

Abstract
Pulmonary hypertension is an uncommon disease that carries a significant morbidity and mortality. Pulmonary arterial hypertension is a subtype of pulmonary hypertension that describes a group of disease entities that lead to an elevation in precapillary pulmonary artery pressure. Despite advances in the diagnosis and treatment of pulmonary arterial hypertension, it remains a difficult disease to recognize and manage. In this review article, we will discuss the definition and diagnosis of pulmonary arterial hypertension. Additionally, we will discuss the ever-expanding management options, their mechanisms and strategies, including combination therapy and the most recent advances and future directions.
Keywords: pulmonary hypertension, pulmonary arterial hypertension, phosphodiesterase-5 inhibitor, prostacyclin analog, endothelin receptor antagonists, combination therapy, calcium channel blockers
Pulmonary hypertension (PH) is an uncommon condition that describes a group of diseases with a seemingly similar yet vastly different array of pathophysiologies, etiologies, and treatments. PH may have been first recognized over a century ago when changes in pulmonary vasculature were seen during an autopsy of a patient who presented with cyanosis. 1 In the following decades, the disease underwent several stages of understanding until the work of Brenner in 1935 laid a foundation for the current diagnosis of PH. 1 Today PH is thought to affect anywhere from 1 to 7% of adults in developed countries. 2 While it was thought that the disease affected primarily young females, it is now understood that the disease is more commonly diagnosed among patients 65 and older. Females are still more affected than males at a younger age though and have better survival. 2
Normal mean pulmonary arterial pressure (PAPm) is currently defined to be 14 mm Hg with an upper limit of normal of 20 mm Hg. 3 PH is defined as PAPm of > 25 mm Hg at rest as measured by right heart catheterization (RHC). The term borderline PH is sometimes used to describe patients with PAPm of greater than 20 but less than 25; however, the term remains controversial. 3 4 5 The most recent world symposium on PH has suggested to adjust this definition to include PAPm >20, and this will most likely be included in the next iteration of the guidelines and some physicians have started using it in their practices. 6 Exercise-induced PH is yet another controversial subtype of PH. It was initially removed from the guidelines due to heterogeneity in testing and definitions; however, a growing body of evidence in the recent years may be restoring it as its own category. 7 8
Additional values obtained via catheterization are used to further classify the category of PH and define the subsequent management. Values such as the pulmonary artery wedge pressure (PAWP), pulmonary vascular resistance (PVR), and left ventricular end diastolic pressure (LVEDP) are most commonly obtained. LVEDP is usually obtained via left heart catheterization. 3 4 5 Perhaps the most valuable is the PAWP. Current clinical practice is accustomed to using the term pulmonary capillary wedge pressure; however, expert groups prefer the term PAWP or pulmonary artery occlusion pressure. This is due to the fact that it is misleading to believe that the pressure measured truly represents the capillary pressure in all of the pulmonary circulation. 3
The differentiation of the subtype of PH is crucial to patient management and prognosis as they all represent unique diseases and have unique treatment plans. The European Society of Cardiology/European Respiratory Society (ERS/ESC) has provided the most updated classification categories for PH. The classification is often determined by both the patient's history and values obtained during catheterization as mentioned above. A summary of the classifications is described in Table 1 . 4 5
Table 1. Clinical classification of pulmonary hypertension.
| 1. Pulmonary arterial hypertension | |
| 1.1 Idiopathic 1.2 Heritable 1.2.1 BMPR2 mutation 1.2.2 Other mutation 1.3 Drugs and toxins induced 1.4 Associated with: 1.4.1 Connective tissue disease 1.4.2 Human immunodeficiency virus (HIV) infection 1.4.4 Portal hypertension 1.4.4 Congenital heart disease 1.4.5 Schistosomiasis |
|
| 1′ Pulmonary Veno-occlusive disease and/or pulmonary capillary hemangiomatosis | |
| 1′.1 idiopathic 1′.2 Heritable 1′.2 1 EIF2AK4 mutation 1′.2 2 Other mutation 1′.3 Drugs, Toxins and radiation induced 1′.4 Associated with: 1′.4.1 Connective tissue disease 1′.4.2 HIV infection | |
| 2. Pulmonary hypertension due to left heart disease | |
| 2.1 Left ventricular systolic dysfunction 2.2 Left ventricular diastolic dysfunction 2.3 Valvular disease 2.4 Congenital/acquired left heart inflow/outflow tract obstruction and congenital cardiomyopathies 2.5 Congenital/acquired pulmonary veins stenosis |
|
| 3. Pulmonary hypertension due to lung disease and/or hypoxia | |
| 3.1 Chronic obstructive pulmonary disease 3.2 Interstitial lung disease 3.3 Other pulmonary disease with mixed restrictive and obstructive pattern 3.4 Sleep-disordered breathing 3.5 Alveolar hypoventilation disorders 3.6 Chronic exposure to high altitude 3.7 Developmental lung disease |
|
| 4. Chronic thromboembolic pulmonary hypertension and other pulmonary artery obstruction | |
| 4.1 Chronic thromboembolic pulmonary hypertension 4.2 Other pulmonary artery obstruction 4.2.1 Angiosarcoma 4.2.2 Other intravascular tumors 4.2.3 Arteritis 4.2.4 Congenital pulmonary arteries stenosis 4.2.5 Parasites (hydatidosis) |
|
| 5. Pulmonary hypertension with unclear and/or multifactorial mechanisms | |
| 5.1 Hematological disorders: chronic hemolytic anemia, myeloproliferative disorders, splenectomy 5.2 Systemic disorders: sarcoidosis, pulmonary histiocytosis, lymphangioleiomyomatosis, neurofibromatosis 5.3 Metabolic disorders, glycogen storage disease, Gaucher disease, thyroid disorders 5.4 Others: Pulmonary tumoral thrombotic microangiopathy, fibrosing mediastinitis, chronic renal failure (with/without dialysis), segmental pulmonary hypertension |
|
Abbreviations: BMPR2, bone morphogenetic protein receptor, type 2; EIF2AK4, eukaryotic translation initiation factor 2 α kinase 4.
