

SUMMARY
The CNS plays a pivotal role in energy homeostasis, but whether oligodendrocytes are involved has been largely unexplored. Here, we show that signaling through GPR17, a G-protein-coupled receptor predominantly expressed in the oligodendrocyte lineage, regulates food intake by modulating hypothalamic neuronal activities. GPR17-null mice and mice with an oligodendrocyte-specific knockout of GPR17 have lean phenotypes on a high-fat diet, suggesting that GPR17 regulates body weight by way of oligodendrocytes. Downregulation of GPR17 results in activation of cAMP-protein kinase A (PKA) signaling in oligodendrocytes and upregulated expression of pyruvate dehydrogenase kinase 1 (PDK1), which promotes lactate production. Elevation of lactate activates AKT and STAT3 signaling in the hypothalamic neurons, leading to increased expression of Pomc and suppression of Agrp. Our findings uncover a critical role of oligodendrocytes in metabolic homeostasis, where GPR17 modulates the production of lactate, which, in turn, acts as a metabolic signal to regulate neuronal activity.
Graphical Abstract
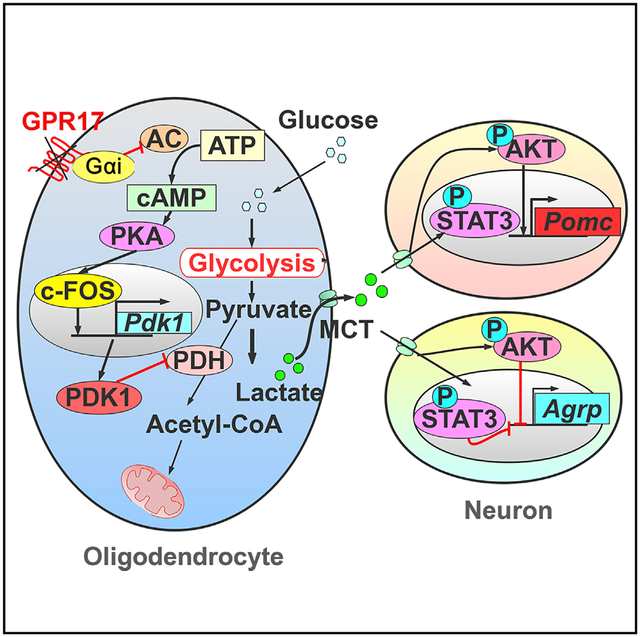
In Brief
Ou et. al. show that Gpr17 in oligodendrocytes contributes to whole-body metabolic regulation. Gpr17-deficient mice exhibit decreased body weight on long-term high-fat feeding by reducing food intake. Loss of Gpr17 increases oligodendrocytic lactate production, which results in the lean phenotype of the KO animals.
INTRODUCTION
The arcuate nucleus in the hypothalamus is critical for regulating whole-body metabolic homeostasis (Bouret et al., 2004; Coppari et al., 2005). The agouti-related protein (AgRP) and neuropeptide Y (NPY), which are synthesized and secreted from AgRP/NPY neurons, are two anabolic neuropeptides that stimulate food intake and decrease energy expenditure. Increased hypothalamic NPY levels in rats lead to hyperphagia and obesity (Zarjevski et al., 1993), whereas loss of the Npy gene attenuates the obesity syndrome in ob/ob mice (Erickson et al., 1996). Conversely, melanocortins, such as a-melanocyte-stimulating hormone (α-MSH) synthesized by proopiomelanocortin (POMC) neurons, inhibit food intake and promote energy expenditure (Fan et al., 1997). Mice lacking the melanocortin receptor MC4R are hyperphagic and severely obese, suggesting the importance of melanocortin signaling in the control of energy homeostasis (Huszar et al., 1997).
Recent studies have demonstrated the importance of metabolic interaction between neurons and other cell types, such as astrocytes(García-Cáceres et al., 2016; Kim et al., 2014), microglia (Valdearcos et al., 2017; Zhang et al., 2008), and tanycytes (Balland et al., 2014; Hofmann et al., 2017) in the regulation of energy homeostasis. Oligodendrocytes have also been suggested to be involved in metabolic homeostasis (Trevisiol and Nave, 2015). Lack of proliferative oligodendrocyte precursor cells (OPCs) in the median eminence impairs hypothalamic leptin sensing and leads to weight gain in mice (Djogo et al., 2016). Oligodendrocytes provide pyruvate and lactate to ensheathed neurons to maintain the integrity and normal neuronal functions (Lee et al., 2012; Saab et al., 2016), but the mechanisms underlying oligodendrocyte regulation of energy metabolic homeostasis remain elusive.
GPR17, a G-protein coupled receptor (GPCR) coupled to the Gi subunit, is predominantly restricted to the oligodendrocyte lineage. It is crucial for the timing of oligodendrocyte myelination (Chen et al., 2009). Overexpression of Gpr17 inhibits myelin sheath development, whereas loss of Gpr17 accelerates it and promotes remyelination upon injury (Chen et al., 2009; Ou et al., 2016). Recent studies suggest that GPR17 might also be involved in the regulation of whole-body energy homeostasis. In experiments in mice, administration of the GPR17 antagonist cangrelor reduced food intake, whereas the GPR17 agonist LTD4 induced food intake (Ren et al., 2012). Moreover, mice with Agrp-cre-mediated depletion of GPR17 are leaner than wild-type (WT) mice (Ren et al., 2015). In contrast, Mastaitis et al. (2015) reported that whole-body Gpr17 knockout mice (Gpr17−/−) did not differ from WT mice in food consumption, glucose homeostasis, or energy expenditure when fed either standard chow or a high-fat diet (HFD) for 10 weeks. Therefore, whether and how GPR17 is involved in the regulation of energy homeostasis remains debated.
In this study, we used multiple approaches and animal models to investigate the effect of oligodendrocytes and GPR17 on energy homeostasis in mice. By analysis of mice that express GFP knocked in at the Gpr17 locus, we showed that this gene is specifically expressed in oligodendrocyte lineage cells but hardly detectable in neurons. Both whole-body and oligodendrocyte-specific Gpr17 knockout mice gained less weight when chronically exposed to an HFD than WT mice. Mechanistically, loss of Gpr17 in oligodendrocytes enhanced glycolysis and lactate production, which then activated AKT and STAT3 signaling in hypothalamic neurons, leading to reciprocal AgRP and POMC neuropeptide regulation, increased energy expenditure, and amelioration of HFD-induced obesity in mice. Our data confirm the important role of GPR17 in oligodendrocytes, but not in neurons, in metabolic control and also demonstrate that the GPR17/cyclic AMP (cAMP)/lactate signaling axis regulates the activities of hypothalamic neurons to maintain energy homeostasis.
RESULTS
Gpr17−/− Mice Are Resistant to HFD-Induced Obesity
To investigate the effect of GPR17 on energy homeostasis, Gpr17−/− mice (Chen et al., 2009) and their WT littermates were fed a normal chow diet (NCD) or an HFD. On the NCD, the body weight of Gpr17−/− mice did not differ from WT litter-mate controls (Figure S1). On the HFD, there was no significant difference in body weight between the two genotypes for the first 12 weeks, but after that, the body weight in Gpr17−/− mice became lower than that of WT controls; the difference was statistically significant beginning at 16 weeks (Figure 1A). When sacrificed after 28 weeks on the HFD, Gpr17−/− mice had significantly less fat mass compared with WT mice (Figure 1B). The serum levels of insulin and leptin were also significantly lower in Gpr17−/− mice (Figures 1C and 1D). H&E staining showed that adipocytes in the epididymal white adipose tissue (eWAT) of Gpr17−/− mice were much smaller than those of WT mice (Figures 1E and 1F). Consistently, there was less lipid accumulation in brown adipose tissue (BAT) of Gpr17−/− mice and improved hepatic steatosis compared with WT mice (Figures 1E, 1G, and 1H). Gpr17−/− mice showed improved capability for glucose clearance and increased insulin sensitivity relative to WT mice (Figures 1I and 1J). As shown in Figure 1K, Gpr17−/− mice consumed less food than their WT counterparts in the dark phase. When normalized to lean mass, the levels of oxygen consumption, carbon dioxide production, heat production, and ambulatory activities were comparable between the two geno-types (Figures 1L–1O).
Figure 1.

Gpr17−/− Mice Are Resistant to HFD-Induced Obesity (A–H) Gpr17−/− and their wild-type (WT) littermates were subjected to 28-week HFD feeding initiating at 4 weeks of age. After feeding, the mice were sacrificed, and tissue samples were collected.
(A) Body weight recorded weekly.
(B) Weight of eWAT.
(C) Levels of serum insulin.
(D) Levels of serum leptin.
(E) Representative images of H&E stained eWAT, BAT, and liver. Scale bar, 50 μm.
(F) The volume of the adipocytes in eWAT.
(G) Lipid droplet-positive area in the BAT.
(H) Triglyceride content in the liver.
(I) Glucose tolerance test (GTT) at 24 weeks of HFD.
(J) Insulin tolerance test (ITT) at 25 weeks of HFD.
(K) The food intake of WT and Gpr17−/− mice.
(L–O) Metabolic parameters of Gpr17−/− and WT mice after 26 weeks of HFD feeding, normalized by lean body mass. Shown are (L) O2 consumption, (M) CO2 production, (N) heat production, and (O) ambulatory activity recorded during 48 h of dark and light cycles.
Open and filled bars represent light and dark phases, respectively. Each value represents mean ± SEM of 7 mice from each genotype. *p < 0.05, **p < 0.01, ***p < 0.001, Student’s t test.
