/j.cell.2006.04.026. [DOI] [PubMed] [Google Scholar]
- Grant B, Hirsh D. Receptor-mediated endocytosis in the Caenorhabditis elegans oocyte. Mol Biol Cell. 1999;10:4311–4326. doi: 10.1091/mbc.10.12.4311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hackam AS, Yassa AS, Singaraja R, Metzler M, Gutekunst CA, Gan L, Warby S, Wellington CL, Vaillancourt J, Chen N, Gervais FG, Raymond L, Nicholson DW, Hayden MR. Huntingtin interacting protein 1 induces apoptosis via a novel caspase-dependent death effector domain. J Biol Chem. 2000;275:41299–41308. doi: 10.1074/jbc.M008408200. [DOI] [PubMed] [Google Scholar]
- Holbert S, Denghien I, Kiechle T, Rosenblatt A, Wellington C, Hayden MR, Margolis RL, Ross CA, Dausset J, Ferrante RJ, Neri C. The Gln-Ala repeat transcriptional activator CA150 interacts with huntingtin: neuropathologic and genetic evidence for a role in Huntington's disease pathogenesis. Proc Natl Acad Sci USA. 2001;98:1811–1816. doi: 10.1073/pnas.041566798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holtzman DA, Yang S, Drubin DG. Synthetic-lethal interactions identify two novel genes, SLA1 and SLA2, that control membrane cytoskeleton assembly in Saccharomyces cerevisiae. J Cell Biol. 1993;122:635–644. doi: 10.1083/jcb.122.3.635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalchman MA, Koide HB, McCutcheon K, Graham RK, Nichol K, Nishiyama K, Kazemi-Esfarjani P, Lynn FC, Wellington C, Metzler M, Goldberg YP, Kanazawa I, Gietz RD, Hayden MR. HIP1, a human homologue of S. cerevisiae Sla2p, interacts with membrane-associated huntingtin in the brain. Nat Genet. 1997;16:44–53. doi: 10.1038/ng0597-44. [DOI] [PubMed] [Google Scholar]
- Kamath RS, Martinez-Campos M, Zipperlen P, Fraser AG, Ahringer J. Effectiveness of specific RNA-mediated interference through ingested double-stranded RNA in Caenorhabditis elegans. Genome Biol. 2001;2:RESEARCH0002. doi: 10.1186/gb-2000-2-1-research0002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamiya H, Ozawa S. Kainate receptor-mediated inhibition of presynaptic Ca2+ influx and EPSP in area CA1 of the rat hippocampus. J Physiol (Lond) 1998;509:833–845. doi: 10.1111/j.1469-7793.1998.833bm.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khatchadourian K, Smith CE, Metzler M, Gregory M, Hayden MR, Cyr DG, Hermo L. Structural abnormalities in spermatids together with reduced sperm counts and motility underlie the reproductive defect in HIP1−/− mice. Mol Reprod Dev. 2007;74:341–359. doi: 10.1002/mrd.20564. [DOI] [PubMed] [Google Scholar]
- Koushika SP, Schaefer AM, Vincent R, Willis JH, Bowerman B, Nonet ML. Mutations in Caenorhabditis elegans cytoplasmic dynein components reveal specificity of neuronal retrograde cargo. J Neurosci. 2004;24:3907–3916. doi: 10.1523/JNEUROSCI.5039-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lackner MR, Nurrish SJ, Kaplan JM. Facilitation of synaptic transmission by EGL-30 Gqalpha and EGL-8 PLCbeta: DAG binding to UNC-13 is required to stimulate acetylcholine release. Neuron. 1999;24:335–346. doi: 10.1016/s0896-6273(00)80848-x. [DOI] [PubMed] [Google Scholar]