Pulmonary arterial hypertension (PAH) is used to describe a subset of patient with PH that is characterized by having a precapillary elevation in the PAPm. This subset is also known as group 1 PH. 3 4 5 While this subgroup sufficiently categorizes those with similar hemodynamics, it still contains a diverse variety of physiologies within itself. This is driven by different disease processes with each having their own management strategy. 3 4 5
Diagnostic Approaches
PH may present with subtle symptoms. These symptoms are nonspecific but most commonly begin with exertional dyspnea. Fatigue, weakness, and shortness of breath are also common. Patients who present later in their course may have manifestation of right ventricular failure. 5 Making the diagnosis usually requires a stepwise approach that involves various screening modalities. Many of these modalities may indicate the presence of PH; however, an RHC is always needed to definitively confirm the diagnosis. Before a diagnosis of PAH is made, other causes of PH need to be ruled out. Current guidelines recommend further workup including pulmonary function testing, ventilation–perfusion (V/Q) scanning, and polysomnography. 1
Echocardiography
Echocardiography is often used as a screening tool. Its broad utilization in a variety of clinical scenarios has increased the incidental finding of PH. This may lead to further investigation depending on the severity of the findings. Transthoracic echocardiography (TTE) can evaluate and monitor the effects of PH on the heart and is often used for surveillance on patients receiving vasodilator therapy. TTE offers the ability to measure the right atrial pressure and calculate the pulmonary artery systolic pressure, which are commonly used values in monitoring of response or deterioration. 5 Being commonly available, inexpensive and noninvasive, TTE has become the preferred means of follow-up of right ventricular function (RVF) and hemodynamics. 9 In the surgical setting, transesophageal echocardiography (TEE) has been used to evaluate patients with PH, who may need further perioperative interventions prior to their anticipated surgery. 10 TEE has also been used in lung transplant surgeries intraoperatively to evaluate right heart function and to monitor for acute processes that may ultimately affect the long-term outcome of the transplant, such as left to right shunting. 10
Ventilation–Perfusion Scanning
V/Q scanning is a useful tool in ruling out chronic thromboembolic pulmonary hypertension (CTEPH) as a cause of PH once confirmed on RHC. The current ESC/ERS guidelines recommend V/Q scanning due to its increased sensitivity (90–100%) and specificity (94–100%) in ruling out CTEPH with a normal or low probability scan as compared with other modalities such as computed tomography (CT) with pulmonary angiography. 5 While V/Q scanning can effectively rule out CTEPH, there is limited evidence for its role in confirming PAH. Data from a recent retrospective study conducted in China has suggested that in patients with confirmed PAH, a patchy perfusion pattern on V/Q scan is associated with a significantly increased PAPm, total pulmonary pressure, and all-cause mortality. 11 While this information may be useful in prognostication and assessing risk in patients with PAH, use of a patchy perfusion pattern on V/Q scan as a screening or diagnostic modality for PAH requires further study.
Cardiac MRI
Cardiac magnetic resonance imaging (MRI) is useful in assessing multiple cardiac parameters including RVF, size, cardiac output, stroke volume, and pulmonary artery distensibility. This can be particularly helpful in patients diagnosed with PAH as this modality has the added benefit of having reliable and reproducible results. 5 Cardiac MRI has been studied as a tool for long-term follow-up and overall prognostication in patients with PAH. A decrease in the parameters measured on cardiac MRI such as right ventricular ejection fraction, stroke volume, and left ventricular end diastolic volume has been associated with a poor prognosis in patients with PAH, and thus it has been useful in monitoring the progression of disease. 12 In patients with PAH who are on vasodilatory therapy, PVR is often used to assess response to therapy. RVF assessed by cardiac MRI has been shown to be progressively reduced in some patients with PAH despite a decrease in PVR, thus it has been proposed that monitoring RVF with cardiac MRI in patients with PAH may be a more useful means of monitoring disease progression. 13
Right Heart Catheterization
While there are other available diagnostic modalities, the diagnosis of PAH ultimately requires RHC. Not only is this used to rule in pre-capillary PH but also is used to assess vasodilatory response to help guide treatment. The current ESC/ERS PH guidelines note that a RHC is a necessary test for PAH and vasoreactivity test should be done for the cases of idiopathic PAH (iPAH), hereditary PAH, and drug-induced PAH to identify the subset of patients that will respond to calcium channel blocker (CCB) therapy. 5 Historically, the definition of PH on RHC has been a PAPm of ≥25 mm Hg. The recent 6th world symposium on PH now is proposing the cutoff be changed to a PAPm of ≥20 mm Hg based on the fact that this number has been found to be two standard deviations above the mean of normal. 6
After PH is confirmed on RHC, it is further characterized into precapillary (PH groups 1, 3, 4, and 5), postcapillary (PH group 2 and sometimes 5), or mixed pre- and postcapillary. This categorization is based on the values of PAPm, PVR, and PAWP. PAH falls into the precapillary PH category and is defined hemodynamically as having the following: PAPm of ≥25 mm Hg, PVR ≥3 wood units, and PAWP of ≤15 mm Hg. 14 Other values that can be extrapolated from the RHC which may be helpful in further characterizing PH include the transpulmonary gradient (TPG) and the diastolic pulmonary gradient (DPG). The DPG is particularly useful in diagnosing “out of proportion” PH, which refers to pulmonary artery pressures that are disproportionately elevated in the setting of group 2 PH. A DPG ≥ 7 mm Hg is suggestive of a mixed pre- and postcapillary PH and could be a clue that the patient may have PAH in the setting of left heart disease. 15 A TPG > 12 mm Hg has also been used to help identify postcapillary PH with a precapillary component; however, this measure is affected by cardiac output as well as recruitment and distensibility of the pulmonary vasculature; therefore, the TPG can over- or underestimate the upstream effects of the left atrial pressure. 15 The vasoreactivity test entails administering inhaled nitric oxide (NO) during RHC at 10 to 20 ppm. A test is considered positive if there is a reduction in the PAPm ≥ 10 mm Hg to reach a PAPm ≤ 40 mm Hg with no change or an increase in cardiac output. 5
CT Angiography
CT angiography (CTA) has several benefits when it comes to evaluating patients with PH. The high-resolution images allow for the evaluation of the lung parenchyma, architecture of the heart, and the structure of the pulmonary vessels which can then help narrow down potential causes of PH. CTA is particularly useful as a noninvasive method of evaluating patients for CTEPH that may be amenable to pulmonary endarterectomy. 5 The pulmonary artery (PA) diameter, the PA-to-ascending aorta ratio, and the segmental artery-to-bronchus ratio are all measurements that have been identified as reliable markers of the presence of PH on CTA. A PA diameter >29 mm has a sensitivity and specificity of 87 and 89%, respectively, and the specificity increases to 100% when combined with a segmental artery-to-bronchus ratio of > 1:1 in three pulmonary lobes. 16 A main PA diameter to ascending aorta diameter ratio of > 1:1 has a 96% positive predictive value for PH and thus can also be used to identify evidence of PH on CTA. 16 While much of the information obtained on CTA is not specific to PAH, there are some parenchymal changes on CT such as centrilobular nodules that are seen more commonly in PAH in comparison to other groups of PH. Patients with PAH are at an increased risk of pulmonary hemorrhage; this leads to phagocytosis of red blood cells by pulmonary macrophages which then develop into cholesterol granulomas that are visible on CT as centrilobular nodules. 17 CTA may provide other clues as to the etiology of PAH; for example, it may reveal a large and dilated esophagus which may be seen in systemic sclerosis which is a known cause of PAH. 5
Pathophysiology
PAH can be idiopathic, or it can be the end result of a wide array of mechanisms ranging from genetic disorders to infections. 18 The resulting pathologic feature regardless of the cause is the remodeling of the pulmonary vasculature and subsequently of the right ventricle (RV). 19 20 Small pulmonary arteries are the most affected. Endothelial dysfunction along with intimal hyperplasia, medial hypertrophy, and subsequent inflammation and thrombosis are often the pathologic findings in disordered, remodeled pulmonary vasculature in patients with PAH. 19 These changes are what leads to progressive partial occlusion of pulmonary vasculature and the elevation in the PVR. 19 20
The RV response to the rise in the PVR and the increased afterload is hypertrophy. Right ventricular hypertrophy (RVH) can be subclassified as adaptive or maladaptive. Adaptive RVH is characterized by minimal eccentric dilatation and fibrosis as well as minimal changes in ejection fraction, and filling pressures. Maladaptive RVH, on the other hand, is characterized by increased eccentric dilatation and fibrosis, thus leading to reduction in ejection fraction and cardiac output, and increase in filling pressure. Patient with maladaptive RVH exhibits increase in sympathetic activation. 19 21 The mechanism by which the transition occurs from adaptive to maladaptive RVH remains unclear. However, it has been observed that patients with connective tissue disease, particularly scleroderma, progress to maladaptive RVH early. 22
The aforementioned vascular remodeling and subsequent pathologic changes in both the vasculature and the RV can be attributed to the disruption and imbalance of factors regulating vasodilation, proliferation, and thrombosis pathways. 19 20 A foundational understanding of these pathways leads to a better appreciation for the mechanism of action of the drugs used to treat PAH ( Fig. 1 ).