Mice Treated with a GPR17 Antagonist Are Resistant to HFD-Induced Obesity
To confirm that GPR17 was required for weight gain on the HFD, WT mice were treated with pranlukast, a GPR17 antagonist that has been shown to effectively antagonize GPR17 biological functions (Ou et al., 2016). Pranlukast-treated WT mice gained less body weight on the HFD than untreated mice (Figure S2A). Pranlukast treatment also modestly decreased the levels of serum insulin, leptin, free fatty acids, and liver triglycerides (Figures S2B–S2E).
Mice Lacking GPR17 in the Oligodendrocyte Lineage Are Resistant to HFD-Induced Obesity
Gpr17 has been shown to be predominantly expressed in the oligodendrocyte lineage (Chen et al., 2009) but has also been reported in other cells types (Ren et al., 2015; Zhao et al., 2012). We performed a study to characterize the expression pattern of Gpr17 in the brain using Gpr17−/− mice, in which the Gpr17 gene was replaced by a nucleus-localized GFP gene (Chen et al., 2009). The majority of GFP-positive cells were oligodendrocytes in the corpus callosum, cortex, and hypothalamus arcuate nucleus area, as indicated by a series of oligodendrocyte lineage markers (Figure 2A; Figure S3A). None of the GFP-positive cells expressed neuronal markers—e.g., NeuN or parvalbumin (Figure 2A; Figure S3B) in the cortex that can express Olig2, and the GFP-positive cells did not overlap with AgRP or POMC expression (Figure 2B; Figure S3C), indicating that parvalbumin, AgRP, or POMC neurons did not express Gpr17. In addition, no GFP-positive cells expressed Iba1 or glial gibrillary acidic protein (GFAP) (Figure S3C), suggesting that Gpr17 is not expressed in microglia or astrocytes.
Figure 2.

Gpr17 Is Expressed Predominantly in the Oligodendrocyte Lineage (A) Immunofluorescent analysis of GFP, Olig2, and NeuN in cryostat sections of the corpus callosum (C.C), arcuate nucleus (ARC), and ventromedial nucleus (VMH) of Gpr17−/− mice as described in STAR Methods. Scale bar, 25 μm. The percentage of Olig2- or NeuN-positive cells was quantified as indicated. N.D., not detected.
(B) Immunofluorescence analysis of GFP and AgRP or POMC in the ARC of Gpr17−/− mice. Scale bar, 25 μm. Arrows indicate GFP-positive cells.
(C) Immunofluorescence analysis of tdTomato and oligodendrocyte lineage markers, including Olig2, CC1, and PDGFRα, and the neuron marker NeuN in cryostat sections from the C.C (Olig2, CC1, and PDGFRα) or ARC (NeuN) in Olig1cre/+;tdTomato mice. Scale bar, 25 μm. Arrows indicate tdTomato-positive cells. The percentages of Olig2-, CC1-, PDGFRα-, and NeuN-positive cells were quantified as indicated.
(D) Immunofluorescence analysis of tdTomato and AgRP or POMC in the ARC of Olig1cre/+;tdTomato mice. Scale bar, 25 mm. Arrows indicate tdTomato-positive cells.
Given these results, we hypothesized that GPR17 produced in oligodendrocytes regulates whole-body metabolic homeostasis. To test this, we generated conditional Gpr17 knockout mice (Olig1cre/+;Gpr17fl/fl, Gpr17cKO) in which Gpr17 was specifically knocked out in the oligodendrocyte lineage by crossing the Gpr17fl/fl and oligodendrocyte lineage-expressing Olig1cre/+ mouse strains (Lu et al., 2002; Xin et al., 2005). To show that Olig1-Cre depleted GPR17 in oligodendrocytes, Olig1cre/+ mice were crossed with Gt(ROSA)26Sortm9(CAG-tdTomato)Hze mice, and immunofluorescence analysis was performed to evaluate the expression of tdTomato. This experiment revealed that the Olig1-Cre recombinase targeted mostly oligodendrocyte lineage cells, which were positive for Olig2, CC1, and platelet-derived growth factor receptor a (PDGFRa) (Figure 2C). tdTomato was completely undetectable in NeuN-positive neurons and in Iba1-positive microglia (Figures 2C; S3D). Importantly, the Olig1-Cre recombinase did not target AgRP- or POMC-positive cells in the hypothalamic arcuate nucleus (Figure 2D). Olig1-Cre recombinase expression was detected in a subset of astrocytes (GFAP-positive cells; Figure S3D), but it should have little or no effect on our analysis because we did not detect GFP expression in astrocytes in the Gpr17 knockin experiment (Figure S3C). These data demonstrate that the Gpr17cKO mouse can be used to investigate the potential effect of Gpr17 deletion, specifically in oligodendrocytes, on whole-body energy homeostasis.
When Gpr17cKO mice and their control littermates (Gpr17fl/fl) were fed the NCD, there were no obvious differences in body weight, fat mass, or lipid accumulation in tissues or metabolic parameters, including food intake, oxygen consumption, carbon dioxide production, heat production, or ambulatory activity (Figure S4). On the HFD, however, Gpr17cKO mice mirrored Gpr17−/− mice. Gpr17cKO mice had lower body weight and less fat mass content than their control littermates after 16 weeks of HFD feeding (Figures 3A and 3B). Serum levels of insulin and leptin were decreased in Gpr17cKO mice relative to controls (Figures 3C and 3D), and lipid accumulation in the liver, WAT, and BAT was also significantly lower in Gpr17cKO mice (Figures 3E–3H). Lack of GPR17 in oligodendrocytes also improved glucose tolerance and insulin sensitivity (Figures 3I and 3J). Moreover, the metabolic cage data also revealed that Gpr17cKO mice had decreased food intake in the dark phase (Figure 3K). When normalized to lean mass, Gpr17cKO mice had similar levels of oxygen consumption, carbon dioxide, heat production, and ambulatory activities as their control littermates (Figures 3L–3O).
Figure 3.

Loss of Oligodendrocytic GPR17 Protects against HFD-Induced Obesity (A–H) Gpr17cKO (Olig1cre/+;Gpr17fl/fl) and control (Gpr17fl/fl) mice were subjected to HFD feeding initiated at 8 weeks of age. The mice were sacrificed, and tissue samples were collected after 30 weeks on the HFD.
(A) Body weight recorded weekly.
(B) Weight of eWAT.
(C) Serum levels of insulin.
(D) Serum levels of leptin.
(E) Representative images of H&E stained eWAT, BAT, and liver. Scale bar, 50 μm.
(F) The volume of the adipocytes in eWAT.
(G) Lipid droplet-positive area in the BAT.
(H) Triglyceride content in the liver.
(I) GTT at 24 weeks of HFD.
(J) ITT at 25 weeks of HFD.
(K) The food intake of control and Gpr17cKO mice.
(L–O) Metabolic parameters of Gpr17cKO and WT mice after 26 weeks of HFD feeding, normalized by lean body mass. Shown are (L) O2 consumption, (M) CO2 production, (N) heat production, and (O) ambulatory activity during 48 h of dark and light cycles.
Open and filled bars represent light and dark phases, respectively. Each value represents mean ± SEM of 11 mice per genotype. *p < 0.05, **p < 0.01, ***p < 0.001, Student’s t test.
Because we have previously reported that loss of Gpr17 promotes precocious myelination and remyelination (Chen et al., 2009), we examined myelin sheaths in Gpr17−/− mice fed an NCD or HFD and their control littermates. We found that the g-ratio and myelinated axon numbers were similar after 30 weeks of NCD feeding and after 60 weeks of HFD feeding (Figure S5). This suggests that myelination does not contribute to the lean phenotype of Gpr17−/− mice.
A previous study by Mastaitis et al. (2015) showed that the expression of Lims2 was decreased by 30% in their Gpr17-null mice relative to WT controls (Mastaitis et al., 2015). This was likely due to the fact that the coding region of Gpr17 is nested in the fourth intron of Lims2 (Figure S6C). We then examined the expression of Lims2 in the corpus callosum and hypothalamus in our Gpr17−/− mice and their littermates. We found that the expression of Lims2 was unaffected after Gpr17 deletion (Figure S6D). Furthermore, there was no difference in the expression of Lims2 between WT and Gpr17-deficient OPCs or mature oligodendrocytes (OLs; Figure S6E). These results suggest that Lims2 expression is not the driving force behind the lean pheno-type in our Gpr17−/− mice.
Loss of Gpr17 Promotes Lactate Production in OLs
Previous studies have shown that OLs can metabolically support ensheathed axons through secretion of metabolites, including lactate and pyruvate, which maintain axonal function and ensure neuronal survival (Lee et al., 2012; Saab et al., 2016). We hypothesized that depletion of GPR17 from OLs resulted in an altered metabolic signal from OLs to nearby neurons and, thus, led to changes in whole-body energy homeostasis.
To examine the effect of GPR17 on the metabolic profile of OLs, we isolated primary OPCs from WT and Gpr17−/− mice and subjected these cells, before and after differentiation into OLs, to the Seahorse assay. GPR17-deficient OPCs displayed dramatically increased extracellular acidification rates (ECARs), a surrogate marker of glycolysis, compared with WT OPCs (Figures 4A and 4B), suggesting increased glycolysis and lactate production in the absence of GPR17. To verify this, we directly measured lactate production by WT and GPR17-deficient OPCs because GPR17 is strongly expressed at the OPC and newly formed OL stages. We found a higher lactate concentration in medium collected from GPR17-deficient OPCs (Figure 4C). Similar observations were made in OLs (Figures 4D–5F). Moreover, the oxygen consumption rates (OCRs) were similar in WT and GPR17-deficient OPCs and OLs (Figure 4G and 4H), indicating that oxidative phosphorylation in mitochondria was unaffected by lack of GPR17. There was no difference in viability between GPR17-deficient and WT OPCs, as indicated by a CCK-8 cell viability assay (Figure 4I) and by the lack of TdT-mediated dUTP nick-end labeling (TUNEL) staining (data not shown).