- Laforet GA, Sapp E, Chase K, McIntyre C, Boyce FM, Campbell M, Cadigan BA, Warzecki L, Tagle DA, Reddy PH, Cepeda C, Calvert CR, Jokel ES, Klapstein GJ, Ariano MA, Levine MS, DiFiglia M, Aronin N. Changes in cortical and striatal neurons predict behavioral and electrophysiological abnormalities in a transgenic murine model of Huntington's disease. J Neurosci. 2001;21:9112–9123. doi: 10.1523/JNEUROSCI.21-23-09112.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Legendre-Guillemin V, Metzler M, Lemaire JF, Philie J, Gan L, Hayden MR, McPherson PS. Huntingtin interacting protein 1 (HIP1) regulates clathrin assembly through direct binding to the regulatory region of the clathrin light chain. J Biol Chem. 2005;280:6101–6108. doi: 10.1074/jbc.M408430200. [DOI] [PubMed] [Google Scholar]
- Li H, Li SH, Johnston H, Shelbourne PF, Li XJ. Amino-terminal fragments of mutant huntingtin show selective accumulation in striatal neurons and synaptic toxicity. Nat Genet. 2000;25:385–389. doi: 10.1038/78054. [DOI] [PubMed] [Google Scholar]
- Lupas A, Van Dyke M, Stock J. Predicting coiled coils from protein sequences. Science. 1991;252:1162–1164. doi: 10.1126/science.252.5009.1162. [DOI] [PubMed] [Google Scholar]
- Magelby KL. Short-term changes in synaptic efficacy. In: Edelman GM, Gall VE, Cowan WM, editors. Synaptic function. New York: Wiley; 1987. pp. 21–56. [Google Scholar]
- Metzler M, Legendre-Guillemin V, Gan L, Chopra V, Kwok A, McPherson PS, Hayden MR. HIP1 functions in clathrin-mediated endocytosis through binding to clathrin and adaptor protein 2. J Biol Chem. 2001;276:39271–39276. doi: 10.1074/jbc.C100401200. [DOI] [PubMed] [Google Scholar]
- Metzler M, Li B, Gan L, Georgiou J, Gutekunst CA, Wang Y, Torre E, Devon RS, Oh R, Legendre-Guillemin V, Rich M, Alvarez C, Gertsenstein M, McPherson PS, Nagy A, Wang YT, Roder JC, Raymond LA, Hayden MR. Disruption of the endocytic protein HIP1 results in neurological deficits and decreased AMPA receptor trafficking. EMBO J. 2003;22:3254–3266. doi: 10.1093/emboj/cdg334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metzler M, Gan L, Wong TP, Liu L, Helm J, Liu L, Georgiou J, Wang Y, Bissada N, Cheng K, Roder JC, Wang YT, Hayden MR. NMDA receptor function and NMDA receptor-dependent phosphorylation of huntingtin is altered by the endocytic protein HIP1. J Neurosci. 2007;27:2298–2308. doi: 10.1523/JNEUROSCI.5175-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mills IG, Gaughan L, Robson C, Ross T, McCracken S, Kelly J, Neal DE. Huntingtin interacting protein 1 modulates the transcriptional activity of nuclear hormone receptors. J Cell Biol. 2005;170:191–200. doi: 10.1083/jcb.200503106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mishra SK, Agostinelli NR, Brett TJ, Mizukami I, Ross TS, Traub LM. Clathrin- and AP-2-binding sites in HIP1 uncover a general assembly role for endocytic accessory proteins. J Biol Chem. 2001;276:46230–46236. doi: 10.1074/jbc.M108177200. [DOI] [PubMed] [Google Scholar]
- Modregger J, DiProspero NA, Charles V, Tagle DA, Plomann M. PACSIN 1 interacts with huntingtin and is absent from synaptic varicosities in presymptomatic Huntington's disease brains. Hum Mol Genet. 2002;11:2547–2558. doi: 10.1093/hmg/11.21.2547. [DOI] [PubMed] [Google Scholar]
- Newpher TM, Idrissi FZ, Geli MI, Lemmon SK. Novel function of clathrin light chain in promoting endocytic vesicle formation. Mol Biol Cell. 2006;17:4343–4352. doi: 10.1091/mbc.E06-07-0606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nonet ML, Grundahl K, Meyer BJ, Rand JB. Synaptic function is impaired but not eliminated in C. elegans mutants lacking synaptotagmin. Cell. 1993;73:1291–1305. doi: 10.1016/0092-8674(93)90357-v. [DOI] [PubMed] [Google Scholar]