Fig. 1.
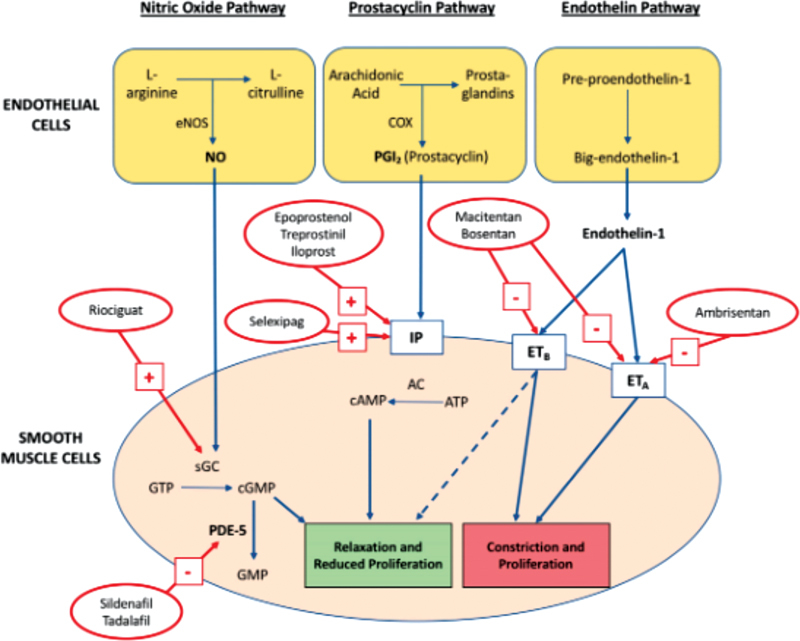
The major pathways and the pharmacological treatments targeting them. ATP, adenosine triphosphate; cAMP, cyclic adenosine monophosphate; ET, endothelin; GTP, guanosine triphosphate; GMP, guanosine monophosphate; IP, I-prostanoid receptors; NO, nitric oxide; PDE-5, phosphodiesterase type 5; sGC, soluble guanylate cyclase. Adapted with permission from Prior et al. update on pharmacotherapy for pulmonary hypertension, Med J Aust 2016. 86
Nitric Oxide
NO is a potent vasodilator and is produced by endothelial cells. It also is an inhibitor of platelet activation and smooth muscle proliferation. 18 19 It performs its functions by diffusing from the endothelial cells to the smooth muscle layer and binds soluble guanylate cyclase (sGC). The activated sGC then converts guanosine triphosphate to cyclic guanosine monophosphate (cGMP) which then activates this pathway. cGMP is broken down by phosphodiesterase type 5 (PDE-5) effectively regulating the function of the pathway controlled by NO. 20 NO is decreased in PAH leading to overall conditions favoring vasoconstriction and smooth muscle cell proliferation. 19 20 These decreased levels were especially observed in patients with iPAH. 18 23
Prostacyclin and Thromboxane A2
Prostacyclin and thromboxane are metabolites of arachidonic acid that are produced by endothelial cells. Once prostacyclin is produced, it is released and bind to receptors on vascular smooth muscle cells called I-prostanoid receptors (IPs). 20 IP receptors then activate a pathway that leads to vasodilation by relaxing smooth muscles. It also plays a role in inhibiting platelet aggregation and smooth muscle proliferation. 19 20 Thromboxane A2, on the other hand, is a potent activator of platelet aggregation. It also stimulates vasoconstriction and smooth muscle proliferation. 19 20 In patients with PAH, there is a notable increase in the ratio of the two factors in favor of thromboxane A2. Patients with PAH have reduced prostacyclin production as well as reduced expression of IP receptor. 24 25 This is especially true in patients with iPAH where the production of prostacyclin synthase was notably decreased. 25
Endothelin-1
Endothelin-1 (ET-1) is produced at the surface of endothelial cells and acts as a vasoconstrictor and a stimulator for smooth artery muscle cells proliferation. 19 20 There are two receptors on smooth muscle cells that are activated by ET-1, which are ETa and ETb. 20 In patients with PAH, there is an increased expression of Eta and ETb, 20 as well as increased concentrations of ET-1, though it is unclear if this elevation is the cause or an effect of PH. 26 It has been observed that concentrations of ET-1 are inversely proportional to pulmonary blood flow and cardiac output, possibly implicating a direct relationship. 18
Serotonin
Serotonin is another vasoconstrictor with pro-hypertrophy and hyperplasia properties. 18 The direct role of serotonin in PAH is yet to be clearly defined. The plasma levels of serotonin, however, were noted to be elevated in patients with PAH, with a concomitant reduction in platelet levels. 27 The level of serotonin in the plasma is not likely to be implicated as selective serotonin inhibitors, which effectively increase serotonin levels in the plasma, do not raise risk of PAH, and could possibly be protective. 28 The more likely cause is abnormalities with serotonin uptake and transporters. It has been observed that incidence of PAH is increased among patients who took medication known to increase serotonin release from platelets and increase its uptake. 29 Recently mutations in the serotonin transporter and receptor or both have been described in patients with PAH. 30
In addition to the molecular pathways that are in array in patients with PAH, PAH could be inheritable. Heritable PAH (hPAH) exists in roughly one-tenth of patients, and is often autosomal dominant. 1 20 A variety of mutations have been implicated in hPAH. Up to 100 families have been identified with hPAH worldwide. 31 It is noted that the inheritance pattern of these mutations follows a pattern described as genetic anticipation in which each generation develops the disease earlier and at a greater severity than the generation prior. 18
Transforming Growth Factor β Family of Mutations
This superfamily of mutations has been associated with PAH. Bone morphogenetic protein receptor (BMPR2) is probably the most common and well-studied, with over 45 mutations identified in patients with hPAH. 31 This mutation is present in up to 75% of patients with hPAH and may also be present in up to 20% of cases of iPAH. 32 BMPR2 regulates vascular smooth muscle cell growth and loss of its function leads to increase in pulmonary artery smooth muscle cells. 19 Activin-like kinase, mothers against decapentaplegic homolog 9, and endoglin are other known mutation, but comparatively less common than BMPR2 as they collectively appear in as little as only in 5% of patients of hPAH. 1
Other Mutations
Caveolin-1 and KCNK3 have been identified in families with hPAH without abnormalities in the transforming growth factor β pathway both of which are involved in cell membrane trafficking. 19
Mutations in Serotonin Pathway
It has been observed that patients with iPAH have increased expression of the serotonin transporter 5 hydroxytryptamine Transporter (5-HTT), and as results, an increase in smooth muscles cells of the pulmonary vessels. 30
Patients with PAH often have multiple comorbidities; however, several conditions have been irrefutably associated with the development of PAH.