Figure 4.

Ablation of Gpr17 Promotes Glycolysis in Oligodendrocytes Primary OPCs from WT or Gpr17−/− mice were isolated, cultured, and differentiated into oligodendrocytes (OLs) as described in the STAR Methods.
(A) ECAR of WT and Gpr17−/− OPCs as a function of time after sequential administrations of 10 mM glucose, 1.5 μM oligomycin, and 50 mM 2-Deoxy-D-glucose (2-DG). Dotted lines indicate the starting point of treatment with the indicated compounds.
(B) Quantifications of the ECAR results in (A).
(C) Lactate concentration in the medium of WT and Gpr17−/− OPCs.
(D) ECAR of WT and Gpr17−/− OLs.
(E) Quantifications of the ECAR results in (D).
(F) Lactate concentrations in the culture medium of WT and Gpr17−/− OLs.
(G and H) OCR was monitored over time after sequential administration of 1.5 μM oligomycin, 1.5 μM carbonyl cyanide-4-(trifluoromethoxy)phenylhydrazone (FCCP), and 0.5 mM rotenone and antimycin A mixture in (G) OPCs and (H) OLs.
(I) Viability of mouse OPCs measured with the CCK-8 assay.
Data are means of at least three independent experiments. Each value represents mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001, Student’s t test.
Figure 5.

Ablation of Gpr17 Increases Pdk1 Expression through cAMP-PKA Signaling (A) Total proteins isolated from primary OPCs and OLs from WT or Gpr17−/− mice were subjected to western blot using the indicated antibodies. The relative band intensity of each protein is shown on the right. The experiments were repeated 3 times.
(B) Total RNAs were isolated, and Pdk1 was quantified by real-time PCR. n = 3 animals/genotype.
(C) Total protein was extracted from the hypothalamus of Gpr17fl/fl and Gpr17cKO mice fed the NCD or HFD for 30 weeks and subjected to western blot using the indicated antibodies. The relative intensity of each band is shown on the right panel. n = 3 animals/genotype.
(D) Intracellular cAMP levels in OPCs and OLs from WT or Gpr17−/− mice.
(E–J) ECAR after sequential administration of 10 mM glucose, 1.5 μM oligomycin, and 50 mM 2-DG of (E) WT OPCs treated with 0.5 mM dibutyl-cAMP (db-cAMP),(F) WT OPCs treated with 10 mM forskolin (FSK), (G) Gpr17−/− OPCs treated with 0.5 μM KT5720, (H) WT OLs treated with 0.5 mM db-cAMP, (I) WT OLs treated with 10 μM FSK, and (J) Gpr17−/− OLs treated with 0.5 μM KT5720. Dotted lines indicate the starting point of treatment with the indicated compounds.
(K) Total RNAs were isolated, and c-Fos was quantified detected by real-time PCR as described in the STAR Methods.
(L) Total proteins isolated from primary OPCs and OLs from WT or Gpr17−/− mice were subjected to western blot using the indicated antibodies. The relative band intensity of each protein is shown on the right. n = 3 animals/genotype.
(M) OLN-93 cells were transfected with an empty vector (Vec) or the c-Fos-expressing construct pBOB-c-Fos (c-Fos) for 48 h. Total mRNA was harvested, and Pdk1 mRNA was quantified by real-time PCR.
(N) HEK293T cells were co-transfected with empty Vec or pBOB-c-Fos and the Pdk1 promoter constructs containing a WT or mutated c-Fos response element. Luciferase activities were assayed 24 h after transfection.
Data were reproduced from at least three independent experiments. Each value represents mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001, Student’s t test.
GPR17 Regulates Expression of PDK1 through cAMP and the Protein Kinase A Signaling Pathway
We reasoned that the increase in lactate production in GPR17-deficient OLs could be due to decreased shuttling of pyruvate into the Krebs cycle. Consistent with this hypothesis, the level of the kinase PDK1 was much higher in GPR17-deficient OPCs and OLs than in controls, and phosphorylation of the E1a subunit of pyruvate dehydrogenase (PDH), a well-established substrate of PDK1, was increased in the GPR17-deficient OPCs and OLs (Figure 5A). Furthermore, the level of Pdk1 mRNA was also significantly higher in the GPR17-deficient OPCs and OLs than in controls (Figures 5B). In both NCD- and HFD-fed Gpr17cKO mice, the levels of PDK1 and phosphorylated PDH were significantly higher than those of WT mice (Figure 5C).
Previous studies have demonstrated that expression of GPR17 results in activation of Gai, reducing the production of intracellular cAMP and, thus, inhibiting downstream signaling pathways involving cAMP, such as protein kinase A (PKA) signaling (Hennen et al., 2013). Similar to what we reported pre viously for rat OPCs deficient in GPR17 (Ou et al., 2016), the cAMP levels were increased in both OPCs and OLs isolated from Gpr17−/− mice compared with those in WT mice (Figure 5D). As a result, the level of phosphorylated PKA was also significantly elevated (Figure 5A).
To further confirm the requirement for PKA signaling in the glycolytic process in OL lineage cells, OPCs and OLs were treated with the PKA signaling activators dibutyl-cAMP and forskolin or the PKA inhibitor KT5720. Treatment of WT OPCs with0.5 mM dibutyl-cAMP or 10 μM forskolin increased the ECAR, indicating increased production of lactate (Figures 5E, 5F, S7A, and S7B). Conversely, when PKA signaling was blocked by treatment of cells with 0.5 μM KT5720, the extracellular acidification level in GPR17-deficient OPCs declined (Figures 5G and S7C). Similar results were obtained at the OL stage with the same treatments (Figures 5H–5J and S7D–S7F).
Consistent with the activation of PKA signaling, c-Fos expression was upregulated at both the mRNA and protein levels in Gpr17−/− cells (Figures 5K and 5L). When c-Fos was overex-pressed in OLN-93 cells, the expression of Pdk1 increased about1.7-fold (Figure 5M). A c-Fos response element was identified in the promoter region of the Pdk1 gene, suggesting that c-Fos directly regulates its expression. To test this, we generated a reporter construct carrying the 1-kb proximal promoter of the Pdk1 gene. When a c-Fos-expressing vector was co-transfected with this reporter, a 3.5-fold induction of the luciferase activity was observed. When the putative c-Fos response element was mutated, c-Fos no longer induced the expression of luciferase (Figure 5N).
Increased Lactate Production in OLs Augments Neuronal AKT and STAT3 Signaling
We then sought to explore whether lactate influences AKT or STAT3 signaling pathways because activation of these pathways in AgRP/NPY and POMC neurons suppresses appetite and accelerates energy expenditure by reciprocally reducing the synthesis of AgRP/NPY peptides and increasing the synthesis of POMC peptide (Djogo et al., 2016; Kim et al., 2017; Yang et al., 2017). We treated primary cultures of hypothalamic neurons from rats with lactate and found that the phosphorylated AKT and STAT3 levels were increased in a time-dependent manner (Figure 6A). Because lactate is transferred from glial cells into the neuronal cytoplasm through monocarboxylate transporters (MCTs) (Lee et al., 2012; Pellerin et al., 1998), we treated cultures with α-cyano-4-hydroxycinnamic acid (4-CIN), a broad-spectrum MCT inhibitor. When the cells were treated with 4-CIN, lactate-mediated activation of AKT and STAT3 was largely blocked (Figure 6B). Notably, when primary hypothalamic neurons were treated with lactate, there was increased expression level of Pomc and decreased expression level of Agrp (Figure 6C), and these alterations in gene expression were not observed when cells were treated with both lactate and 4-CIN.
Figure 6.

Hypothalamic Administration of Lactate Modulates Appetite through Activation of AKT and STAT3 Signaling (A) Primary hypothalamus neurons were treated with 20 mM lactate. Left: levels of phosphorylated AKT (p-AKT) and phosphorylated STAT3 (p-STAT3) were analyzed by western blotting. Right: Relative intensities of p-AKT or p-STAT3 normalized to total AKT or STAT3. n = 3 independent experiments.
(B) Neurons were pretreated with 0.25 mM 4-CIN for 15 min and then treated with 20 mM lactate for 15 min. Left: the levels of p-AKT and p-STAT3 were analyzed by western blotting. Right: relative intensities of p-AKT or pSTAT3 normalized to total AKT or STAT3. n = 3 independent experiments.
(C) The relative mRNA levels of Pomc and Agrp were measured by real-time qPCR in neurons treated with 0 (−) or 20 mM (+) lactate for 1 h with or without a 15-min pretreatment with 0.25 mM 4-CIN.(D and E) WT 8-week-old mice received i.c.v. microinjections of 50 nmol lactate (D) and were sacrificed after the indicated time or (E) received the indicated dose of lactate and were sacrificed after 120 min. Left: total hypothalamic proteins were extracted and subjected to western blot using the indicated antibodies. Right: relative intensities of p-AKT or p-STAT3 normalized to total AKT or STAT3. n = 3 independent experiments.(F) WT 8-week-old mice received i.c.v. microinjections of saline or 25 or 50 nmol of lactate. Hypothalamus samples were collected 6 h after the injections. The relative mRNA levels of Pomc and Agrp were measured by real-time qPCR. (G and H) Mice were injected i.c.v. with 50 nmol of lactate with or without a 1-h pretreatment with 40 nmol 4-CIN.
(G) Left: mice were sacrificed 2 h after injection of lactate, and hypothalamus samples were subjected to western blot using the indicated antibodies. Right: relative intensities of p-AKT or p-STAT3 normalized to total AKT or STAT3. n = 3 independent experiments.
(H) Mice (n = 6/group) were subjected to 24-h food intake measurement.