- Nystuen AM, Schwendinger JK, Sachs AJ, Yang AW, Haider NB. A null mutation in VAMP1/synaptobrevin is associated with neurological defects and prewean mortality in the lethal-wasting mouse mutant. Neurogenetics. 2007;8:1–10. doi: 10.1007/s10048-006-0068-7. [DOI] [PubMed] [Google Scholar]
- Okano A, Usuda N, Furihata K, Nakayama K, Bao Tian Q, Okamoto T, Suzuki T. Huntingtin-interacting protein-1-related protein of rat (rHIP1R) is localized in the postsynaptic regions. Brain Res. 2003;967:210–225. doi: 10.1016/s0006-8993(03)02236-4. [DOI] [PubMed] [Google Scholar]
- Oravecz-Wilson KI, Kiel MJ, Li L, Rao DS, Saint-Dic D, Kumar PD, Provot MM, Hankenson KD, Reddy VN, Lieberman AP, Morrison SJ, Ross TS. Huntingtin interacting protein 1 mutations lead to abnormal hematopoiesis, spinal defects and cataracts. Hum Mol Genet. 2004;13:851–867. doi: 10.1093/hmg/ddh102. [DOI] [PubMed] [Google Scholar]
- Parker JA, Connolly JB, Wellington C, Hayden M, Dausset J, Neri C. Expanded polyglutamines in Caenorhabditis elegans cause axonal abnormalities and severe dysfunction of PLM mechanosensory neurons without cell death. Proc Natl Acad Sci USA. 2001;98:13318–13323. doi: 10.1073/pnas.231476398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker JA, Arango M, Abderrahmane S, Lambert E, Tourette C, Catoire H, Neri C. Resveratrol rescues mutant polyglutamine cytotoxicity in nematode and mammalian neurons. Nat Genet. 2005;37:349–350. doi: 10.1038/ng1534. [DOI] [PubMed] [Google Scholar]
- Rao DS, Hyun TS, Kumar PD, Mizukami IF, Rubin MA, Lucas PC, Sanda MG, Ross TS. Huntingtin-interacting protein 1 is overexpressed in prostate and colon cancer and is critical for cellular survival. J Clin Invest. 2002;110:351–360. doi: 10.1172/JCI15529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao DS, Bradley SV, Kumar PD, Hyun TS, Saint-Dic D, Oravecz-Wilson K, Kleer CG, Ross TS. Altered receptor trafficking in Huntingtin interacting protein 1-transformed cells. Cancer Cell. 2003;3:471–482. doi: 10.1016/s1535-6108(03)00107-7. [DOI] [PubMed] [Google Scholar]
- Senetar MA, Foster SJ, McCann RO. Intrasteric inhibition mediates the interaction of the I/LWEQ module proteins Talin1, Talin2, Hip1, and Hip12 with actin. Biochemistry. 2004;43:15418–15428. doi: 10.1021/bi0487239. [DOI] [PubMed] [Google Scholar]
- Sieburth D, Ch'ng Q, Dybbs M, Tavazoie M, Kennedy S, Wang D, Dupuy D, Rual JF, Hill DE, Vidal M, Ruvkun G, Kaplan JM. Systematic analysis of genes required for synapse structure and function. Nature. 2005;436:510–517. doi: 10.1038/nature03809. [DOI] [PubMed] [Google Scholar]
- Singaraja RR, Hadano S, Metzler M, Givan S, Wellington CL, Warby S, Yanai A, Gutekunst CA, Leavitt BR, Yi H, Fichter K, Gan L, McCutcheon K, Chopra V, Michel J, Hersch SM, Ikeda JE, Hayden MR. HIP14, a novel ankyrin domain-containing protein, links huntingtin to intracellular trafficking and endocytosis. Hum Mol Genet. 2002;11:2815–2828. doi: 10.1093/hmg/11.23.2815. [DOI] [PubMed] [Google Scholar]
- Sittler A, Walter S, Wedemeyer N, Hasenbank R, Scherzinger E, Eickhoff H, Bates GP, Lehrach H, Wanker EE. SH3GL3 associates with the Huntingtin exon 1 protein and promotes the formation of polygln-containing protein aggregates. Mol Cell. 1998;2:427–436. doi: 10.1016/s1097-2765(00)80142-2. [DOI] [PubMed] [Google Scholar]