Human Immunodeficiency Virus
Human immunodeficiency virus (HIV) has been shown to be associated with an increased incidence of PH. The incidence has been noted to be roughly 0.5% which is up to 12 times greater than the general population. The development of PAH appears to be independent of CD4 cell count; however, it is directly proportional to the duration of HIV infection. The mechanism, is yet to be completely understood. 18
Rheumatologic and Connective Tissue Disease
Features of PAH are sometimes observed in patients with systemic lupus erythematosus, rheumatoid arthritis, and connective tissue disease; however, the most commonly associated condition is scleroderma. This is particularly evident in the CREST variant where up to 15% of patients will have clinically significant PAH, and changes within the pulmonary vasculature are noted in up to 80% of patients on autopsy. 33
Sickle Cell Anemia
PAH is present in up to 30% of patients with sickle cell anemia. The suggested mechanism is through the consumption of NO by free hemoglobin or via increased levels of reactive oxygen species, which are increased in patients with sickle cell anemia. 34 It was previously thought that recurrent episodes of acute chest syndrome were responsible for increased incidence of PAH in patients with sickle cell; however, that has since been refuted. The presence of PAH increases the mortality in patients with sickle cell and the relationship is inversely proportional to the degree of increase in mPAP. 35
Management
Patient with PAH requires close follow-up and frequent monitoring. While targeted therapy is the main focus of treatment for these patients, symptomatic and supportive treatment remains a large component of their care. Their treatment is often complex and is applied in a stepwise approach. 5 These treatment strategies are paired with lifestyle changes and are tailored to several metrics that are used to prognosticate and measure severity for patients with PAH.
The most widely used test is the 6-minute walking test, as it is easy to perform and inexpensive. It is also recommended to use Borg score at the end of the test to provide additional information about the degree of effort. 5 36 Cardiopulmonary Exercise Testing (CPET) can also be used to provide information regarding prognostication and therapeutic choices, it is much less utilized. 37
While there is no specific biomarker for PAH, the most commonly accepted markers are brain natriuretic peptide (BNP) and N-terminal proBNP (NT-proBNP). The levels of these markers correlate to level of cardiac dysfunction, and while they are not specific to PH by any means, they have been helpful in providing prognostic information. 38 It has been noted that BNP correlates more accurately to the pulmonary vasculature status and is less affected by renal dysfunction, while NT-proBNP may be more accurate in prognostication; however, overall one is not superior to the other in the management of PAH. 38
Lifestyle changes are key components of the management of PAH. Pregnancy is not only associated with high morbidity and mortality but also limits the treatment options available and thus should be avoided. 5 While the risk remains high, patients with PAH have improved outcomes with pregnancies in recent years, especially in patients who respond to CCBs. 5 39 While Progesterone-only options are potentially safe, Barrier contraceptives are arguably the safest for these patients. 40 There remains, however, controversy regarding the most appropriate option. 5
Supportive measure also plays an important role in the treatment plans for patients of PAH. The use of diuretics is common with the goal of prevention of edema and the potential deleterious effects of volume overload on RV function and remodeling. There are, however, no randomized controlled trial (RCT) to evaluate the diuretic use in PAH, and their use remains entirely based on clinical status. 41 Oxygen use is also common in patients with PAH as a means to avoid hypoxia-induced pulmonary vascular constriction. It has been shown that the hypoxia can increase PVR in patients with PAH and oxygen reduces that. 42 Current recommendations include supplemental oxygen to maintain saturations above 90%; however, there remains a lack of RCTs to evaluate its effects 4 .
Anticoagulants remains an important component of the treatment of patients with PAH, though specifically limited to patients with iPAH, hPAH, and PAH due to anorexigens. 5 This recommendation is based on the findings of increased number of thrombotic lesions in autopsies of patients with iPAH, as well an increased risk of venous thromboembolism risk. 43 Prospective RCTs as well as registry data offer controversial and inconsistent results, and thus the supporting data are limited retrospective evaluation. 5 43
Targeted Therapy
Calcium Channel Blockers
CCBs act directly on vascular smooth muscle cells and lead to vasodilation. 20 While they are commonly used in the treatment of PAH, only a small subset of patients demonstrates the vasoreactivity needed for their efficacy. 44 Vasoreactivity is defined as a drop in mean PAPm of >10 mm Hg from baseline to a value <40 mm Hg without a decrease in cardiac output. 5 CCB should be avoided in patients who have negative vasoreactivity testing as the potential adverse events include syncope, hypotension, and progression of RV failure. 45 CCB, along with vasoreactivity testing, should only be utilized in the treatment of iPAH, hPAH, and drug-associated PAH. This is due to the fact that vasoreactive testing may be misleading in other forms of PAH and those patients do not benefit from long-term CCB therapy. 46
Patients with iPAH, hPAH, and drug-associated PAH who do show positive vasoreactivity should be started on either nifedipine, diltiazem, or amlodipine. 5 The most commonly used are diltiazem and nifedipine. The choice of CCB is dependent on patient's characteristics such as baseline blood pressure and heart rate. 44 47 Doses used are often higher than those commonly used for other conditions, but it is recommended to start at a low dose and escalate slowly with close follow-up. 5 Patients who do not have an adequate response should have additional PAH therapy added. An adequate response is defined by the ERS/ESC to be near normalization or marked improvement in hemodynamics, As well as improvement in symptoms to WHO functional class I or II. 5
Endothelin Receptor Antagonists
As discussed earlier, ET-1 is responsible for proliferation and vasoconstrictor and exerts its functions by acting on ET-A and ET-B receptors. Thus, inhibitors of these two receptors were developed to target this physiological process. As this class of medications shares the same mechanism of action, they also share the adverse effects. Endothelin receptor antagonists (ERA) are potent teratogens and thus all female patients of childbearing age are typically enrolled in risk evaluation and mitigation strategy programs. 1 ERAs are also known to cause fluid retention which could be a major barrier to their utilization in patients with tenuous fluid status.