Each value represents mean ± SEM from three independent experiments. *p < 0.05, ***p < 0.001, Student’s t test.
To confirm these results in vivo, we administered lactate into the third ventricle of WT mice through intracerebroventricular (i.c.v.) injection. Both phosphorylated AKT and STAT3 levels increased in a time- and dose-dependent manner after lactate injection (Figures 6D and 6E). Lactate also induced the expression of Pomc and suppressed Agrp (Figure 6F). Moreover, when the mice were pretreated with 4-CIN, lactate-induced changes in the levels of phosphorylated AKT and STAT3 were not observed (Figure 6G). Physiologically, mice given lactate had reduced food intake relative to untreated mice. Although administration of 4-CIN only elicited a tendency toward increased food intake, which might be due to the complicated network of appetite control, pre-treatment with 4-CIN largely restored the lactate-suppressed appetite (Figure 6H).
These results clearly demonstrated that excessive lactate activates neuronal AKT and STAT3 signaling pathways. Considering that Gpr17−/− OLs produced more lactate than WT OLs (Figure 4), we hypothesized that the elevated lactate in the environment of the hypothalamic neurons in Gpr17cKO mice might have the same effects on these signaling pathways. To test this hypothesis, we quantified lactate in the hypothalami of control Gpr17fl/fl and Gpr17cKO mice. Consistent with the in vitro findings, loss of Gpr17 increased the lactate concentration in the hypothalamus (Figure 7A).
Figure 7.

Depletion of Gpr17 or Activation of OLs Regulates AgRP and POMC Neuron Activity (A) Lactate was quantified in hypothalamus samples from Gpr17fl/fl (control [Ctrl]) and Gpr17cKO mice subjected to either NCD or HFD feeding for 30 weeks (n = 6 animals/genotype).
(B) Left: hypothalamus samples from mice fed the NCD were analyzed by western blot using the indicated antibodies. Right: relative intensities of p-AKT or p-STAT3 normalized to total AKT or STAT3. n = 3 animals/genotype.
(C) Left: hypothalamus samples from mice fed the HCD were analyzed by western blot using the indicated antibodies. Right: relative intensities of p-AKT or p-STAT3 normalized to total AKT or STAT3. n = 3 animals/genotype.
(D) Left: immunofluorescent staining for p-AKT or p-STAT3 of cryostat sections from representative hypothalamus samples of Gpr17fl/fl and Gpr17cKO mice after 30 weeks of HFD feeding. Scale bar, 50 μm. Right: quantification of samples from 3 mice.
(E) NCD-fed Gpr17cKO mice were injected i.c.v. with 4-CIN or oxamate (Oxa). Left: after 12 h, total protein samples were collected from the hypothalamus and subjected to western blot using the indicated antibodies. Right: the relative band intensity of each protein. n = 3 animals/group.
(F) After 30 weeks of HFD feeding, the levels of Pomc and Agrp in the hypothalamus of Gpr17fl/fl and Gpr17cKO mice were measured by real-time qPCR (n = 9 animals/genotype).
(G) Immunofluorescent staining for POMC and c-Fos in the hypothalamus of Gpr17fl/fl and Gpr17cKO mice after 30 weeks of HFD. Scale bar, 25 μm.(H and J) Lactate content (H) and mRNA (J) levels of Pomc and Agrp in the hypothalamus of Gi-DREADD or Olig1cre/+;Gi-DREADD mice treated with CNO (1 mg/kg) at P12 for 2 h (n = 5 animals/genotype).
(I) Relative lactate content of isolated OPCs from Gi-DREADD or Olig1cre/+;Gi-DREADD mice with CNO treatment (5 μM) for 2 h (n = 3 independent experiments). Each value represents mean ± SEM from three independent experiments.
All quantifications of immunofluorescent staining were obtained from at least nine images from three mice. Each value represents mean ± SEM. *p < 0.05, **p < 0.01, Student’s t test.
We then compared the hypothalamus samples from control and Gpr17cKO mice after 30 weeks on either the NCD or HFD and found that the levels of phosphorylated AKT and phosphorylated STAT3 were higher in the Gpr17cKO mice (Figures 7B and 7C). Immunofluorescence showed that the population of cells that were positive for both phosphorylated AKT and STAT3 were increased in the hypothalamus of HFD-fed Gpr17cKO relative to Gpr17fl/fl mice (Figure 7D). When the Gpr17cKO mice were treated with 4-CIN or oxamate, a lactate dehydrogenase (LDH) inhibitor, the levels of phosphorylated AKT and phosphorylated STAT3 were decreased concordantly in control Gpr17fl/fl mice (Figure 7E). Moreover, the levels of Agrp were decreased and the levels of Pomc were increased in HFD-fed Gpr17cKO mice relative to the HFD-fed controls (Figure 7F). To further demonstrate the consequences of AKT and STAT3 activation in hypothalamic neurons, we the evaluated levels and localization of c-Fos, which serves as a surrogate indicator for neuron activities (Day et al., 2008; Dragunow and Faull, 1989; VanElzakker et al., 2008). As shown in Figure 7G, compared with control mice, Gpr17cKO mice fed the HFD showed a dramatic upregulation of c-Fos expression in POMC neurons.
To further interrogate the effect of Gi-coupled GPR17 signaling activation in OLs on lactate production and hypothalamic neuronal responses, we utilized a chemogenetic DREADD (designer receptors exclusively activated by designer drugs) system (Zhu et al., 2014) by breeding Olig1cre/+ mice with the R26-LSL-Gi-DREADD transgenic line to generate Olig1cre/+; Gi-DREADD mice. In these mice, expression of hM4Di, a mutant GPCR that activates the canonical Gi pathway upon treatment with clozapine-N-oxide (CNO) in OLs. CNO might exhibit some off-target effects (MacLaren et al., 2016; Manvich et al., 2018). To minimize any potential non-specific effect of CNO, both Gi-DREADD and Olig1cre/+;Gi-DREADD mice were treated with CNO at post-natal day 12 (P12) with a dose of 1 mg/kg of body weight for 2 h, and then hypothalamus samples were harvested for analysis. The lactate content in the hypothalamus of Olig1cre/+;Gi-DREADD mice decreased by approximately 25% compared with that of vehicle-treated control mice (Figure 7H). In vitro, OPCs isolated from Olig1cre/+; Gi-DREADD mice showed a lower lactate content compared with those from Gi-DREADD mice (Figure 7I). In addition, CNO treatment resulted in a significant decrease in Pomc expression and a moderate increase in Agrp expression (Figure 7J). These data indicate that activation of GPR17 signaling in OLs leads to reduced lactate production in the hypothalamus, leading to the alteration in Agrp and Pomc expression in neurons that eventually affects appetite.
DISCUSSION
GPR17-Mediated OL Activity Controls Metabolic Homeostasis
OLs have been implicated in the regulation of energy homeostasis, although the mechanisms were incompletely understood. A recent report indicated that depletion of NG2-positive glia resulted in increased food consumption and body weight in mice by impairing leptin sensing in the hypothalamus (Djogo et al., 2016), suggesting a potential role of OL precursors in the regulation of metabolic homeostasis. Here we showed that Gpr17−/− and Gpr17cKO mice are both resistant to HFD-induced obesity. Considering that GPR17 is primarily expressed in OLs and restricted to them among cells undergoing Olig1-promoter-driven ablation, we conclude that the GPR17 expressed by OLs regulates the activity of downstream effectors intercellularly to ultimately control energy metabolism through neurons.
Mastaitis et al. (2015) found that weight gain by Gpr17−/− mice did not differ from that of WT mice fed on the NCD for 13 weeks and then the HFD for an additional 10 weeks. Consistent with this, we found that the weights of Gpr17−/− mice did not differ from those of control littermates fed the NCD for up to 30 weeks or the HFD for 12 weeks. We continued to monitor body weight for 28 weeks on the HFD. From week 12, the body weight of Gpr17−/− mice gradually fell behind that of their WT littermates, and the differences were statistically significant from week 16.
By generating OL-specific Gpr17 knockout alleles, we found that Gpr17cKO mice exhibited lower body weights than WT littermate controls starting from week 16 on the HFD. In addition, treating WT mice with pranlukast, an antagonist of GPR17, slowed the gain in body weight of mice fed the HFD. Therefore, our data demonstrate that GPR17-mediated signaling modulates OL activity to control energy metabolism.
A previous study indicated that the expression of Lims2 in a Gpr17−/− mouse line was downregulated by approximately 30%, according to RNA sequencing fragments per kilobase of transcript per million fragments mapped (FPKM) data (Mastaitis et al., 2015); however, this change was not confirmed by qRT-PCR analyses. In contrast, in our Gpr17−/− mice, which were generated by removing the Gpr17 coding exon without affecting Lims2 exons, when measuring the Lims2 mRNA level in brain tissues as well as isolated OPCs or OLs by qRT-PCR, we did not observe any significant difference between Gpr17−/− mice and their control littermates. The exact mechanism behind the potential discrepancy between these Gpr17-null mice is not known; it is possible that the difference in the genetic knockout strategies or different genetic backgrounds may contribute to the changes in neighboring Lims2 gene expression (Figure S6A). Nonetheless, the lack of significant alteration of Lims2 expression suggests that Lims2 not likely to be the major factor contributing to the energy metabolism phenotype observed in our Gpr17 knockout mice.
