

Abstract
Excitotoxicity is generally studied in dissociated neurons, cultured hippocampal slices, or intact animals. However, the requirements of dissociated neurons or cultured slices to use prenatal or juvenile rats seriously limit the advantages of these systems, whereas the complexity of intact animals prevents detailed molecular investigations. In the present experiments, we studied developmental changes in NMDA neurotoxicity in acute hippocampal slices with lactate dehydrogenase (LDH) release in medium, propidium iodide (PI) uptake, and Nissl staining as markers of cell damage. Calpain-mediated spectrin degradation was used to test calpain involvement in NMDA neurotoxicity. NMDA treatment produced increased LDH release, PI uptake, and spectrin degradation in slices from juvenile rats but not adult rats. NMDA-induced changes in slices from young rats were blocked completely by the NMDA receptor antagonist (+)-5-methyl-10,11-dihydro-5H-dibenzo [a,d] cyclohepten-5,10-imine maleate (MK-801) and by the antagonists of NR2B receptor ifenprodil and R-(R, S)-α-(4-hydroxyphenyl)-β-methyl-4-(phenylmethyl)-1-piperidine propranol and were partly blocked by calpain inhibitor III but were not affected by the NR2A-specific antagonist [(R)-[(S)-1-(4-bromo-phenyl)-ethylamino]-(2,3-dioxo-1,2,3,4-tetrahydroquinoxalin-5-yl)-methyl]-phosphonic acid. NMDA-induced changes in Nissl staining were also different in slices from young and adult rats and blocked by NR2B but not NR2A antagonists. In contrast to NMDA treatment, oxygen/glucose deprivation (OGD) induced neurotoxicity in slices from both young and adult rats, although OGD-induced toxicity was attenuated by MK-801 only in slices from young rats. Our results are consistent with the idea that NMDA-mediated toxicity is caused by activation of NR2B- but not NR2A-containing NMDA receptors leading to calpain activation and that developmental changes in NMDA toxicity reflect developmental changes in NMDA receptor subunit composition.
Keywords: hippocampus, excitotoxicity, development, NR2A, NR2B, calpain
Introduction
Glutamate is the principal excitatory neurotransmitter in the CNS (Collingridge and Lester, 1989). Ionotropic glutamate receptors consist of three subtypes: the NMDA receptors (NMDARs), AMPA receptors, and kainate receptors. NMDARs play a crucial role in neuronal development, plasticity, and survival; in addition, overactivation of NMDARs is involved in the pathophysiology of epileptic seizures, hypoxic–ischemic neuronal damage, and excitotoxic cell death (Dingledine et al., 1999; Lynch and Guttmann, 2002). The intracellular signaling pathways mediating NMDA excitotoxicity are still debated (Skaper et al., 2001; Waxman and Lynch, 2005). Most researchers agree that NMDARs produce neurotoxicity via rapid Ca2+ influx leading to cellular Ca2+ overload. Several studies have revealed developmental changes in brain susceptibility to excitotoxicity (Kuroiwa and Okeda, 1994; Towfighi and Mauger, 1998), although the mechanisms responsible for these changes have not yet been elucidated.
Several NMDAR subtypes, differing in kinetic properties, sensitivity to various ligands, permeability to divalent ions, and interactions with intracellular proteins have been identified (Cull-Candy et al., 2001). NMDARs are composed of two NR1 subunits and at least one type of NR2 subunits with predominantly NR2A or NR2B subunits in adult rat hippocampus (Wenzel et al., 1997). During the postnatal period, cortical neurons exhibit changes in kinetics of NMDAR-mediated EPSCs (Barth and Malenka, 2001; Lu et al., 2001), corresponding to a developmental change in the composition of the NMDARs from predominantly NR1/NR2B to NR1/NR2A oligomers (Liu et al., 2004). It has been proposed recently that NR2A- and NR2B-containing NMDA receptors are linked to different intracellular cascades and participate in different functions in synaptic plasticity and pathological conditions (Krapivinsky et al., 2003; Liu et al., 2004; Kim et al., 2005).
Calpains are calcium-activated neutral proteases participating in excitotoxic neuronal death (Saido et al., 1994; Brorson et al., 1995; Vanderklish and Bahr, 2000; Araujo et al., 2005) and in several neurodegenerative conditions (Chan and Mattson, 1999; Vanderklish and Bahr, 2000). Calpains are activated by calcium permeating through NMDA receptors during intense synaptic activity (Seubert et al., 1988; Lankiewicz et al., 2000) and have been implicated in synaptic modification both during neuronal development and in adult synaptic plasticity (Lynch and Baudry, 1987; Vanderklish et al., 1995). The cytoskeletal protein spectrin is a calpain substrate, and quantification of calpain-generated spectrin breakdown products (SBDPs) of molecular weight of 150 and 145 kDa is often used to assess endogenous calpain activation (Nath et al., 1996; Bi et al., 1997; Chan and Mattson, 1999). PSD-95 (postsynaptic density-95) is a major postsynaptic protein that binds to the C-terminal domains of NMDA receptors (Kornau et al., 1995; Wechsler and Teichberg, 1998) and is also a calpain substrate (Lu et al., 2000). In the present studies, we determined the role of NR2A- and NR2B-containing NMDA receptors in calpain activation and in excitotoxicity. We used both the known developmental changes in the relative abundance of these two types of NMDA receptors as well as blockers selective for each type of receptors to determine the degree of neuronal damage and calpain activation elicited by NMDA and oxygen/glucose deprivation in acute hippocampal slices prepared from animals at different postnatal ages.
Materials and Methods
Hippocampal slice preparation
Hippocampi were rapidly dissected from postnatal 7-d-old to 3-month-old Sprague Dawley rats, submerged in chilled cutting medium containing (in mm) 220 sucrose, 20 NaCl, 2.5 KCl, 1.25 NaH2PO4, 26 NaHCO3, 10 glucose, 2 ascorbic acid, and 2 MgSO4, bubbled with 95% O2–5% CO2, and cut into transverse slices (400 μm thick) using a McIlwain tissue chopper. After isolation, hippocampal slices were placed in incubation baskets in an artificial CSF (aCSF) containing the following (in mm): 124 NaCl, 2.5 KCl, 1.25 NaH2PO4, 24 NaHCO3, 2 ascorbic acid, 10 glucose, 1.5 MgSO4, and 2.5 CaCl2, saturated with 95% O2–5% CO2 and incubated for a 1 h recovery period at 37°C.
NMDA treatment and oxygen/glucose deprivation
NMDA treatment.