- Steffan JS, Bodai L, Pallos J, Poelman M, McCampbell A, Apostol BL, Kazantsev A, Schmidt E, Zhu YZ, Greenwald M, Kurokawa R, Housman DE, Jackson GR, Marsh JL, Thompson LM. Histone deacetylase inhibitors arrest polyglutamine-dependent neurodegeneration in Drosophila. Nature. 2001;413:739–743. doi: 10.1038/35099568. [DOI] [PubMed] [Google Scholar]
- Sudhof TC. The synaptic vesicle cycle revisited. Neuron. 2000;28:317–320. doi: 10.1016/s0896-6273(00)00109-4. [DOI] [PubMed] [Google Scholar]
- Swayne LA, Chen L, Hameed S, Barr W, Charlesworth E, Colicos MA, Zamponi GW, Braun JE. Crosstalk between huntingtin and syntaxin 1A regulates N-type calcium channels. Mol Cell Neurosci. 2005;30:339–351. doi: 10.1016/j.mcn.2005.07.016. [DOI] [PubMed] [Google Scholar]
- Thomas JH. Genetic analysis of defecation in Caenorhabditis elegans. Genetics. 1990;124:855–872. doi: 10.1093/genetics/124.4.855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trushina E, Singh RD, Dyer RB, Cao S, Shah VH, Parton RG, Pagano RE, McMurray CT. Mutant huntingtin inhibits clathrin-independent endocytosis and causes accumulation of cholesterol in vitro and in vivo. Hum Mol Genet. 2006;15:3578–3591. doi: 10.1093/hmg/ddl434. [DOI] [PubMed] [Google Scholar]
- Usdin MT, Shelbourne PF, Myers RM, Madison DV. Impaired synaptic plasticity in mice carrying the Huntington's disease mutation. Hum Mol Genet. 1999;8:839–846. doi: 10.1093/hmg/8.5.839. [DOI] [PubMed] [Google Scholar]
- Vetter JM, Jehle T, Heinemeyer J, Franz P, Behrens PF, Jackisch R, Landwehrmeyer GB, Feuerstein TJ. Mice transgenic for exon 1 of Huntington's disease: properties of cholinergic and dopaminergic pre-synaptic function in the striatum. J Neurochem. 2003;85:1054–1063. doi: 10.1046/j.1471-4159.2003.01704.x. [DOI] [PubMed] [Google Scholar]
- Waelter S, Scherzinger E, Hasenbank R, Nordhoff E, Lurz R, Goehler H, Gauss C, Sathasivam K, Bates GP, Lehrach H, Wanker EE. The huntingtin interacting protein HIP1 is a clathrin and alpha-adaptin-binding protein involved in receptor-mediated endocytosis. Hum Mol Genet. 2001;10:1807–1817. doi: 10.1093/hmg/10.17.1807. [DOI] [PubMed] [Google Scholar]
- Wanker EE, Rovira C, Scherzinger E, Hasenbank R, Walter S, Tait D, Colicelli J, Lehrach H. HIP-I: a huntingtin interacting protein isolated by the yeast two-hybrid system. Hum Mol Genet. 1997;6:487–495. doi: 10.1093/hmg/6.3.487. [DOI] [PubMed] [Google Scholar]
- Wu LG, Saggau P. Presynaptic calcium is increased during normal synaptic transmission and paired-pulse facilitation, but not in long-term potentiation in area CA1 of hippocampus. J Neurosci. 1994;14:645–654. doi: 10.1523/JNEUROSCI.14-02-00645.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao PJ, Bushlin I, Petralia RS. Partially overlapping distribution of epsin1 and HIP1 at the synapse: analysis by immunoelectron microscopy. J Comp Neurol. 2006;494:368–379. doi: 10.1002/cne.20810. [DOI] [PubMed] [Google Scholar]
- Ybe JA, Mishra S, Helms S, Nix J. Crystal structure at 2.8 A of the DLLRKN-containing coiled-coil domain of huntingtin-interacting protein 1 (HIP1) reveals a surface suitable for clathrin light chain binding. J Mol Biol. 2007;367:8–15. doi: 10.1016/j.jmb.2006.12.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeron MM, Hansson O, Chen N, Wellington CL, Leavitt BR, Brundin P, Hayden MR, Raymond LA. Increased sensitivity to N-methyl-d-aspartate receptor-mediated excitotoxicity in a mouse model of Huntington's disease. Neuron. 2002;33:849–860. doi: 10.1016/s0896-6273(02)00615-3. [DOI] [PubMed] [Google Scholar]