GPR17 Modulates Glycolysis and Lactate Productionin OLs
We showed that loss of Gpr17 led to increased lactate production through the cAMP-PKA pathway. Consistent with our previous findings in rat OLs (Ou et al., 2016), loss of Gpr17 in mouse OLs caused a significant increase in cellular cAMP levels and PKA activity, which, through c-Fos, upregulated Pdk1 and, hence, increased phosphorylation of the E1α subunit of PDH. Both Seahorse analysis and direct measurements indicated that lactate production was significantly increased in Gpr17−/− OLs both at the OPC and OL stages. Furthermore, the cAMP-elevating agent forskolin and the cAMP mimetic dibutyl (db)-cAMP both dramatically increased lactate production in OLs, suggesting that GPR17 regulates lactate production through cAMP-PKA signaling in OLs.
To show the hypothalamic specificity of the GPR17-cAMP-lactate axis, we found that i.c.v. injection of lactate activated AKT and STAT3 signaling, upregulated Pomc, and downregulated Agrp. Importantly, administration of 4-CIN prior to lactate injection almost completely blocked activation of these pathways and resulted in a normal appetite. Moreover, administration of 4-CIN and oxamate to the hypothalamus of NCD-fed Gpr17cKO mice significantly decreased AKT and STAT3 activation. Consistent with our findings, Djogo et al. (2016) reported that removal of NG2-positive OLs from the hypothalamus resulted in increased food intake and body weight gain (Djogo et al., 2016). These results suggest that OLs in the hypothalamus play a role in the regulation of whole-body metabolism.
GPR17-Mediated Signaling Modulates Neuronal Activity through a Lactate Shuttle between OLs and Neurons
Lactate has been reported previously to provide nutritional support to neurons (Schurr et al., 1988) and has been shown to serve as a signal transmitter in the brain (Bozzo et al., 2013; Lauritzen et al., 2014). Recent studies demonstrated that OLs provide pyruvate and lactate, necessary for normal function of ensheathed axons (Lee et al., 2012; Saab et al., 2016). Activation of the AKT and STAT3 pathways in the hypothalamus leads to increased Pomc but decreased Agrp, which then results in reduced appetite (Djogo et al., 2016; Kim et al., 2017; Yang et al., 2017). In our studies, lactate treatment of primary cultured neurons caused increased phosphorylation of both AKT and STAT3, upregulated Pomc, and downregulated Agrp. Importantly, these effects of lactate were largely abolished by pretreatment with 4-CIN, a monocarboxylate transport inhibitor. Interestingly, injection of lactate into the third ventricle also caused increased levels of phosphorylated AKT (p-AKT) and p-STAT3. Furthermore, depletion of GPR17 in the OLs caused a dramatic increase in p-AKT and p-STAT3 in the hypothalamus and activation of anorexigenic POMC neurons with concomitant and reciprocal reduced activation of orexigenic AgRP neurons, which could explain the resistance of Gpr17−/− and Gpr17cKO mice to HFD-induced obesity. Therefore, we hypothesize that the lactate produced by OLs functions as a metabolic signal to modulate hypothalamic neuronal activity. Consistent with this hypothesis, lactate has been reported to increase the action potential frequency of the glucose-excited neurons in the ventromedial nucleus (VMH) area (Yang et al., 1999). Although we showed that lactate activates AKT and STAT3 signaling pathways through MCTs to regulate the expression of Pomc and Agrp, the molecular mechanism by which lactate acts remains to be determined.
The Regulatory Effect of GPR17 on Energy Homeostasis Acts through OLs
Our studies clarified the effect of GPR17 on the regulation of energy metabolism upstream of lactate secretion by way of OLs. Using agonists and antagonists of GPR17, Ren et al. (2015) showed that GPR17 is positively related to food intake and showed that, when Gpr17 was knocked out in AgRP neurons, mice were resistant to weight gain. Our previous studies (Chen et al., 2009; Ou et al., 2016) and the current data (Figures 2 and S3) indicate that Gpr17 is specifically expressed in the OL lineage (Olig2+, PDGFRα+, NG2+, CC1+, proteolipid protein [PLP]+, and myelin basic protein [MBP]+) but not in GFAP+ astrocytes, NeuN+ neurons, or Iba1+ microglia. When fed with an HFD, Gpr17−/− mice were less obese compared with control WT mice, showing improved capacity for glucose clearance and increased insulin sensitivity. In addition, Gpr17−/− mice exhibited reduced food intake. Moreover, the reduced obesity in Gpr17−/− mice can be recapitulated in Gpr17cKO mice in terms of HFD effects. These observations suggest a specific role of oligodendrocytic GPR17 in mediating the protection of diet-induced obesity (DIO).
It is worth noting that the Agrp-cre line used by Ren et al. (2015) often mimics germline deletion (Kaelin et al., 2004; Miura et al., 2003). It is therefore possible that the Gpr17 deletion driven by Agrp-cre could potentially take place in OL lineage cells, leading to the phenotypes observed in our OL-specific Gpr17 knockout mice.
Collectively, based on the data described here and previous research, we suggest that loss of GPR17 function increases cellular cAMP levels and activates PKA signaling, which upregulates the expression of c-Fos and Pdk1, leading to elevated lactate production. Lactate released from GPR17-deficient OLs enters nearby neuropeptidergic hypothalamic neurons to regulate metabolism and food intake. Thus, the cascade initiated by loss of GPR17 in OLs regulates whole-body metabolic homeostasis. Our finding that GPR17 signaling in OLs regulates food intake and appetite by modulating neuronal activities in the hypothalamus suggests that modulation of GPR17 signaling might be a potential therapeutic avenue for treating obesity.
STAR⋆METHODS
CONTACT FOR REAGENT OR RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Ying Chen (yingchen@xmu.edu.cn).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Animals
Gpr17−/− mice were generated as described previously (Chen et al., 2009). The Gpr17 fl/fl and Olig1-Cre mice were generated as previously described (Ou et al., 2016; Xin et al., 2005). The Gt(ROSA)26Sortm9(CAG-tdTomato)Hze mice were kindly provided by Dr. Liang Zhang (Xiamen University). The Gt(ROSA)26Sortm1(CAG-CHRM4*,-mCitrine)Ute/J mice were purchased from the Jackson laboratory (Cat# 026219) and were genotyped according to the protocol from the Jackson laboratory. C57BL/6 mice and Sprague-Dawley rats were purchased from and maintained in the Xiamen University Laboratory Animal Center. All mice were maintained in the Xiamen University Laboratory Animal Center. Male mice were used in the study, and littermates were used as controls. Gpr17−/− mice and Gpr17cKO mice were subjected to HFD feeding at 4 and 8 weeks of age, respectively. Mice at P7-P8 were used to collect OPCs for the primary culture. Rats at P0 were used to collect hypothalamus neurons for the primary culture. All of the animal experiments were approved by and performed according to the experimental guidelines of the Animal Care and Use Committee of Xiamen University.
METHOD DETAILS
Metabolic cage analysis
Both HFD-treated Gpr17−/− and Gpr17cKO mice were subjected to metabolic cage analysis after 26 weeks of feeding, and NCD-fed Gpr17cKO mice were subjected to analysis at 31 weeks of age. Metabolic parameters were measured using a TSE Labmaster Platform (TSE System). Cages were maintained at 20–22°C under a 12:12 h light-dark cycle (light period 07:00–19:00). Mice were singly housed for 5 days and were allowed to acclimatize to the TSE metabolic cages 3 days before metabolic experiments.
Glucose tolerance test (GTT) and insulin tolerance test (ITT)
Both HFD-treated Gpr17−/− and Gpr17cKO mice were subjected to GTT after 24 weeks of HFD-feeding and to ITT after 25 weeks of HFD-feeding. For GTT, food was removed 16 h prior to assay from 17:00 to 09:00. Mice received intraperitoneal injections (i.p.) of D-glucose (2 g/kg, Sigma). For ITT, mice were fasted for 4 h from 09:00 to 13:00 followed by i.p. injections of insulin (0.5 units/kg). Blood glucose concentrations from tail vein were measured at the indicated time points using a glucometer (Sinocare, Inc.).
Serum components and liver triglyceride analysis
Mice serum samples were collected from eyeballs and clotted for 30 min at room temperature before centrifuging at 3000 g for 10 min. Serum levels of insulin and leptin were determined using enzyme immunoassay kits from Millipore and R&D Systems, respectively, according to the manufacturers’ instructions. Serum-free fatty acid levels were determined by the ACS-ACOD method (Wako, Cat# 294–63601). Liver triglyceride was extracted by trichloromethane, and measured using a Triglyceride Assay Kit by the GPODAOS method as suggested by the manufacturer (Wako, Cat# 290–63701).
H&E Staining
After deparaffinization and rehydration, 6-mm sections were stained with hematoxylin solution (Sigma Cat# HHS16) for 8 min and rinsed in distilled water for 3 min. Then the sections were stained with eosin solution (Sigma Cat# HT110316) for 2 min and followed by dehydration with graded alcohol and clearing in xylene. The mounted slides were then examined and photographed using an Olympus BX53 microscope. The lipid droplet-positive areas of WAT, BAT, and liver were calculated by ImageJ software.
Electron microscopy
The corpus callosum regions from Gpr17−/− mice and their control littermates, with either 30 weeks of NCD feeding or 60 weeks of HFD feeding were dissected and fixed in a solution of 4% paraformaldehyde, pH 7.4, 2.5% glutaraldehyde, and 0.08 M sodium cacodylate, pH 7.4. The ultrathin sections were investigated under a Transmission Electron Microscope (TEM) (Hitachi HT-7800). A total number of 20 fields per section were randomly chosen and photographed at a magnification of X2000. The percentages of the myelinated axons and the G ratio (the ratio between the axonal diameter and the fibers diameter) were quantified.