Bosentan was the first to be developed of the ERA class and has been available since 2001. 48 It is nonselective and thus binds to both subtypes of the ET receptor. Multiple studies have been done to evaluate the efficacy of bosentan. 49 The earliest was Channick et al who demonstrated in an RCT referred to as Study 351 that bosentan improved hemodynamics, exercise capacity, time to clinical worsening, and Borg dyspnea score. 50 The study, however, was very small and subsequent studies, the largest being BREATHE-1, showed mostly similar findings. 49 51 These studies include BREATHE-2, Breathe-5, and EARLY. 52 53 54 The most recent study is COMPASS-2; a large long term trial that compared bosentan with sildenafil to sildenafil alone, and it failed to show benefits with the exception of improved 6MWT distance. 55 A common finding in almost all the trials involving bosentan noted an elevation in hepatic aminotransferases to be present in as high as 10% of patients, as a result liver function monitoring is recommended in these patients. This effect was noted to be dose dependent and reversible upon the discontinuation of bosentan. 5 Due to this side effect profile, as well as more recent disappointing long-term data, bosentan is not recommended to be part of first-line therapy for newly diagnosed PAH patients. 48
Ambrisentan was the second of the ERAs to be produced. It is a more selective ERA antagonist, as it only binds ET-A. It is associated with less hepatotoxicity than bosentan and thus liver function monitoring is not mandated with treatment with ambrisentan. It is, however, associated with an increased incidence of peripheral edema. 5 Ambrisentan was initially introduced in 2005 after a pilot study showed that treatment with ambrisentan at a dose of up to 10 mg daily improved 6MWT distance, Borg dyspnea index, WHO functional class, and hemodynamics in patients with iPAH or PAH associated with connective tissue disease. 56 These findings were confirmed by two larger RCTs, namely ARIES 1 and 2. 57
Macitentan is the most recent of the ERAs. It is nonselective and acts similarly to bosentan by blocking both ET-A and ET-B receptors, except it was designed to be more lipophilic. 1 Its use was shown to be efficacious in 2013 in the SERAPHIN trial which included 742 patients with PAH and showed that macitentan reduced morbidity, as well as improved exercise capacity. There was a reported mortality benefit, but only when combined with hospitalization as a composite end point. Mortality alone was not significantly reduced but only showed a trend, though the authors note that the study was not powered to show such benefit. These benefits were seen in both treatment-naive patients and patients with other background treatments, primarily sildenafil, but also inhaled prostanoids and CCBs. 58 No hepatotoxicity was observed; however, anemia was a notable adverse effect, especially at the higher dose ranges of macitentan. 5
Phosphodiesterase Type 5 Inhibitors
cGMP, a substrate for the enzyme PDE-5, is the main target of activation of the NO pathway discussed earlier. Inhibition of phosphodiesterase function prolongs the half-life of cGMP and as a result potentiates the effects of NO. 5 Phosphodiesterase type 5 inhibitors (PDE-5is) were initially investigated in erectile dysfunction, but they were noted to have a potent vasodilatory effect on the pulmonary vasculature and were later instituted in the treatment of PAH after a series of RCTs that initially showed benefit with sildenafil. 5 All PDE-5is have similar side effect profile that include flushing, headaches, epistaxis, and diarrhea. Since these drugs are potent vasodilators, they should be used with caution when combined with other vasodilating medications such as CCB and nitrates. 20
Sildenafil was the first PDE-5i to be approved and it is available in both intravenous (IV) and per os formulation. The first trial to show the benefits of sildenafil was the SUPER-1 trial. While it was a short-term trial, at only 12 weeks, it showed improvements in 6MWT distance, WHO functional class, and hemodynamics. 59 The current dose is usually 80 mg three times a day (TID), though it is only approved at 20 mg TID. 5 The maximum tolerated dose has not yet been investigated. 48 Further trials, such as the SUPER-2, showed similar benefits when extended to longer follow-up periods. 60 Later studies, led by Iversen et al and Pfizer, failed to show benefits in the 6MWT distance when sildenafil was studied in combination with bosentan, though the Iverson study recruited only patients who had developed Eisenmenger syndrome. 61
Tadalafil is a long-acting version of sildenafil and can be administered as a once daily dose. It was first introduced in 2009 when the PHIRST trial showed that tadalafil with a dose of up to 40 mg showed significant improvement in 6MWT distance, time, and incidence of clinical worsening, and hemodynamics. 62 This initial trial showed similar results to those shown by sildenafil. 48 A follow-up long-term trial tested the safety and efficacy of tadalafil over a 52-week period and showed well tolerability and sustained benefit. 63 Tadalafil also showed benefit in combination therapy (see Table 2 ).
Table 2. Summary of clinical trial for combination therapy in PAH.
| Randomized, controlled short-term studies | ||||
| Study N design key results | ||||
| PACES 87 | 267 | Sildenafil in patients previously treated with epoprostenol | Significant improvement in 6MWT distance, hemodynamics, and time to clinical worsening | |
| STEP 88 | 37 | Inhaled iloprost in patients previously treated with bosentan | Trend toward increase in 6MWT distance, but significant improvement in WHO-FC and clinical deterioration | |
| COMBI 89 | 40 | Inhaled iloprost in patients previously treated with bosentan | Study prematurely ended after interim analysis; no clinical improvement observed. | |
| TRIUMPH 80 | 255 | Inhaled iloprost in patients previously treated with bosentan | Significant improvement in 6MWT distance, but no improvement in WHO-FC or time to clinical worsening. | |
| FREEDOM-C 82 | 350 | Oral treprostinil in addition to ERAs and/or PDE-5 inhibitors | No significant improvement in 6MWT distance, WHO-FC, or time to clinical deterioration | |
| FREEDOM-C2 83 | 310 | Oral treprostinil in addition to ERAs and/or PDE-5 inhibitors | No significant improvement in 6MWT distance, WHO-FC, or time to clinical deterioration | |
| PHIRST 63 | 216 | Subgroup analysis of patients who were given tadalafil + bosentan | No significant improvement in 6MWT distance. No improvement in WHO-FC or time to clinical deterioration in patients previously treated with bosentan | |
| IMPRES 90 | 202 | Imatinib in addition to dual or triple combination therapy | Significant improvement in 6MWT distance, PVR and NT-proBNPNo effect on time to clinical worsening. High dropout rate because of adverse effectsUnexpectedly high rate of subdural hematoma | |
| PATENT 66 | 222 | Riociguat in addition to ERAs and/or prostacyclin analogues | Significant improvement in 6MWT distance and PVR in previously treated patients | |
| PATENT-plus 91 | 18 | Riociguat in addition to sildenafil | No change in systolic blood pressure (primary end point), no efficacy signals, increased hypotensive episodes in the extension phase of the study | |
| Event-driven randomized, controlled studies | ||||
| Study N design key results | ||||
| COMPASS-2 56 | 334 | Bosentan in addition to sildenafil | • No significant improvement in progression-free survival (primary end point) • Significant improvement in 6MWT distance and NT-proBNP after 16 weeks |
|
| SERAPHIN 59 | 471 | Macitentan in previously treated patients (predominantly with PDE-5 inhibitors) |
• Significant improvement in progression-free survival (HR 0.62;
p
= 0.009)
• Significant improvement in 6MWT distance, WHO-FC and PVR after 6 months |
|
| AMBITION 86 | 500 | Initial combination therapy with ambrisentan and tadalafil versus monotherapy with one of these substances | • 50% risk reduction in time to therapy failure with initial combination therapy • Significant improvement in 6MWT distance and NT-proBNP after 6 months |
|
| GRIPHON 84 | 925 | Selexipag in addition to ERAs and/or PDE-5 inhibitors | • 40% reduction in risk for disease progression, almost independently of pre-existing therapy • No improvement in WHO-FC |
|
Abbreviations: 6MWT, 6-minute walking test; ERA, endothelin receptor antagonist; NT-proBNP, N-terminal pro-brain natriuretic peptide; PDE-5, phosphodiesterase type 5; PVR, pulmonary vascular resistance; WHO-FC, World Health Organization functional class.