After 1 h recovery, hippocampal slices were washed twice with fresh aCSF, gently transferred into individual vials in 2 ml of aCSF containing (in mm) 124 NaCl, 2.5 KCl, 1.25 NaH2PO4, 24 NaHCO3, 10 glucose, 1.5 MgSO4, and 2.5 CaCl2, saturated with 95% O2–5% CO2, and further incubated in the absence or presence of NMDA (100 μm) for 1 or 3 h at 34°C. For NMDA receptor subunit antagonist treatment, slices were incubated with ifenprodil, R-(R,S)-α-(4-hydroxyphenyl)-β-methyl-4-(phenylmethyl)-1-piperidine propranol (Ro25-6981), or [(R)-[(S)-1-(4-bromo-phenyl)-ethylamino]-(2,3-dioxo-1,2,3,4-tetrahydroquinoxalin-5-yl)-methyl]-phosphonic acid (NVP-AAM077) for 20 min before adding NMDA.
Oxygen and glucose deprivation followed by reoxygenation.
After 1 h recovery, hippocampal slices were washed twice with oxygen/glucose deprivation (OGD) solution containing (in mm) 124 NaCl, 2.5 KCl, 1.25 NaH2PO4, 24 NaHCO3, 1.5 MgSO4, and 2.5 CaCl2, pH 7.5, and transferred into individual vials with two slices per vial in 2 ml of OGD solution, previously bubbled with nitrogen for 20 min. Hippocampal slices were incubated in OGD solution at 34°C for 1 h in anaerobic vials saturated with nitrogen; in some cases, slices were then collected and processed for Western blots. For lactate dehydrogenase (LDH) assay, incubation medium was collected after the 1 h incubation period, and slices were washed with aCSF and further incubated for 2 h in 2 ml of fresh aCSF solution saturated with 95% O2–5% CO2. The final LDH release was the combination of LDH release during the 1 and 2 h incubation.
Western blots.
Hippocampal slices were sonicated with a tip sonicator on ice for 6 s twice in a lysis buffer containing 150 mm NaCl, 5 mm EDTA, 1% Triton X-100, 10 mm Tris-HCl, pH 7.4, 0.5 mm phenylmethylsulphonyl fluoride, 2 mg/ml leupeptin, and 1:1000 protease inhibitor mixture. After sample preparation, 20 μg of total proteins was loaded to each lane of 6% or 10% SDS-PAGE gels, and, after separation, proteins were transferred onto polyvinylidene difluoride (PVDF) membranes. The PVDF membranes were blocked with 5% nonfat milk at room temperature for 1 h and probed with different primary antibodies (spectrin and β-actin, 1:10,000 dilution; other primary antibodies, 1:1000 dilution) at 4°C overnight. Membranes were then incubated with secondary antibodies for 1 h and developed with ECL solutions. Western blots were scanned and analyzed quantitatively by densitometry with NIH ImageJ software. Data were generally calculated as fold of control and expressed as means ± SEM from at least four independent experiments. A number of control experiments were performed in which the blots were also labeled with antibodies against β-actin, and the results were then expressed relative to the levels of actin. No significant differences were observed between the two methods, indicating that the results were not caused by unequal amounts of protein loading. Student’s t test was used for statistical analyses, and only p values <0.05 were considered as statistically significant.
Cell viability assay.
Neuronal damage was assessed by measurement of LDH released into the incubation solution (Koh and Choi, 1987; Bruce et al., 1995). At the end of the various treatments, 0.2 ml of medium solution was mixed with 0.8 ml of potassium phosphate buffer (100 mm K2HPO4, adjusted to pH 7.5 with KH2PO4). After 20 min, 0.5 ml of freshly made sodium pyruvate and nicotinamide adenine dinucleotide (NADH) solutions was added to this solution immediately followed by measuring absorbance at 340 nm. LDH activity is expressed as units/ml, with one unit of activity representing the amount of LDH that causes a decrease of 0.001 absorbance units of NADH per minute in the presence of sodium pyruvate. LDH release was normalized to protein concentration and results are shown as fold of controls. Neuronal damage was also assessed by propidium iodide (PI) uptake as described previously (Laake et al., 1999). PI (4.6 μg/ml) was added to bath solution together with different treatment, and after 3 h incubation, slices were fixed overnight in 4% paraformaldehyde in 0.1 m phosphate buffer at pH 7.4. Slices were then transferred to 0.1 m phosphate buffer with 20% sucrose for 24 h and sectioned at 20 μm using a freezing microtome. PI uptake was visualized using a 5× objective with a microscope fitted with fluorescence detection, and images of PI-labeled slices were captured with a CCD camera; at this magnification, one image was sufficient to analyze an entire hippocampal slice. To obtain the best intensity of images and to avoid saturation, all acute hippocampal slices were exposed for 50 ms, and the camera gain was kept constant throughout each experiment. Fluorescence intensity was estimated by the following method: first, images were adjusted to gray levels and captured with Adobe Photoshop (Adobe Systems, San Jose, CA), with the background of images in white and PI-stained structures in black; second, modified images were analyzed quantitatively by densitometry with ImageJ software. Data are generally shown as means ± SEM from the indicated number of independent experiments.
Nissl staining.
After sectioning (20 μm), sections were mounted and blotted onto slides before being processed through different baths in the following order: 100% ethyl alcohol (EtOH), 2 min; xylene, 2 min; 100% EtOH, 2 min; 70% EtOH, 2 min; distilled water, 5 min; cresyl violet, 3 min; distilled water, two dips; 70% EtOH, 5 min; 80% EtOH, 2 min; 90% EtOH, 2 min; 95% EtOH, 2 min; 100% EtOH, 5 min; xylene, 5 min; and then mounted with permount. After 24 h, they were observed under the microscope.
Results
Changes in NMDA-induced toxicity in acute hippocampal slices from young and adult rats
Acute hippocampal slices from rats of various postnatal ages were treated with NMDA (100 μm) for 1 or 3 h. Because LDH release was relatively low in the first hour, 1 h of NMDA treatment was followed by a 3 h recovery period in fresh aCSF to allow enough LDH release into the incubation solution. A 1 h NMDA treatment induced a significant release of LDH in slices from 1- to 3-week-old rats but not in slices from 2- or 3-month-old rats (Fig. 1A). A similar pattern was observed after a 3 h treatment with NMDA (Fig. 1B). In both cases (1 or 3 h NMDA treatment), NMDA-induced LDH release was partly blocked by APV (50 μm) and completely blocked by (+)-5-methyl-10,11-dihydro-5H-dibenzo [a,d] cyclohepten5,10-imine maleate (MK-801; 10 μm) (Fig. 1A,B). Note that basal LDH release was higher in slices from 3 month-old rats compared with that in slices from 1-week-old rats; however, NMDA treatment did not result in higher LDH release than in control slices (Fig. 1C).