Mouse OL primary culture
Mouse cortices were isolated from postnatal day 7 to 8 mouse as described previously (Chan et al., 2004) with slight modifications. Briefly, Cortical tissues were diced thoroughly (~1 mm3 pieces) and transferred into 20 units/ml papain buffer (Worthington, Cat# 3126) to digest for 45 min. After digestion, tissues were dispersed into single cells in an ovomucoid trypsin inhibitor buffer (Worthington, Cat# A003085). The cell suspension was then subjected to immunopanning with antibodies against GalC and O4 sequentially. The enriched Galc-negative O4-positive OPCs were plated into PDL-coated culture dishes and culture with OPC growth medium. OPC growth medium was DMEM/F-12 (GIBCO, Cat# 11330–032) containing 1% N2 supplement (GIBCO Cat# A1370701), 2% B27 supplement (GIBCO Cat# A3582801), penicillin-streptomycin solution (MP Biomedicals Cat# 0916700), 1% sodium pyruvate (GIBCO Cat# 11360070), 1% L-glutamine (Hyclone Cat# SH30034), 10 ng/ml platelet-derived growth factor-aa (Peprotech Cat# 100–13A), 10 ng/ml ciliary neurotrophic factor (Peprotech Cat# 450–13), 20 ng/ml human basic fibroblast growth factor (Sino Biological Cat# 10014HNAE), 0.5 mg/ml insulin (Sigma Cat# 91077), 5 mg/ml N-acetyl cysteine (Sigma Cat# A8199), 10 ng/ml D-biotin (Sigma Cat# B4639), 5 mM forskolin (Sigma Cat# F3917), 0.1% Trace Elements B (Corning Cat# 25–022-CI). OPC differentiation medium contained the same components above except that 40 ng/ml triiodo-thyronine (Sigma Cat# T2877) without human basic fibro-blast growth factor and platelet-derived growth factor-aa as previously described (Zhao et al., 2018).
Hypothalamic neuron primary culture
A mixed primary culture of hypothalamic neurons and glia were prepared from Sprague-Dawley rats pups at P0 and maintained in culture at 37°C and 5% CO2 as previously described (Loktev and Jackson, 2013) with slight modifications. Briefly, the hypothalamic region of the brain was dissected in ice cold HBSS (HyClone Cat# SH30268.01) containing penicillin-streptomycin solution (MP Biomedicals Cat# 0916700) and sodium pyruvate (GIBCO Cat# 11360070), and was subjected to enzymatic desegregation for 20 min at 37°C in papain buffer containing 20 units/ml papain (Worthington Cat# 3126) and 250 units/ml DNaseI (Worthington Cat# 2006). The tissue was then mechanically disaggregated into isolated cells in a Neurobasal medium (GIBCO Cat# A1371201) with 5% (v/v) horse serum (GIBCO Cat# 26050088), Glutamax-I (GIBCO Cat# 35050–061), B27 supplement (GIBCO Cat# A3582801), and 250 units/ml DNaseI. The cells (1.0 × 106 cells/cm2) were plated in wells of 6-well plates that were coated with 0.1 mg/mL poly-D-lysine (Sigma-Aldrich Cat# P7280). After 2 h, medium was replaced with Neurobasal medium supplemented with B27, Glutamax-I, and penicillin-streptomycin solution. The medium was replaced every 3 days, and the cells were maintained in culture for 5–7 days.
Immunohistology
Mice were anesthetized by chloral hydrate and perfused with 4% paraformaldehyde (PFA). Tissues were post-fixed overnight in 4% PFA, transferred to 30% sucrose, and 30-μm frozen sections were prepared. The sections were incubated in blocking solution (0.4% Triton X-100, 3% BSA in PBS) for 1 h at room temperature. In the case of POMC staining, the sections were pretreated with0.5% Triton X-100, 0.5% H2O2. Primary antibodies were then applied overnight at 4°C. The next day, sections were washed three times (5 min each time) in PBS and incubated with the secondary antibodies for 2 h at room temperature.
Real-time quantitative PCR
Total cellular RNAs were extracted using TRIzol (Takara Cat# 9109) according to the manufacturer’s instructions, and RNA purity was confirmed by analysis of the ratio of absorbance at 260 nm to that at 280 nm absorption. cDNAs were prepared from 1 g total RNAs using Hifair™ II 1st Strand cDNA Synthesis SuperMix for qPCR (Yeasen Cat# 11123ES60) according to the manufacturer’s instructions. Quantitative PCRs were performed using Hieff™ qPCR SYBR Green Master Mix (Yeasen Cat# 11201ES08). The primers used were as follows (5′-3′): Mouse primers: GAPDH (forward: AACAGCAACTCCCACTCTTC; reverse: CCTGTTGCTGTAGCCGTATT), AgRP (forward: TGTGTAAGGCTGCACGAGTC; reverse: ATTGAAGAAGCGGCAGTAGC), POMC (forward: GCTTGCAAACTCGA CCTCTC; reverse: GTACTTCCGGGGGTTTTCAG), PDK1 (forward: GGACTTCGGGTCAGTGAATGC; reverse: TCCTGAGAAGATT GTCGGGGA), c-Fos (forward: CTCCTGAAGAGGAAGAGAAACG; reverse: GTGTATCTGTCAGCTCCCTCCT), Lims2 I (forward: GTCAACAGCAATGGGGAACT; reverse: AGAAGAGTCCCTCGGGAAAT), Lims2 II (forward: GCCAAGTGTGAGAAGCCATT; reverse: CAGACATCCCCAAAGAGCTG), Lims2 III (forward: GTTCGATATGAAGCCCGTGT; reverse: CGGACAGCTTCTTCAACCTC), Gpr17 (forward: GGATAGAGAAGCACCTCAAGAA; reverse: CACGGTAGTGAAGCACATAGA), Rat primers: GAPDH (forward: ACTCC CATTCTTCCACCTTTG; reverse: CCCTGTTGCTGTAGCCATATT), AgRP (forward: CTGTGTAAGGCTGCACGAGTC; reverse: AGG TATTGAAGAAGCGGCAGT), POMC (forward: TGCCGAGATTCTGCTACAGTC; reverse: CACCAGCTCCACACGTCTATG), PDK1 (forward: GAACTGTTCAAGAACGCAATGA, reverse: CCAGTGTGACGTGAACTTGAAT).
Western blotting analysis
The hypothalamus was isolated from brain and then lysed in RIPA buffer (50 mM Tris-HCl, pH 7.5, 150 mM NaCl, 1 mM EDTA, 1% Triton X-100, 1% deoxycholate, 0.1% SDS). The primary neurons were lysed in cell lysis buffer (20 mM Tris-HCl, pH 7.5, 150 mM NaCl, 1 mM EDTA, 1 mM EGTA, 1% Triton X-100, 2.5 mM sodium pyrophosphate, 1 mM β-glycerophosphate, 1 mM NaF, 1 mM PMSF), supplemented with protease inhibitor cocktail (MCE Cat# HY-K0010), phosphatase inhibitor cocktail I (MCE Cat# HY-K0021), and phosphatase inhibitor cocktail II (MCE Cat# HY-K0022) on ice for 30 min followed by centrifugation at 12,000 rpm for 30 min. Protein concentrations were measured using the BCA Protein Assay Kit (Sangon Biotech Cat# C503021). Proteins were resolved on 12% SDS-PAGE and transferred to PVDF membrane (GE Healthcare Cat# 10600023). Membranes were blocked with 3% BSA in TBS (50 mM Tris-HCl, pH 7.5, 150 mM NaCl) for 1 h and then incubated overnight at 4°C with primary antibodies. After 3 washes with TBS plus 0.05% Tween 20, membranes were incubated with secondary antibodies for 1 h and developed using an ECL kit (Millipore Cat# WBKLS0500).
Intracellular cAMP concentration assessment
To assess intracellular cAMP concentration, primary mouse OPCs were cultured in poly-D-lysine-coated wells of 96-well plates at a density of 2 3 104/well. cAMP was quantified using the cAMP-Glo Assay Kit (Promega Cat# V1501) following the manufacturer’s instructions.
Transfection and reporter gene assays
293T cells in 24-well plates were transfected with either the pBOB-GFP or pBOB-c-Fos and co-transfected with luciferase reporter plasmid PGL3-PDK1 or the mutant plasmid with Lipofectamine 3000 (Thermo Fisher Scientific Cat# L3000–015). The Renilla lucif-erase construct pRL-TK (Promega Cat# E2241) was co-transfected as a normalization control. After 24 h, the cells were harvested and lysed, and 20 mL of each lysate was incubated with luciferase substrates for firefly and Renilla (Promega Cat# E151A). The PDK1-dependent firefly luciferase activity was normalized to Renilla-dependent luciferase activity.
Lactate colorimetric assay
To determine the lactate release, OPCs were seeded into 24-well plates at a density of 5 × 104 cells/well and cultured until the desired stage in appropriate medium. The culture medium was first changed to 250 μL of glucose-free DMEM medium (GIBCO Cat# 11966025) to starve the cells for 1 h, then 25 mM D-glucose (Sigma Cat# G8270) was added into the medium to initiate glycolysis. For the hM4Di OPCs, 5 μM CNO was added at the beginning of glucose starvation. The medium was collected 1 h after glucose treatment and treated with two volumes of acetone immediately to precipitate protein. Samples were then lyophilized to remove acetone and re-dissolved in sterilized deionized water. For tissue lactate content detection, mouse brains were dissected, and the hypothalamus was snapfrozen in liquid nitrogen within 20 s. The isolated hypothalamus samples were weighed and homogenized in 50% methanol to precipitate protein. After centrifuging at 4°C, the homogenate was lyophilized and re-dissolved by sterilized deionized water. Lactate levels were measured using the L-lactate assay kit according to the manufacturer’s protocol (Biovision Cat# K627–100).