Adapted with permission from Hoeper MM et al, Targeted therapy of pulmonary arterial hypertension, Int J cardiol, 2018. 49
Vardenafil is most recent iteration of the PDE-5i family and is currently available in a twice daily formulation. Despite being shown to be safe and efficacious in a 12-week RCT, with benefits similar to those of sildenafil, it is currently not US Food and Drug Administration approved for the treatment of PH in the United States. 64
Guanylate Cyclase Stimulators
sGC, the binding molecule for NO, is the gateway to the NO pathway for vasodilation. 20 Riociguat activates sGC directly, bypassing the need for NO secretion by endothelial cells and causing production of cGMP and thus vasodilation. 5 Its use was validated in an RCT of 443 patients named PATENT-1, where it was shown to improve 6MWT distance, exercise capacity, time to clinical worsening, and hemodynamics. Up to 50% of patients enrolled in PATENT-1 were already on therapy, mostly with an ERA. Analysis of this group of patients on other therapies showed sustain benefit from the addition of riociguat. 65 The most common side effects were hypotension and syncope. 65 The long-term safety and whether it provides sustained efficacy is yet to be determined and is currently being evaluated in PATENT-2. 66
Prostacyclin Analogues
Given the importance of prostacyclin and its pathway in the regulation of pulmonary vasculature tone and prevention of thrombosis, it was one of the first pathways to be targeted for treatment. 5
Epoprostenol was one of the first drugs to be commonly implicated in patients with severe PAH. 20 It can only be administered as a continuous IV infusion due to its very short half-life, and to make matters more difficult, it is also only stable in room temperature for roughly 8 hours. 5 The latter, however, has recently changed with the introduction of a new formulation of epoprostenol that is more stable at room temperature. 67 It currently remains the only medication used in PAH that has been shown to improve mortality, though this was only demonstrated by one RCT by Barst et al. 68 This mortality benefit was confirmed by meta-analysis of the three available RCTs. 69 All three RCTs demonstrated that epoprostenol improved symptoms, 6MWT distance, exercise capacity, and hemodynamics. This benefit was observed in patients with iPAH, as well as patients with PAH related to CTD, namely scleroderma. 68 70 71 Side effects include flushing, headache, diarrhea, and leg pain. Certain adverse events are unique to epoprostenol therapy as they are related to the infusion and delivery system, such as site infection and sepsis. 5 Guidelines have been developed to minimize infections related to venous access with epoprostenol therapy, and the solution has been developed to be bactericidal in nature. 72 Abrupt discontinuation of infusion of epoprostenol is extremely dangerous as it can lead to rebound elevation in pulmonary pressure which may lead to return of symptoms or even death. 5 Currently, ERS/ESC recommends the use of epoprostenol in patients with WHO functional class III or IV symptoms, especially those who are failing symptoms. 5 Despite the known benefits, epoprostenol may be underutilized in patients with severe PAH, possibly due to the presence of other comorbidities precluding the use of this agent. 1
Beraprost was the first oral prostacyclin analog to become available. 5 It has only been shown to improve exercise capacity in an RCT in 2003. 73 Despite this result being sustained during long-term follow-up, the lack of effect on hemodynamics or outcomes has resulted in beraprost being seldom used. 5
Iloprost is available in IV, inhaled, and oral formulations. It is, however, most commonly utilized in the inhaled formulation. This is due to the fact that the oral route has not been evaluated in PAH patients, and aside from small observational studies, there is a lack of data on the use of IV Iloprost. 74 Inhaled iloprost was validated in the AIR study, where 203 patients were randomized for iloprost versus placebo and demonstrated to improve symptoms, quality of life, WHO functional class, and hemodynamics. 75 It is generally well tolerated with the most common side effects being flushing and jaw pain. 5
Treprostinil is a more stable formulation similar to epoprostenol with longer half-life and stability at room temperature. It is also available in oral, inhaled, IV, and subcutaneous forms. 5 The subcutaneous formulation was the first to become available, in the form of a continuous infusion via a subcutaneous catheter. 76 In a 2002 study, it was shown that a subcutaneous infusion of treprostinil in patients with PAH led to modest, but statistically significant improvements in 6MWT distance, symptoms, and hemodynamics. Side effects were flushing, headaches, and most importantly local site pain, which caused up to 8% of patients to withdraw from the study. 76 It is thought that the modest improvements were due to the use of lower than average doses limited by infusion site pain; however, new strategies have been implemented that allow for use of higher dose ranges with good tolerance. 76 77
The IV formulation was developed next, thought to be an alternative to the more painful subcutaneous infusion approach, but an RCT designed to study the drug was prematurely terminated due to safety concerns. 78 The data that was collected, however, showed promising result with improvement in symptoms, 6MWT distance, and WHO functional class. 78 Observational studies in the following years have shown that IV Treprostinil is relatively well tolerated. 5 Due to higher risk of infection related to IV administration, an implantable pump is being investigated for treprostinil delivery, which would decrease the risk. 1
The inhaled formulation was introduced in 2010 when the TRIUMPH trial showed that adding inhaled treprostinil to therapy with either bosentan or sildenafil led to significant improvement in 6MWT distance and quality of life, but failed to change WHO functional class or time to clinical worsening. 79
Oral treprostinil was the last to be introduced. When used in patients without other therapy, it led to only modest improvement in 6MWT distance. 80 However, when used in patients with background therapy with either bosentan or sildenafil, it failed to generate any significant results. 81 82
Prostacyclin Receptor Agonists
Selexipag is a recent addition to the therapeutic options for PAH. It acts on the prostacyclin pathway by binding and activating the prostacyclin receptor directly. While similar to prostacyclin analogue in function, selexipag is pharmacologically distinct. 5 In GRIPHON, the use of selexipag was investigated in patients receiving therapy with single or multiple agents showed improvement in combined mortality and morbidity end point by up to 40%. This benefit was most significant when defined by clinical worsening or hospitalization, as mortality alone did not show significant improvements. 83 It also improved 6MWT distance. The main side effects were similar to other drugs in this class and included headache, nausea, diarrhea, and jaw pain. 83