Figure 1.

Effects of NMDA treatment on LDH release in acute hippocampal slices from rats of different ages. Hippocampal slices were prepared from rats of the indicated ages and were preincubated for 1 h before adding NMDA. A, B, NMDA-induced LDH release in acute hippocampal slices from rats of various postnatal ages. Slices were incubated with NMDA (100 μm) in the absence or presence of APV (50 μm) or MK-801 (10 μm) for 1 h (A) or 3 h (B). Results are expressed as fold of increase over the respective control values and are means ± SEM of 12 experiments. C, LDH release expressed as fold of values measured in control slices from 1-week-old rats (means ± SEM of 10 experiments). D, Effects of ifenprodil, Ro25-6981, and NVP-AAM077 on NMDA-induced LDH release in slices prepared from 1-week-old, 3-week-old, and 3-month-old rats. Slices were incubated with or without NMDA in the absence or presence of ifenprodil (5 μm), Ro25-6981 (R025; 0.5 μm), or NVP-AAM077 (NVP; 0.5 μm) for 3 h. Results are expressed as fold of increase over the respective control values and are means ± SEM of eight experiments. *p < 0.05 compared with control; †p < 0.05 compared with NMDA-treated slices.
PI staining also indicated that NMDA treatment induced higher cell death in slices from 1-week-old rats than in slices from adults (Fig. 2). Quantitative analysis of staining intensity confirmed the visual impression and indicated that, compared with control, NMDA treatment induced a 3.11 ± 0.3-fold increase in PI staining; this effect could also be completely blocked by MK-801 (1.32 ± 0.15).
Figure 2.

Effects of NMDA treatment on PI staining in acute hippocampal slices from rats of different ages. A, Representative images of PI staining in sections (20 μm thick) from acute slices of 1-week-old and 3-month-old rats treated for 3 h in the absence [control (Cont)] or presence of NMDA, NMDA plus ifenprodil (5 μm), or NMDA plus NVP-AAM077 (NVP; 0.5 μm). B, Quantification of PI staining. Results are expressed as fold of increase over the respective control values and are means ± SEM of five experiments. *p < 0.05 compared with control; †p < 0.05 compared with NMDA-treated slices.
With Nissl staining, NMDA treatment also resulted in a different pattern of staining in slices from 1-week-old rats than in slices from adults (Fig. 3). In the former, Nissl staining was increased in CA1 pyramidal cells and the cells appeared more shrunk. In slices from adult rats, NMDA treatment induced a loss of staining. In both cases, the effect of NMDA treatment was blocked by MK-801 (Fig. 3).
Figure 3.

Effects of NMDA treatment on Nissl staining in acute slices from 1-week-old and 3-month-old rats. Acute hippocampal slices from 1-week-old or 3-month-old rats were incubated with 100 μm NMDA for 3 h in the absence or presence of ifenprodil (Ifen; 5 μm), NVP-AAM077 (NVP; 0.5 μm), or MK-801 (MK; 10 μm). At the end of incubation, sections were fixed and processed for Nissl staining as indicated in Materials and Methods. Note the marked increase in staining in NMDA-treated slices from 1-week-old rats, which contrasts with the decreased staining in slices from 3-month-old rats.
NMDA-induced excitotoxicity in acute hippocampal slices is mediated through NR2B-containing NMDA receptors
NMDA-induced LDH release in slices from 1-week-old rats was almost completely blocked by ifenprodil (5 μm) and by Ro25-6981 (0.5 μm), antagonists with higher affinity for NR2B-containing NMDA receptors. In contrast, application of NVP-AAM077 (0.5 μm), an antagonist selective for NR2A-containing NMDA receptors, exhibited no significant protection. In slices from 3-week-old rats, although both ifenprodil and NVP-AAM077 attenuated NMDA-induced LDH release, only the effect of ifenprodil reached statistical significance (Fig. 1D).
Similarly, NMDA-induced increase in PI uptake in slices from 1-week-old rats was blocked by ifenprodil (1.25 ± 0.16) but not by NVP-AAM077 (Fig. 2). In slices from adult rats, the effect of NMDA treatment was not modified in the presence of MK-801, ifenprodil, or NVP-AAM077. NMDA-induced changes in Nissl staining in slices from both 1-week-old and adult rats were also blocked by ifenprodil but not by NVP-AAM077 (Fig. 3).
Calpain activation is necessary for NMDA-induced excitotoxicity in acute hippocampal slices of young rats
Spectrin is one of the preferred substrates of calpain, and calpain-mediated spectrin degradation is now widely used as a marker for calpain activation. NMDA treatment induced a 3.87 ± 0.46-fold increase in spectrin degradation compared with control in slices of 1-week-old rats. The enhanced spectrin degradation was blocked by MK-801 (0.41 ± 0.06) and markedly decreased by ifenprodil (1.56 ± 0.30) and Ro25-6981 but was not significantly affected by NVP-AAM077 (3.64 ± 0.42) (Fig. 4). Note that MK-801 resulted in a decrease in spectrin degradation below control values, suggesting that some NMDA receptor activation takes place during the incubation of slices even in the absence of exogenous NMDA. In slices from 3-week-old rats, NMDA treatment induced a 1.92 ± 0.13-fold increase in spectrin degradation, which was partly blocked by ifenprodil (1.55 ± 0.09) and by NVP-AAM077 (1.47 ± 0.07) and completely blocked by MK-801 (1.21 ± 0.17). In contrast, NMDA treatment had no effect on spectrin degradation in slices from 3-month-old rats (Fig. 4A).
Figure 4.

Effects of NMDA on calpain-mediated spectrin degradation in acute hippocampal slices from rats of different postnatal ages. Acute hippocampal slices were prepared from 1-week-old, 3-week-old, and 3-month-old rats and were incubated under various experimental conditions. At the end of incubation, slices were sonicated and aliquots were processed for immunoblotting with spectrin antibodies. A, Representative images of Western blots indicating the levels of the calpain-generated SBDPs at 150 and 145 kDa (arrows) in slices from rats of the indicated ages incubated in the absence [control (Cont)] or presence of NMDA, NMDA plus ifenprodil, NMDA plus NVP-AAM077 (NVP), or NMDA plus MK-801. B, Quantitative analysis of blots similar to those shown in A. Blots were scanned and the intensities of SBDP bands were quantified and expressed as fold increase over the respective control values, and the data represent means ± SEM of four experiments. *p < 0.05 compared with control; †p < 0.05 compared with NMDA-treated slices.