Cellular metabolism measurement
Cellular glycolysis and oxidative phosphorylation were monitored with a Seahorse XFe96 extracellular flux analyzer (Seahorse Bioscience XFe96) by measuring the ECAR and OCR, respectively, in real time as described by the manufacturer. Briefly, 1.5 × 104 to 2 × 104 OPCs were seeded in specific 96-well plates designed for XF96 with appropriate growth medium. Prior to measurements, cells were washed three times with the XF base medium (Agilent, Cat# 102353) and incubated in the absence of CO2 at 37°C for 1 h in the XF base medium. The OCR and ECAR values were measured in a 9-min cycle of mix (3 min), dwell (3 min), and measure (3 min). The basal levels of OCR and ECAR were recorded first, and then the OCR and ECAR levels were recorded after sequential injection of compounds that inhibit the respiratory mitochondrial electron transport chain, ATP synthesis, or glycolysis as described previously (Xing et al., 2017).
Stereotaxic microinjection
Mice were starved for 16 h, followed by a 4 h refeeding before experiments, and then anesthetized by chloral hydrate (300 mg/kg, i.p.) and placed in a stereotactic device. A stainless-steel microliter syringe was inserted into the third ventricle (−1.7 mm posterior and 0 mm lateral to bregma, and −5.0 mm ventral from the dura), then 5 μL of vehicle (sterile saline), 5 or 10 mM lactate, 4 mM 4-CIN, or 20 mM oxamate was injected at a rate of 0.5 μL/min. When 4-CIN pretreatment was performed, 4-CIN or saline was administrated 1 h before the subsequent vehicle or lactate injection. The hypothalamus samples were collected and flash frozen by liquid nitrogen at indicated time points after injection. For food intake measurement after stereotaxic microinjection, mice were separately housed and fasted with water supply for 4h (9:00 to 13:00) before surgery, and then refed after injection. The remaining food pellets were weighed at 24 hr after surgery as described previously (Nonogaki et al., 2016).
Clozapine-N-oxide (CNO) treatment
The Gi-DREADD or Olig1cre/+;Gi-DREADD mice at P12 were intraperitoneally injected with CNO (1 mg/kg) for 2 h, and then their hypothalamus samples were collected for total mRNA analysis or lactate colorimetric assay.
QUANTIFICATION AND STATISTICAL ANALYSIS
The statistical analyses are performed with Graphpad Prism 5. The data for two-group comparisons were analyzed for statistical significance using two-tailed Student’s t tests. Error bars represent standard error of measurement (s.e.m.). For multiple comparisons, which were performed using one-way analyses of variance (ANOVAs), the Tukey’s multiple-comparison test was used for post-tests. P values are indicated with single asterisk (* < 0.05), double asterisks (** < 0.01) and triple asterisks (*** < 0.001) on graphs. The n number for each experiment has been stated in figure legends.
DATA AND SOFTWARE AVAILABILITY
All data used in this manuscript are available upon request. The original data are available at https://data.mendeley.com/datasets/wdmx6trhnr/4
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Olig2 | Millipore | Cat# AB9610; RRID:AB_570666 |
| CC1 | Calbiochem | Cat# OP80; RRID:AB_2057371 |
| PDGFRα | Santa Cruz Biotechnology | Cat# SC-338; RRID:AB_631064 |
| GFAP | Cell Signaling Technology | Cat# 3670; RRID:AB_561049 |
| Iba-1 | Wako | Cat# 019–19744; RRID: AB_839504 |
| NeuN | Abcam | Cat# Ab177487; RRID:AB_2532109 |
| c-Fos | Abcam | Cat# Ab7693; RRID: AB_306177 |
| phosphorylated PKA | Santa Cruz Biotechnology | Cat# SC-32968; RRID:AB_2237593 |
| PKA | Santa Cruz Biotechnology | Cat# SC-98951; RRID:AB_2170328 |
| GAPDH | Proteintech | Cat# 60004–1-lg; RRID: AB_2107436 |
| POMC | Phoenix Pharmaceuticals | Cat# H-029–30; RRID:AB_2307442 |
| AGRP | R&D Systems | Cat# AF634; RRID:AB_2273824 |
| phosphorylated STAT3 | Cell Signaling Technology | Cat# 9131; RRID:AB_331586 |
| STAT3 | Cell Signaling Technology | Cat# 12640; RRID:AB_2629499 |
| phosphorylated AKT | Cell Signaling Technology | Cat# 4060; RRID:AB_2315049 |
| AKT | Cell Signaling Technology | Cat# 9272; RRID:AB_329827 |
| PDK1 | Cell Signaling Technology | Cat# 3820; RRID:AB_1904078 |
| phosphorylated PDH | Abcam | Cat# ab177461; RRID:AB_2756339 |
| PDH | Cell Signaling Technology | Cat# 3205; RRID:AB_2162926 |
| MBP | Biolegend | Cat# 808401; RRID:AB_2564741 |
| NG2 | Millipore | Cat# AB5320; AB_11213678 |
| Parvalbumin | Abcam | Cat# ab11427; RRID:AB_298032 |
| Critical Commercial Assays | ||
| cAMP-Glo Assay Kit | Promega | Cat# V1501 |
| L-lactate assay kit | Biovision | Cat# K627–100 |
| Deposited Data | ||
| Original pictures | This paper | DOI: https://data.mendeley.com/datasets/wdmx6trhnr/4 |
| Experimental Models: Organisms/Strains | ||
| Gt(ROSA)26Sortm1(CAG-CHRM4*,-mCitrine)Ute/J mice | Jackson laboratory | Cat# 026219 |
| Gt(ROSA)26Sortm9(CAG-tdTomtao)Hze mice | Kindly provided by Dr. Liang Zhang (Xiamen University) | N/A |
| Gpr17−/− mice | Chen et, al. 2009 | N/A |
| Olig1cre/+;Gpr17fl/fl mice | Ou et, al. 2016 | N/A |
| C57BL/6 mice | Xiamen University Laboratory Animal Center | N/A |
| Sprague-Dawley rats | Xiamen University Laboratory Animal Center | N/A |
Highlights.
GPR17-deficient mice are resistant to high-fat-diet-induced obesity
Oligodendrocytic Gpr17 signaling controls metabolic homeostasis and food intake
Oligodendrocyte lactate is a metabolic signal for neuronal AKT/STAT3 signaling
Oligodendrocytic GPR17-cAMP-lactate axis regulates neuronal activity
ACKNOWLEDGMENTS
This work was supported by grants from the National Natural Science Foundation of China (31471039, 31771122, and 81601080), the National Key R&D Program of China (2016YFA0502003), and Project 111 sponsored by the State Bureau of Foreign Experts and Ministry of Education of China (BP2018017).
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found with this article online at https://doi.org/10.1016/j.celrep.2019.02.060.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Balland E, Dam J, Langlet F, Caron E, Steculorum S, Messina A, Rasika S, Falluel-Morel A, Anouar Y, Dehouck B, et al. (2014). Hypothalamic tanycytes are an ERK-gated conduit for leptin into the brain. Cell Metab 19, 293–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouret SG, Draper SJ, and Simerly RB (2004). Formation of projection pathways from the arcuate nucleus of the hypothalamus to hypothalamic regions implicated in the neural control of feeding behavior in mice.J. Neurosci 24, 2797–2805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bozzo L, Puyal J, and Chatton JY (2013). Lactate modulates the activity of primary cortical neurons through a receptor-mediated pathway. PLoS ONE 8, e71721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan JR, Watkins TA, Cosgaya JM, Zhang C, Chen L, Reichardt LF, Shooter EM, and Barres BA (2004). NGF controls axonal receptivity to myelination by Schwann cells or oligodendrocytes. Neuron 43, 183–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Wu H, Wang S, Koito H, Li J, Ye F, Hoang J, Escobar SS, Gow A, Arnett HA, et al. (2009). The oligodendrocyte-specific G protein-coupled receptor GPR17 is a cell-intrinsic timer of myelination. Nat. Neurosci 12, 1398–1406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coppari R, Ichinose M, Lee CE, Pullen AE, Kenny CD, McGovern RA, Tang V, Liu SM, Ludwig T, Chua SC Jr., et al. (2005). The hypothalamic arcuate nucleus: a key site for mediating leptin’s effects on glucose homeostasis and locomotor activity. Cell Metab 1, 63–72. [DOI] [PubMed] [Google Scholar]
- Day HE, Kryskow EM, Nyhuis TJ, Herlihy L, and Campeau S (2008). Conditioned fear inhibits c-fos mRNA expression in the central extended amygdala. Brain Res 1229, 137–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Djogo T, Robins SC, Schneider S, Kryzskaya D, Liu X, Mingay A, Gillon CJ, Kim JH, Storch KF, Boehm U, et al. (2016). Adult NG2-Glia Are Required for Median Eminence-Mediated Leptin Sensing and Body Weight Control. Cell Metab 23, 797–810. [DOI] [PubMed] [Google Scholar]
- Dragunow M, and Faull R (1989). The use of c-fos as a metabolic marker in neuronal pathway tracing. J. Neurosci. Methods 29, 261–265. [DOI] [PubMed] [Google Scholar]
- Erickson JC, Hollopeter G, and Palmiter RD (1996). Attenuation of the obesity syndrome of ob/ob mice by the loss of neuropeptide Y. Science 274, 1704–1707. [DOI] [PubMed] [Google Scholar]
- Fan W, Boston BA, Kesterson RA, Hruby VJ, and Cone RD (1997). Role of melanocortinergic neurons in feeding and the agouti obesity syndrome. Nature 385, 165–168. [DOI] [PubMed] [Google Scholar]