Combination Therapy
In the treatment of PAH, there are many drugs available to choose from and tailor for each particular patient's needs. It is common, however, that patients will not respond to monotherapy. Patients often need a combination of two or more treatment agents. Combination therapy is easily achieved given the separate signaling pathways affecting the pathophysiology of PH. Clinical outcome and 6MWT distance were noted to improve in a 2010 meta-analysis evaluating escalation to combination therapy after suboptimal response to monotherapy. A trend toward a mortality benefit was also noted, but the study was not powered to detect a statistically significant result. 84
Combination therapy may be instituted as the initial regimen, or medications can be added in sequence to achieve the needed clinical response. Sequential escalation of therapy is the most common, however, in both daily practice and RCTs. 5 The BREATHE-2 trial was the first of its kind to attempt to combine treatments on initial enrolment, and while it was a negative study, it set the stage for the following trials. 52 The AMBITION trial was the most promising as it showed that an initial combination of ambrisentan and tadalafil was better than either agent alone. The combination of the two medication reduced the occurrence of clinical failure by 50% greater than monotherapy. 85 Recent trials have also included rising numbers of patients with existing background therapy. Up to 80% of patients in the GRIPHON trial with selexipag had existing background therapy. Same pattern is seen with 50% of patients in PATENT-1 with riociguat and 64% in the SERAPHIN trial with macitentan, thus adding to the body of evidence for efficacy and safety of combination therapy. 58 65 83 TRITON is a large trial comparing double versus triple combination therapy as initial therapy and is currently underway. This trial will shed light on the importance of combination therapy as an initial strategy. 86 Table 2 summarizes the current body of evidence for the use of combination therapy.
The ESC/ERS guidelines from 2015 currently provide the most standard accepted guidelines for the management of patients with combination therapy. This includes both treatment-naive patients and patients on existing background therapy. The treatment guidelines are based on patient's risk class at the time of the assessment. Sequential therapy is recommended based on a stepwise, goal-directed therapy that calls for escalation of therapy if clinical goals are not met. 5 These guidelines were updated with recommendations from the Cologne consensus conference by Hoeper et al. These updated guidelines recommend more aggressive treatment plans that include upfront treatment with double and even triple regimens 48 ( Table 2).
Novel Medications and Targets
There are a wide variety of biologic targets and novel therapies which are being investigated as treatments for PAH. Cellular mechanisms being researched currently are aimed at treating PAH through targeting various vasoactive peptides, inhibition of cell proliferation, induction of apoptosis, and mitochondrial modulation of angiogenesis. 87 There is also an effort to identify genes that may play a role in pathogenic pulmonary artery smooth muscle cell (PASMC) proliferation and creating therapies that are targeted directly to these genes. 90 There are also newer medications from the already well-described PDE-5i class that are being investigated for use in PAH.
Recent attention has been paid to inhibitors of Rho-kinase, tyrosine kinase, elastase, survivin, and HMG-COA reductase that have been shown to reverse PH in rodent models by inhibition of cell proliferation and apoptosis. 87 Mitochondrial permeability transition pore (MPTP) is a protein that when activated results in termination of mitochondrial function and triggers cell death. 88 MPTP has been thought to play a role in PAH both in vivo and in vitro and thus it has been proposed that targeted activation of MPTP may have a role in PAH therapy; however, much of this research has been done in animal models and needs further investigation. 87
It is thought that the characteristics of PASMCs in patients with PAH (PAH-PASMCs) have a unique profile when compared with healthy controls. 89 Activation of Rho-kinase appears to have many downstream effects on the development of PAH through its role in activation of AMP-activated protein kinase (AMPK), or secretion cyclophilin A (CyPA) from vascular smooth muscle cells. 90 91 The basigin (Bsg) extracellular CyPA receptor plays a role in endothelial activation of Rho-Kinase through disruption of NO metabolism. 92 A novel pathogenic protein called selenoprotein P (SeP) is thought to play a role in the development PAH through a variety of mechanisms including causing mitochondrial dysfunction, inhibiting apoptosis, and through its negative effect on antioxidative stress signaling. 90 Thus, Rho-kinase, AMPK, CyPA, Bsg, and SeP are all currently being investigated as potential targets of therapy for PAH.
Udenafil is a PDE-5i that has been evaluated previously for the use in the treatment of congestive heart failure with reduced ejection fraction, erectile dysfunction, and portal hypertension. 93 A recent phase IIa study published in the Korean Circulation Journal has demonstrated that udenafil had favorable effects on hemodynamics in patients with PAH. Udenafil was found to have an effect on hemodynamics that lasted around 4 hours, which is longer than other medications in the same group, and this study also noted a relatively low number of adverse events with the 50mg dose of the medication. 93 Larger studies are required to further evaluate the role of Udenafil in PAH.
Conclusion
There has been a rapid growth of our ability to diagnose, manage, and better understand PH in the recent years. Many diagnostic modalities are available for both diagnosis and follow-up, as well as early detection of deterioration. Management options are ever expanding and several pathways have been explored and exploited for the treatment options. The use of these medications is supported by many long-term trials, though some of the evidence is heterogenous and sometimes inconclusive. Combination therapy is now the standard of care with many trials supporting the safety and efficacy of multiple agents. Ongoing research is on the cusp of unlocking more pathways and therapeutic option, as well further validating the use of existing therapy as either monotherapy or combination therapy.
Footnotes
Conflict of Interest None declared.