When NMDA was applied together with the membrane-permeable calpain inhibitor calpain inhibitor III, or with EGTA in the absence of calcium, spectrin degradation was decreased below control levels in slices from rats of all ages (Fig. 5A). In slices from 1-week-old rats, calpain inhibitor III also significantly reduced NMDA-induced LDH release from 4.03 ± 0.22 to 2.62 ± 0.19 (Fig. 5C).
Figure 5.
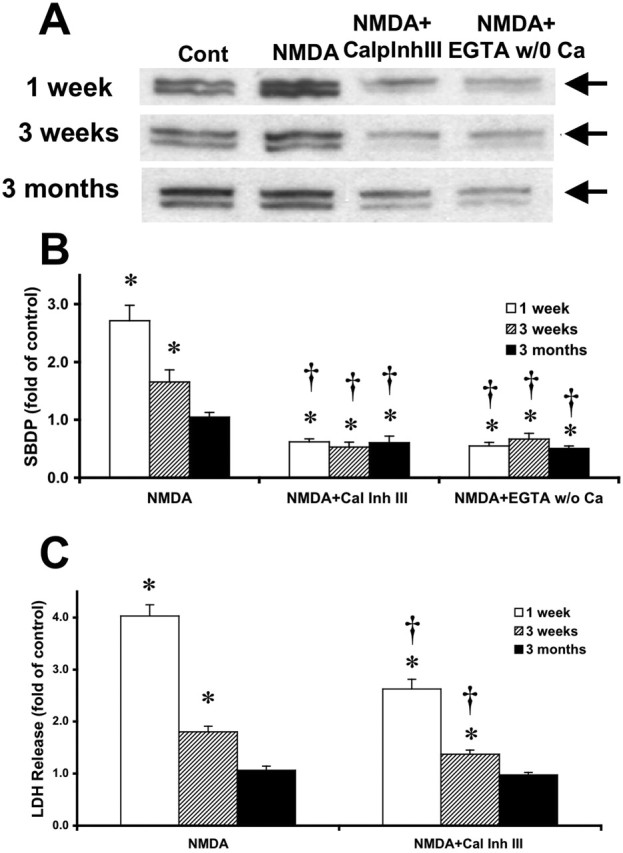
Effects of calpain inhibitor III on NMDA-induced calpain activation and toxicity in acute hippocampal slices from rats of different postnatal ages. A, Representative images of Western blots showing calpain-mediated spectrin breakdown products at 150 and 145 kDa (arrows) in slices treated in the absence [control (Cont)] or presence of NMDA, NMDA plus calpain inhibitor III (10 μm), or NMDA plus EGTA (2 mm) without calcium. B, Quantitative analysis of blots similar to those shown in A. Blots were scanned and the intensities of SBDP bands were quantified and expressed as fold increase over the respective control values, and the data represent means ± SEM of five experiments. C, LDH release in the medium of slices treated in the absence (control) or presence of NMDA or NMDA plus calpain inhibitor III (10 μm) in slices from rats of different ages. Results are expressed as fold of increase over the respective control values and are means ± SEM of six experiments. *p < 0.05 compared with control; †p < 0.05 compared with NMDA-treated slices. CalpInhIII or Cal Inh III, Calpain inhibitor III; w/o, without.
The lack of NMDA-induced toxicity in slices from adult rats is not attributable to a priming effect
It has been reported previously that low levels of NMDA receptor activation could produce a refractory period to subsequent NMDA receptor activation (Vyklicky, 1993). To test the hypothesis that the lack of NMDA toxicity observed in slices from adult rats could be caused by the release of glutamate and the activation of NMDA receptors during the preparation of slices from adult rats and/or the preincubation period, APV (50 μm) or calpain inhibitor III (10 μm) was added to both cutting medium and recovery solution before adding NMDA (100 μm). When APV was present in the cutting medium and during the preincubation period, neither LDH release nor spectrin degradation were increased after NMDA treatment for periods ranging from 10 min to 3 h when compared with control (Fig. 6). With calpain inhibitor III present in the cutting medium and during the preincubation period, there were similarly no obvious changes in NMDA-induced LDH release compared with control (Fig. 6C). However, under these conditions, spectrin degradation was greatly reduced (by ∼60%) in the absence or presence of NMDA for 10 min or 1 h. However, spectrin degradation levels increased to control (no calpain inhibitor III during the 1 h preincubation period) levels after 3 h incubation in the absence of calpain inhibitor III.
Figure 6.
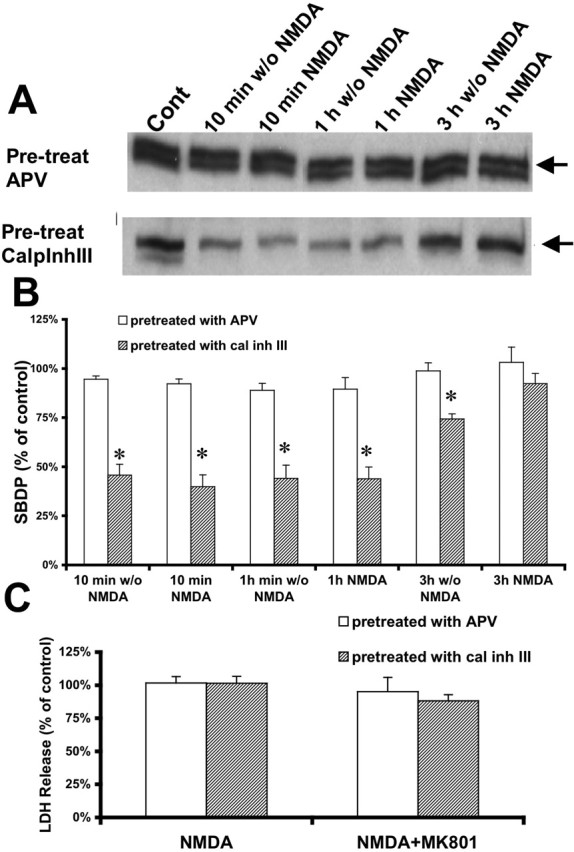
Effects of pretreatment with APV or calpain inhibitor III on NMDA-induced calpain activation and toxicity in acute hippocampal slices of 3-month-old rats. A, Representative images of Western blots showing calpain-mediated spectrin breakdown products at 150 and 145 kDa (arrows) in slices from 3-month-old rats preincubated with APV (50 μm) or calpain inhibitor III (10 μm) during slice preparation and recovery and then treated with NMDA (100 μm) for different periods of time. B, Quantitative analysis of blots similar to those shown in A. Blots were scanned and the intensities of bands were quantified and expressed as percentage of the respective control values, and the data represent means ± SEM of four experiments. C, LDH release in the medium of slices treated in the absence (control) or presence of NMDA and NMDA plus MK-801, after pretreatment with APV or calpain inhibitor III during slice preparation and recovery. Results are expressed as percentage of the respective control values and are means ± SEM of six experiments. *p < 0.05 compared with control. Pre-treat, Pretreated; Cont, control; w/o, without; CalpInhIII or cal inh III, calpain inhibitor III.