- García-Cáceres C, Quarta C, Varela L, Gao Y, Gruber T, Legutko B, Jastroch M, Johansson P, Ninkovic J, Yi CX, et al. (2016). Astrocytic Insulin Signaling Couples Brain Glucose Uptake with Nutrient Availability. Cell 166, 867–880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hennen S, Wang H, Peters L, Merten N, Simon K, Spinrath A, Blätter-mann S, Akkari R, Schrage R, Schröder R, et al. (2013). Decoding signaling and function of the orphan G protein-coupled receptor GPR17 with a small-molecule agonist. Sci. Signal 6, ra93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofmann K, Lamberz C, Piotrowitz K, Offermann N, But D, Scheller A, Al-Amoudi A, and Kuerschner L (2017). Tanycytes and a differential fatty acid metabolism in the hypothalamus. Glia 65, 231–249. [DOI] [PubMed] [Google Scholar]
- Huszar D, Lynch CA, Fairchild-Huntress V, Dunmore JH, Fang Q, Berkemeier LR, Gu W, Kesterson RA, Boston BA, Cone RD, et al. (1997). Targeted disruption of the melanocortin-4 receptor results in obesity in mice. Cell 88, 131–141. [DOI] [PubMed] [Google Scholar]
- Kaelin CB, Xu AW, Lu XY, and Barsh GS (2004). Transcriptional regulation of agouti-related protein (Agrp) in transgenic mice. Endocrinology 145, 5798–5806. [DOI] [PubMed] [Google Scholar]
- Kim JG, Suyama S, Koch M, Jin S, Argente-Arizon P, Argente J, Liu ZW, Zimmer MR, Jeong JK, Szigeti-Buck K, et al. (2014). Leptin signaling in astrocytes regulates hypothalamic neuronal circuits and feeding. Nat. Neurosci 17, 908–910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JD, Toda C, Ramírez CM, Fernández-Hernando C, and Diano S (2017). Hypothalamic Ventromedial Lin28a Enhances Glucose Metabolism in Diet-Induced Obesity. Diabetes 66, 2102–2111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauritzen KH, Morland C, Puchades M, Holm-Hansen S, Hagelin EM, Lauritzen F, Attramadal H, Storm-Mathisen J, Gjedde A, and Bergersen LH (2014). Lactate receptor sites link neurotransmission, neurovascular coupling, and brain energy metabolism. Cereb. Cortex 24, 2784–2795. [DOI] [PubMed] [Google Scholar]
- Lee Y, Morrison BM, Li Y, Lengacher S, Farah MH, Hoffman PN, Liu Y, Tsingalia A, Jin L, Zhang PW, et al. (2012). Oligodendroglia metabolically support axons and contribute to neurodegeneration. Nature 487, 443–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loktev AV, and Jackson PK (2013). Neuropeptide Y family receptors traffic via the Bardet-Biedl syndrome pathway to signal in neuronal primary cilia. Cell Rep 5, 1316–1329. [DOI] [PubMed] [Google Scholar]
- Lu QR, Sun T, Zhu Z, Ma N, Garcia M, Stiles CD, and Rowitch DH (2002). Common developmental requirement for Olig function indicates a motor neuron/oligodendrocyte connection. Cell 109, 75–86. [DOI] [PubMed] [Google Scholar]
- MacLaren DA, Browne RW, Shaw JK, Krishnan Radhakrishnan S, Khare P, España RA, and Clark SD (2016). Clozapine N-Oxide Administration Produces Behavioral Effects in Long-Evans Rats: Implications for Designing DREADD Experiments. eNeuro 3, ENEURO.0219–16.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manvich DF, Webster KA, Foster SL, Farrell MS, Ritchie JC, Porter JH, and Weinshenker D (2018). The DREADD agonist clozapine N-oxide (CNO) is reverse-metabolized to clozapine and produces clozapine-like interoceptive stimulus effects in rats and mice. Sci. Rep 8, 3840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mastaitis J, Min S, Elvert R, Kannt A, Xin Y, Ochoa F, Gale NW, Valenzuela DM, Murphy AJ, Yancopoulos GD, and Gromada J (2015). GPR17 gene disruption does not alter food intake or glucose homeostasis in mice. Proc. Natl. Acad. Sci. USA 112, 1845–1849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miura GI, Froelick GJ, Marsh DJ, Stark KL, and Palmiter RD (2003). The d subunit of the vacuolar ATPase (Atp6d) is essential for embryonic development. Transgenic Res. 12, 131–133. [DOI] [PubMed] [Google Scholar]
- Nonogaki K, Kaji T, Yamazaki T, and Murakami M (2016). Pharmacologic stimulation of central GLP-1 receptors has opposite effects on the alterations of plasma FGF21 levels induced by feeding and fasting. Neurosci. Lett 612, 14–17. [DOI] [PubMed] [Google Scholar]
- Ou Z, Sun Y, Lin L, You N, Liu X, Li H, Ma Y, Cao L, Han Y, Liu M, et al. (2016). Olig2-Targeted G-Protein-Coupled Receptor Gpr17 Regulates Oligodendrocyte Survival in Response to Lysolecithin-Induced Demyelination.J. Neurosci 36, 10560–10573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pellerin L, Pellegri G, Bittar PG, Charnay Y, Bouras C, Martin JL, Stella N, and Magistretti PJ (1998). Evidence supporting the existence of an activity-dependent astrocyte-neuron lactate shuttle. Dev. Neurosci 20, 291–299. [DOI] [PubMed] [Google Scholar]
- Ren H, Orozco IJ, Su Y, Suyama S, Gutiérrez-Juárez R, Horvath TL, Wardlaw SL, Plum L, Arancio O, and Accili D (2012). FoxO1 target Gpr17 activates AgRP neurons to regulate food intake. Cell 149, 1314–1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren H, Cook JR, Kon N, and Accili D (2015). Gpr17 in AgRP Neurons Regulates Feeding and Sensitivity to Insulin and Leptin. Diabetes 64, 3670–3679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saab AS, Tzvetavona ID, Trevisiol A, Baltan S, Dibaj P, Kusch K, Möbius W, Goetze B, Jahn HM, Huang W, et al. (2016). Oligodendroglial NMDA Receptors Regulate Glucose Import and Axonal Energy Metabolism. Neuron 91, 119–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schurr A, West CA, and Rigor BM (1988). Lactate-supported synaptic function in the rat hippocampal slice preparation. Science 240, 1326–1328. [DOI] [PubMed] [Google Scholar]
- Trevisiol A, and Nave KA (2015). Brain Energy Metabolism: Conserved Functions of Glycolytic Glial Cells. Cell Metab 22, 361–363. [DOI] [PubMed] [Google Scholar]
- Valdearcos M, Douglass JD, Robblee MM, Dorfman MD, Stifler DR, Bennett ML, Gerritse I, Fasnacht R, Barres BA, Thaler JP, et al. (2017). Microglial Inflammatory Signaling Orchestrates the Hypothalamic Immune Response to Dietary Excess and Mediates Obesity Susceptibility. Cell Metab 26, 185–197.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VanElzakker M, Fevurly RD, Breindel T, and Spencer RL (2008). Environmental novelty is associated with a selective increase in Fos expression in the output elements of the hippocampal formation and the perirhinal cortex. Learn. Mem 15, 899–908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xin M, Yue T, Ma Z, Wu FF, Gow A, and Lu QR (2005). Myelinogenesis and axonal recognition by oligodendrocytes in brain are uncoupled in Olig1-null mice. J. Neurosci 25, 1354–1365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xing F, Luan Y, Cai J, Wu S, Mai J, Gu J, Zhang H, Li K, Lin Y, Xiao X, et al. (2017). The Anti-Warburg Effect Elicited by the cAMP-PGC1a Pathway Drives Differentiation of Glioblastoma Cells into Astrocytes. Cell Rep. 18, 468–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang XJ, Kow LM, Funabashi T, and Mobbs CV (1999). Hypothalamic glucose sensor: similarities to and differences from pancreatic beta-cell mechanisms. Diabetes 48, 1763–1772. [DOI] [PubMed] [Google Scholar]
- Yang Y, Choi PP, Smith WW, Xu W, Ma D, Cordner ZA, Liang NC, and Moran TH (2017). Exendin-4 reduces food intake via the PI3K/AKT signaling pathway in the hypothalamus. Sci. Rep 7, 6936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zarjevski N, Cusin I, Vettor R, Rohner-Jeanrenaud F, and Jeanrenaud B (1993). Chronic intracerebroventricular neuropeptide-Y administration to normal rats mimics hormonal and metabolic changes of obesity. Endocrinology 133, 1753–1758. [DOI] [PubMed] [Google Scholar]
- Zhang X, Zhang G, Zhang H, Karin M, Bai H, and Cai D (2008). Hypothalamic IKKbeta/NF-kappaB and ER stress link overnutrition to energy imbalance and obesity. Cell 135, 61–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao B, Zhao CZ, Zhang XY, Huang XQ, Shi WZ, Fang SH, Lu YB, Zhang WP, Xia Q, and Wei EQ (2012). The new P2Y-like receptor G protein-coupled receptor 17 mediates acute neuronal injury and late microgliosis after focal cerebral ischemia in rats. Neuroscience 202, 42–57. [DOI] [PubMed] [Google Scholar]
- Zhao C, Dong C, Frah M, Deng Y, Marie C, Zhang F, Xu L, Ma Z, Dong X, Lin Y, et al. (2018). Dual Requirement of CHD8 for Chromatin Landscape Establishment and Histone Methyltransferase Recruitment to Promote CNS Myelination and Repair. Dev. Cell 45, 753–768.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu H, Pleil KE, Urban DJ, Moy SS, Kash TL, and Roth BL (2014). Chemogenetic inactivation of ventral hippocampal glutamatergic neurons disrupts consolidation of contextual fear memory. Neuropsychopharmacology 39, 1880–1892. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data used in this manuscript are available upon request. The original data are available at https://data.mendeley.com/datasets/wdmx6trhnr/4