OGD induces excitotoxicity in acute slices from both young and adult rats
To determine whether acute slices from adult rats were refractory to toxicity, we compared the toxic effects of another insult, OGD, in hippocampal slices from young and adult rats. In contrast to the lack of effect of NMDA treatment on LDH release in slices from adult rats, OGD treatment induced significant LDH release in slices from adult rats. Increasing the duration of OGD treatment from 20 to 60 min induced increased LDH release compared with control conditions (Fig. 7A). In addition, in slices from 1-week-old rats, MK-801 (10 μm) or calpain inhibitor III (10 μm) significantly reduced OGD-induced LDH release by 37 or 52%; however, this effect was not observed in slices from 3-month-old rats (Fig. 7B).
Figure 7.

Effects of OGD on LDH release in acute hippocampal slices from 1-week-old and 3-month-old rats. A, Acute slices from 3-month-old rats were incubated for different periods of time (20, 40, and 60 min) in the absence of oxygen and glucose without or with MK-801 (10 μm). Results are expressed as fold of increase over the respective control values and are means ± SEM of five experiments. B, Acute slices from 1-week-old and 3-month-old rats were subjected to OGD (1 h), OGD plus MK-801 (10 μm), OGD plus ifenprodil (5 μm), OGD plus NVP-AAM077 (NVP; 0.5 μm), or OGD plus calpain inhibitor III (Cal Inh III; 10 μm). Results are expressed as fold of increase over the respective control values and are means ± SEM of seven experiments. *p < 0.05 compared with controls; †p < 0.05 compared with OGD-treated slices.
One hour of OGD treatment induced a 2.73 ± 0.19-fold increase in spectrin degradation in slices from 1-week-old rats, an effect that was markedly attenuated by MK-801 (1.31 ± 0.20) or completely blocked by calpain inhibitor III (0.75 ± 0.08) (Fig. 8). The same OGD treatment induced a small increase in spectrin degradation in slices from 3-month-old rats (1.34 ± 0.14), and this effect was also blocked by calpain inhibitor III (0.51 ± 0.07) but not by MK-801 (Fig. 8B). NVP-AAM077 (0.5 μm) had no effect on OGD-induced spectrin degradation in slices from either young or adult rats, whereas ifenprodil (5 μm) slightly reduced OGD-induced spectrin degradation in slices from 1-week-old rats (Fig. 8A).
Figure 8.

Effects of OGD on calpain-mediated spectrin degradation in acute hippocampal slices from 1-week-old and 3-month-old rats. A, B, Representative images of Western blots showing calpain-mediated spectrin breakdown products at 150 and 145 kDa (arrows) in slices from 1-week-old and 3-month-old rats subjected to OGD (1 h), OGD plus ifenprodil (5 μm), OGD plus NVP-AAM077 (NVP; 0.5 μm), OGD plus MK-801 (10 μm), or OGD plus calpain inhibitor III (10 μm). C, D, Quantitative analysis of blots similar to those shown in A and B. Blots were scanned and the intensities of bands were quantified and expressed as fold increase over the respective control values, and the data represent means ± SEM of five experiments. *p < 0.05 compared with control; †p < 0.05 compared with OGD-treated slices. Cont, Control; CalpInhIII or Cal Inh III, calpain inhibitor III.
NMDA treatment induces calpain-mediated PSD-95 truncation
PSD-95 is a major postsynaptic density protein and plays an important role in the assembly and organization of postsynaptic components of excitatory synapses. PSD-95 levels were much higher in hippocampal slices from 3-month-old than 1-week-old rats. NMDA treatment induced significant PSD-95 degradation in slices from 1-week-old rats but had little effect on PSD-95 levels in slices from 3-month-old rats (Fig. 9). Surprisingly, in slices from 1-week-old rats, neither NVP-AAM077 nor ifenprodil modified NMDA-induced PSD-95 degradation, although calpain inhibitor III totally blocked NMDA-induced PSD-95 degradation, and in fact slightly increased PSD-95 levels, suggesting that some degree of PSD-95 degradation might take place during the incubation period under control conditions (Fig. 9).
Figure 9.

Effects of NMDA treatment on PSD-95 levels in acute hippocampal slices from 1-week-old and 3-month-old rats. A, Representative Western blots of PSD-95 (arrow) in acute slices from 1-week-old and 3-month-old rats incubated in the absence or presence of NMDA for 1 h. B, Representative Western blots of PSD-95 in acute slices from 1-week-old and 3-month-old rats incubated in the absence or presence of NMDA for 1 h, NMDA plus ifenprodil (5 μm), NMDA plus NVP-AAM077 (NVP; 0.5 μm), or NMDA plus calpain inhibitor III (10 μm). C, D, Quantitative analysis of blots in similar to those shown in A and B. Blots were scanned, and the intensities of bands were quantified and expressed as percentage of 1-week-old (C) and respective (D) control values, and the data represent means ± SEM of four experiments. *p < 0.05 compared with controls; †p < 0.05 compared with NMDA-treated slices. Cont, Control; CalpInhIII or Cal Inh III, calpain inhibitor III.
Discussion
Our results indicate that several changes in the mechanisms of NMDA-mediated neurotoxicity take place during the postnatal period. First, NMDA elicits a rapid LDH release, PI uptake, and calpain activation in acute slices from young but not adult rat hippocampus. NMDA treatment also produced different morphological alterations in CA1 pyramidal neurons in slices from 1-week-old rats and from adult rats. In young rats, Nissl staining was darker and labeled cells appeared to be smaller, suggesting that the cells had shrunk and the Nissl substance was more concentrated. In contrast, in slices from adult rats, NMDA treatment resulted in a loss of Nissl staining, indicating a loss of neurons and/or a decreased density of Nissl substance. This could be because of neuronal swelling and cell death, as was reported previously (Siman and Card, 1988). Few studies have investigated developmental changes in NMDA toxicity. Using cultured hippocampal slices, we did not find significant changes in NMDA-mediated toxicity with increasing periods of cultures ranging from 1 to 4 weeks (Bruce et al., 1995). In contrast, McDonald et al. (1988) reported a large decrease in NMDA toxicity during the developmental period using direct in vivo injection of NMDA in striatum and hippocampus. Our current results therefore indicate that acute hippocampal slices provide a useful model to study mechanisms of NMDA toxicity because it reproduces these in vivo changes.
Second, NMDA treatment rapidly activates the calcium-dependent protease calpain in slices from neonatal but not adult rats. We showed previously that calpain levels are highest in neonatal rats and decline during the postnatal period (Simonson et al., 1985), and this effect could account for the observed decrease in NMDA-mediated calpain activation. In addition, it is possible that adult neurons have a higher capacity to regulate intracellular calcium levels than neonatal ones and that this limits the ability of NMDA treatment to activate calpain. We previously discussed the possibility that the switch from anaerobic to aerobic metabolism during the postnatal period plays an important role in developmental changes in synaptic plasticity and responses to injury, because it provides for increased mitochondrial capacity to synthesize ATP and to absorb large calcium loads (Baudry and Lynch, 1985). In any event, as blockade of calpain activity significantly reduced NMDA-mediated neurotoxicity in slices from neonatal rats, our results clearly establish that calpain activation plays a significant role in neurotoxicity in neonatal rat brain. The role of calpain in excitotoxicity has been controversial, with studies reporting clear evidence for the involvement of this protease, whereas others report the opposite (Bizat et al., 2003; Korhonen et al., 2005). Recent studies using transgenic mice overexpressing human calpastatin, the endogenous inhibitor of calpain, provide clear evidence for a critical role of calpain in excitotoxicity (Higuchi et al., 2005). It is also important to mention that in vivo administration of NMDA results in calpain activation (Siman et al., 1989). However, the time course of this activation appears to be somewhat slower because the peak of activation was observed 24 h after NMDA injection.
Several mechanisms could account for the observed developmental changes in NMDA toxicity. First, the expression of NMDA receptors undergoes subunit- and region-related changes during postnatal development, with high NR2B and low NR2A expression at postnatal day 2, and increased expression of NR1 and NR2A during postnatal development (Sans et al., 2000; Liu et al., 2004). This switch in NMDA receptors from predominantly NR2B-containing receptors to predominantly NR2A-containing receptors matches well with our results, because NMDA-induced toxicity in slices from 1-week-old rats was completely abolished by ifenprodil, an antagonist of NR2B-containing receptors, and not affected by NVP-AAM077, an NR2A-containing receptor antagonist. Moreover, the developmental profile of NMDA-mediated toxicity also matches well the developmental changes in the ratio NR2B/NR2A (Liu et al., 2004). Furthermore, NR2A- and NR2B-containing NMDA receptors have been shown to activate different intracellular cascades, although the results have not been consistent between in vitro and in vivo experiments (Krapivinsky et al., 2003; Liu et al., 2004; Kim et al., 2005). Such differences may relate to the previously reported differences in the stimulation of synaptic (mostly NR2A-containing receptors) and extrasynaptic (mostly NR2B-containing receptors) NMDA receptors, with the former leading to cAMP response element-binding protein activation and increased BDNF expression and neuronal survival and the latter leading to neuronal death (Hardingham et al., 2002). Interestingly, blockade of NR2B-containing receptors almost completely blocked NMDA-mediated spectrin degradation, indicating that activation of NR2B-containing receptors leads to calpain activation. Furthermore, results obtained in slices from 3-week-old rats indicated that blockade of either NR2A-containing or NR2B-containing receptors elicited a significant decrease in NMDA-mediated spectrin degradation, suggesting that activation of either type of receptors leads to calpain stimulation. Nevertheless, blockade of NR2B-containing but not NR2A-containing receptors was neuroprotective in slices from 3-week-old rats. Similarly, blockade of NR2B-containing but not NR2A-containing receptors prevented the changes in Nissl staining induced with NMDA treatment in slices from adult rats.
These results strongly support the idea that the activation of NR2B-containing NMDA receptors is more critical for NMDA-mediated neurotoxicity and that the postnatal decrease in these receptors is responsible for the developmental changes in NMDA toxicity. Surprisingly, blocking either receptor did not prevent calpain-mediated NMDA-mediated degradation of PSD-95 in slices from 1-week-old rats. This result suggests that there might exist additional subtypes of NMDA receptors that are linked to calpain activation at this developmental stage and closely associated with PSD-95.
Several experiments were performed to eliminate alternative explanations for the changes in NMDA toxicity in slices from adult rats. We first showed that the lack of NMDA-induced LDH release and PI uptake was not attributable to the priming of the receptors during slice preparation and preincubation, because previous studies have shown that activation of NMDA receptors is followed by a refractory period (Izumi et al., 1992). We also compared the effects of another type of insult, OGD, in slices from neonatal and adult rats (Taylor et al., 1999). In our experiment, 1 h of OGD treatment produced equivalent and high levels of LDH release in acute hippocampal slices from 1-week-old and 3-month-old rats. However, OGD-induced toxicity in slices of young or adult rats also involved different mechanisms, because MK-801 and calpain inhibitor III could partly block OGD-induced LDH release in slices from 1-week-old rats but had no effect on slices from adult rats. These results indicate that activation of NMDA receptors and calpain is involved in OGD-induced cell death in slices from young, but not adult, rats. Although synaptic glutamate release is increased by OGD treatment and OGD-induced cell death could be reduced by NMDA receptor inhibitors (Beck et al., 2003; Fujimoto et al., 2004), recent studies have indicated that in ischemic brain, acidosis might play a key role. It appears more likely that ischemia-induced neuronal injury is mediated by acidosis and activation of Ca2+-permeable acid-sensing ion channels (Xiong et al., 2004). This might explain why glutamate antagonists have failed to show effective neuroprotection in stroke in multiple human trials.
In conclusion, our results show that NMDA treatment exhibited developmental differences in excitotoxicity in acute hippocampal slices. In slices from neonatal rats, NMDA-induced excitotoxicity was associated with LDH release, PI uptake, and calpain activation as a result of the activation of NR2B-containing receptors. In contrast, NMDA-induced toxicity in slices from adult rats was not associated with LDH release, PI uptake, or calpain activation. Although one could argue that the higher levels of background for calpain-mediated spectrin breakdown product might have masked calpain activation in slices from adult rats, several reasons suggest that this is not the likely explanation. First, we were still able to detect an increase in spectrin breakdown after OGD treatment in slices from adult rats. Second, we were not able to detect NMDA-mediated calpain activation under conditions in which the basal levels of spectrin breakdown were markedly decreased (such by including calpain inhibitor in the cutting medium, a treatment that was shown not to modify NMDA-induced calpain activation in juvenile slices). In contrast, a 1 h OGD treatment elicited a similar degree of neurotoxicity in acute hippocampal slices from rats of different ages, although the mechanisms by which OGD induced neurotoxicity were also different between young and old rats. Finally, our results indicate that the use of acute hippocampal slices provide an interesting model to study mechanisms of NMDA toxicity that can be useful to better understand mechanisms of neurodegeneration.
Footnotes
This work was supported by National Institute on Aging Grant AG-14751 (principal investigator, Dr. C. Finch) and National Institute of Neurological Disorders and Stroke Grant NS048521-02 (M.B.). We thank Novartis for the generous gift of NVP-AAM077.
References
- Araujo IM, Xapelli S, Gil JM, Mohapel P, Petersen A, Pinheiro PS, Malva JO, Bahr BA, Brundin P, Carvalho CM (2005). Proteolysis of NR2B by calpain in the hippocampus of epileptic rats. NeuroReport 16:393–396. [DOI] [PubMed] [Google Scholar]
- Barth AL, Malenka RC (2001). NMDAR EPSC kinetics do not regulate the critical period for LTP at thalamocortical synapses. Nat Neurosci 4:235–236. [DOI] [PubMed] [Google Scholar]
- Baudry M, Lynch G (1985). Regulation of calcium transport in rat hippocampal mitochondria during development and following denervation. Prog Brain Res 63:107–119. [DOI] [PubMed] [Google Scholar]
- Beck J, Lenart B, Kintner DB, Sun D (2003). Na–K–Cl cotransporter contributes to glutamate-mediated excitotoxicity. J Neurosci 23:5061–5068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bi X, Chen J, Baudry M (1997). Developmental changes in calpain activity, GluR1 receptors and in the effect of kainic acid treatment in rat brain. Neuroscience 81:1123–1135. [DOI] [PubMed] [Google Scholar]
- Bizat N, Hermel JM, Boyer F, Jacquard C, Creminon C, Ouary S, Escartin C, Hantraye P, Kajewski S, Brouillet E (2003). Calpain is a major cell death effector in selective striatal degeneration induced in vivo by 3-nitropropionate: implications for Huntington’s disease. J Neurosci 23:5020–5030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brorson JR, Marcuccilli CJ, Miller RJ (1995). Delayed antagonism of calpain reduces excitotoxicity in cultured neurons. Stroke 26:1259–1266. discussion 1267. [DOI] [PubMed] [Google Scholar]
- Bruce AJ, Sakhi S, Schreiber SS, Baudry M (1995). Development of kainic acid and N-methyl-d-aspartic acid toxicity in organotypic hippocampal cultures. Exp Neurol 132:209–219. [DOI] [PubMed] [Google Scholar]
- Chan SL, Mattson MP (1999). Caspase and calpain substrates: roles in synaptic plasticity and cell death. J Neurosci Res 58:167–190. [PubMed] [Google Scholar]
- Collingridge GL, Lester RA (1989). Excitatory amino acid receptors in the vertebrate central nervous system. Pharmacol Rev 41:143–210. [PubMed] [Google Scholar]
- Cull-Candy S, Brickley S, Farrant M (2001). NMDA receptor subunits: diversity, development and disease. Curr Opin Neurobiol 11:327–335. [DOI] [PubMed] [Google Scholar]
- Dingledine R, Borges K, Bowie D, Traynelis SF (1999). The glutamate receptor ion channels. Pharmacol Rev 51:7–61. [PubMed] [Google Scholar]
- Fujimoto S, Katsuki H, Kume T, Kaneko S, Akaike A (2004). Mechanisms of oxygen glucose deprivation-induced glutamate release from cerebrocortical slice cultures. Neurosci Res 50:179–187. [DOI] [PubMed] [Google Scholar]
- Hardingham GE, Fukunaga Y, Bading H (2002). Extrasynaptic NMDARs oppose synaptic NMDARs by triggering CREB shut-off and cell death pathways. Nat Neurosci 5:405–414. [DOI] [PubMed] [Google Scholar]
- Higuchi M, Tomioka M, Takano J, Shirotani K, Iwata N, Masumoto H, Maki M, Itohara S, Saido TC (2005). Distinct mechanistic roles of calpain and caspase activation in neurodegeneration as revealed in mice overexpressing their specific inhibitors. J Biol Chem 280:15229–15237. [DOI] [PubMed] [Google Scholar]
- Izumi Y, Clifford DB, Zorumski CF (1992). Low concentrations of N-methyl-d-aspartate inhibit the induction of long-term potentiation in rat hippocampal slices. Neurosci Lett 137:245–248. [DOI] [PubMed] [Google Scholar]
- Kim MJ, Dunah AW, Wang YT, Sheng M (2005). Differential roles of NR2A- and NR2B-containing NMDA receptors in Ras-ERK signaling and AMPA receptor trafficking. Neuron 46:745–760. [DOI] [PubMed] [Google Scholar]
- Koh JY, Choi DW (1987). Quantitative determination of glutamate mediated cortical neuronal injury in cell culture by lactate dehydrogenase efflux assay. J Neurosci Methods 20:83–90. [DOI] [PubMed] [Google Scholar]
- Korhonen L, Hansson I, Kukkonen JP, Brannvall K, Kobayashi M, Takamatsu K, Lindholm D (2005). Hippocalcin protects against caspase-12-induced and age-dependent neuronal degeneration. Mol Cell Neurosci 28:85–95. [DOI] [PubMed] [Google Scholar]
- Kornau HC, Schenker LT, Kennedy MB, Seeburg PH (1995). Domain interaction between NMDA receptor subunits and the postsynaptic density protein PSD-95. Science 269:1737–1740. [DOI] [PubMed] [Google Scholar]
- Krapivinsky G, Krapivinsky L, Manasian Y, Ivanov A, Tyzio R, Pellegrino C, Ben-Ari Y, Clapham DE, Medina I (2003). The NMDA receptor is coupled to the ERK pathway by a direct interaction between NR2B and RasGRF1. Neuron 40:775–784. [DOI] [PubMed] [Google Scholar]
- Kuroiwa T, Okeda R (1994). Neuropathology of cerebral ischemia and hypoxia: recent advances in experimental studies on its pathogenesis. Pathol Int 44:171–181. [DOI] [PubMed] [Google Scholar]
- Laake JH, Haug FM, Wieloch T, Ottersen OP (1999). A simple in vitro model of ischemia based on hippocampal slice cultures and propidium iodide fluorescence. Brain Res Brain Res Protoc 4:173–184. [DOI] [PubMed] [Google Scholar]
- Lankiewicz S, Marc LC, Truc BN, Krohn AJ, Poppe M, Cole GM, Saido TC, Prehn JH (2000). Activation of calpain I converts excitotoxic neuron death into a caspase-independent cell death. J Biol Chem 275:17064–17071. [DOI] [PubMed] [Google Scholar]
- Liu XB, Murray KD, Jones EG (2004). Switching of NMDA receptor 2A and 2B subunits at thalamic and cortical synapses during early postnatal development. J Neurosci 24:8885–8895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu HC, Gonzalez E, Crair MC (2001). Barrel cortex critical period plasticity is independent of changes in NMDA receptor subunit composition. Neuron 32:619–634. [DOI] [PubMed] [Google Scholar]
- Lu X, Rong Y, Baudry M (2000). Calpain-mediated degradation of PSD-95 in developing and adult rat brain. Neurosci Lett 286:149–153. [DOI] [PubMed] [Google Scholar]
- Lynch DR, Guttmann RP (2002). Excitotoxicity: perspectives based on N-methyl-d-aspartate receptor subtypes. J Pharmacol Exp Ther 300:717–723. [DOI] [PubMed] [Google Scholar]
- Lynch G, Baudry M (1987). Brain spectrin, calpain and long-term changes in synaptic efficacy. Brain Res Bull 18:809–815. [DOI] [PubMed] [Google Scholar]
- McDonald JW, Silverstein FS, Johnston MV (1988). Neurotoxicity of N-methyl-d-aspartate is markedly enhanced in developing rat central nervous system. Brain Res 459:200–203. [DOI] [PubMed] [Google Scholar]
- Nath R, Raser KJ, Stafford D, Hajimohammadreza I, Posner A, Allen H, Talanian RV, Yuen P, Gilbertsen RB, Wang KK (1996). Non-erythroid alpha-spectrin breakdown by calpain and interleukin 1 beta-converting-enzyme-like protease(s) in apoptotic cells: contributory roles of both protease families in neuronal apoptosis. Biochem J 319:683–690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saido TC, Sorimachi H, Suzuki K (1994). Calpain: new perspectives in molecular diversity and physiological-pathological involvement. FASEB J 8:814–822. [PubMed] [Google Scholar]
- Sans N, Petralia RS, Wang YX, Blahos J, Hell JW, Wenthold RJ (2000). A developmental change in NMDA receptor-associated proteins at hippocampal synapses. J Neurosci 20:1260–1271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seubert P, Ivy G, Larson J, Lee J, Shahi K, Baudry M, Lynch G (1988). Lesions of entorhinal cortex produce a calpain-mediated degradation of brain spectrin in dentate gyrus. I. Biochemical studies. Brain Res 459:226–232. [DOI] [PubMed] [Google Scholar]
- Skaper SD, Facci L, Strijbos PJ (2001). Neuronal protein kinase signaling cascades and excitotoxic cell death. Ann NY Acad Sci 939:11–22. [DOI] [PubMed] [Google Scholar]
- Siman R, Card JP (1988). Excitatory amino acid neurotoxicity in the hippocampal slice preparation. Neuroscience 26:433–447. [DOI] [PubMed] [Google Scholar]
- Siman R, Noszek JC, Kegerise C (1989). Calpain I activation is specifically related to excitatory amino acid induction of hippocampal damage. J Neurosci 9:1579–1590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simonson L, Baudry M, Siman R, Lynch G (1985). Regional distribution of soluble calcium activated proteinase activity in neonatal and adult rat brain. Brain Res 327:153–159. [DOI] [PubMed] [Google Scholar]
- Taylor CP, Weber ML, Gaughan CL, Lehning EJ, LoPachin RM (1999). Oxygen/glucose deprivation in hippocampal slices: altered intraneuronal elemental composition predicts structural and functional damage. J Neurosci 19:619–629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Towfighi J, Mauger D (1998). Temporal evolution of neuronal changes in cerebral hypoxia-ischemia in developing rats: a quantitative light microscopic study. Brain Res Dev Brain Res 109:169–177. [DOI] [PubMed] [Google Scholar]
- Vanderklish P, Saido TC, Gall C, Arai A, Lynch G (1995). Proteolysis of spectrin by calpain accompanies theta-burst stimulation in cultured hippocampal slices. Brain Res Mol Brain Res 32:25–35. [DOI] [PubMed] [Google Scholar]
- Vanderklish PW, Bahr BA (2000). The pathogenic activation of calpain: a marker and mediator of cellular toxicity and disease states. Int J Exp Pathol 81:323–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vyklicky L Jr (1993). Calcium-mediated modulation of N-methyl-d-aspartate (NMDA) responses in cultured rat hippocampal neurones. J Physiol (Lond) 470:575–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waxman EA, Lynch DR (2005). N-methyl-d-aspartate receptor subtypes: multiple roles in excitotoxicity and neurological disease. The Neuroscientist 11:37–49. [DOI] [PubMed] [Google Scholar]
- Wechsler A, Teichberg VI (1998). Brain spectrin binding to the NMDA receptor is regulated by phosphorylation, calcium and calmodulin. EMBO J 17:3931–3939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wenzel A, Fritschy JM, Mohler H, Benke D (1997). NMDA receptor heterogeneity during postnatal development of the rat brain: differential expression of the NR2A, NR2B, and NR2C subunit proteins. J Neurochem 68:469–478. [DOI] [PubMed] [Google Scholar]
- Xiong ZG, Zhu XM, Chu XP, Minami M, Hey J, Wei WL, MacDonald JF, Wemmie JA, Price MP, Welsh MJ, Simon RP (2004). Neuroprotection in ischemia: blocking calcium-permeable acid-sensing ion channels. Cell 118:687–698. [DOI] [PubMed] [Google Scholar]