References
- 1.Dodson M W, Brown L M, Elliott C G. Pulmonary arterial hypertension. Heart Fail Clin. 2018;14(03):255–269. doi: 10.1016/j.hfc.2018.02.003. [DOI] [PubMed] [Google Scholar]
- 2.Hoeper M M, Humbert M, Souza R et al. A global view of pulmonary hypertension. Lancet Respir Med. 2016;4(04):306–322. doi: 10.1016/S2213-2600(15)00543-3. [DOI] [PubMed] [Google Scholar]
- 3.Hoeper M M, Bogaard H J, Condliffe Ret al. Definitions and diagnosis of pulmonary hypertension J Am Coll Cardiol 201362(25, Suppl):D42–D50. [DOI] [PubMed] [Google Scholar]
- 4.McLaughlin V V, Archer S L, Badesch D B et al. ACCF/AHA 2009 expert consensus document on pulmonary hypertension: a report of the American College of Cardiology Foundation Task Force on Expert Consensus Documents and the American Heart Association: developed in collaboration with the American College of Chest Physicians, American Thoracic Society, Inc., and the Pulmonary Hypertension Association. Circulation. 2009;119(16):2250–2294. doi: 10.1161/CIRCULATIONAHA.109.192230. [DOI] [PubMed] [Google Scholar]
- 5.Galiè N, Humbert M, Vachiery J-L et al. 2015 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension: the Joint Task Force for the Diagnosis and Treatment of Pulmonary Hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS): Endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC), International Society for Heart and Lung Transplantation (ISHLT) Eur Heart J. 2016;37(01):67–119. doi: 10.1093/eurheartj/ehv317. [DOI] [PubMed] [Google Scholar]
- 6.Galiè N, McLaughlin V V, Rubin L J, Simonneau G.An overview of the 6th World Symposium on Pulmonary Hypertension Eur Respir J 20195301):••• 10.1183/13993003.02148-2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Naeije R, Saggar R, Badesch D et al. Exercise-induced pulmonary hypertension: translating pathophysiological concepts into clinical practice. Chest. 2018;154(01):10–15. doi: 10.1016/j.chest.2018.01.022. [DOI] [PubMed] [Google Scholar]
- 8.Herve P, Lau E M, Sitbon O et al. Criteria for diagnosis of exercise pulmonary hypertension. Eur Respir J. 2015;46(03):728–737. doi: 10.1183/09031936.00021915. [DOI] [PubMed] [Google Scholar]
- 9.Wright L M, Dwyer N, Celermajer D, Kritharides L, Marwick T H. Follow-up of pulmonary hypertension with echocardiography. JACC Cardiovasc Imaging. 2016;9(06):733–746. doi: 10.1016/j.jcmg.2016.02.022. [DOI] [PubMed] [Google Scholar]
- 10.Gorcsan J, III, Edwards T D, Ziady G M, Katz W E, Griffith B P. Transesophageal echocardiography to evaluate patients with severe pulmonary hypertension for lung transplantation. Ann Thorac Surg. 1995;59(03):717–722. doi: 10.1016/0003-4975(94)01054-4. [DOI] [PubMed] [Google Scholar]
- 11.Wang M, Ma R, Wu D et al. Value of lung perfusion scintigraphy in patients with idiopathic pulmonary arterial hypertension: a patchy pattern to consider. Pulm Circ. 2019;9(01):2.045894018816968E15. doi: 10.1177/2045894018816968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.van Wolferen S A, Marcus J T, Boonstra A et al. Prognostic value of right ventricular mass, volume, and function in idiopathic pulmonary arterial hypertension. Eur Heart J. 2007;28(10):1250–1257. doi: 10.1093/eurheartj/ehl477. [DOI] [PubMed] [Google Scholar]
- 13.van de Veerdonk M C, Kind T, Marcus J T et al. Progressive right ventricular dysfunction in patients with pulmonary arterial hypertension responding to therapy. J Am Coll Cardiol. 2011;58(24):2511–2519. doi: 10.1016/j.jacc.2011.06.068. [DOI] [PubMed] [Google Scholar]
- 14.Grignola J C. Hemodynamic assessment of pulmonary hypertension. World J Cardiol. 2011;3(01):10–17. doi: 10.4330/wjc.v3.i1.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Naeije R, Vachiery J-L, Yerly P, Vanderpool R. The transpulmonary pressure gradient for the diagnosis of pulmonary vascular disease. Eur Respir J. 2013;41(01):217–223. doi: 10.1183/09031936.00074312. [DOI] [PubMed] [Google Scholar]
- 16.Ascha M, Renapurkar R D, Tonelli A R. A review of imaging modalities in pulmonary hypertension. Ann Thorac Med. 2017;12(02):61–73. doi: 10.4103/1817-1737.203742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Peña E, Dennie C, Veinot J, Muñiz S H. Pulmonary hypertension: how the radiologist can help. Radiographics. 2012;32(01):9–32. doi: 10.1148/rg.321105232. [DOI] [PubMed] [Google Scholar]
- 18.Farber H W, Loscalzo J. Pulmonary arterial hypertension. N Engl J Med. 2004;351(16):1655–1665. doi: 10.1056/NEJMra035488. [DOI] [PubMed] [Google Scholar]
- 19.Prins K W, Thenappan T. World Health Organization Group I pulmonary hypertension: epidemiology and pathophysiology. Cardiol Clin. 2016;34(03):363–374. doi: 10.1016/j.ccl.2016.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lan N SH, Massam B D, Kulkarni S S, Lang C C. Pulmonary arterial hypertension: pathophysiology and treatment. Diseases. 2018;6(02):E38. doi: 10.3390/diseases6020038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ryan J J, Archer S L. The right ventricle in pulmonary arterial hypertension: disorders of metabolism, angiogenesis and adrenergic signaling in right ventricular failure. Circ Res. 2014;115(01):176–188. doi: 10.1161/CIRCRESAHA.113.301129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tedford R J, Mudd J O, Girgis R E et al. Right ventricular dysfunction in systemic sclerosis-associated pulmonary arterial hypertension. Circ Heart Fail. 2013;6(05):953–963. doi: 10.1161/CIRCHEARTFAILURE.112.000008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Giaid A, Saleh D. Reduced expression of endothelial nitric oxide synthase in the lungs of patients with pulmonary hypertension. N Engl J Med. 1995;333(04):214–221. doi: 10.1056/NEJM199507273330403. [DOI] [PubMed] [Google Scholar]
- 24.Christman B W, McPherson C D, Newman J H et al. An imbalance between the excretion of thromboxane and prostacyclin metabolites in pulmonary hypertension. N Engl J Med. 1992;327(02):70–75. doi: 10.1056/NEJM199207093270202. [DOI] [PubMed] [Google Scholar]
- 25.Tuder R M, Cool C D, Geraci M W et al. Prostacyclin synthase expression is decreased in lungs from patients with severe pulmonary hypertension. Am J Respir Crit Care Med. 1999;159(06):1925–1932. doi: 10.1164/ajrccm.159.6.9804054. [DOI] [PubMed] [Google Scholar]
- 26.Stewart D J, Levy R D, Cernacek P, Langleben D. Increased plasma endothelin-1 in pulmonary hypertension: marker or mediator of disease? Ann Intern Med. 1991;114(06):464–469. doi: 10.7326/0003-4819-114-6-464. [DOI] [PubMed] [Google Scholar]
- 27.Hervé P, Launay J M, Scrobohaci M L et al. Increased plasma serotonin in primary pulmonary hypertension. Am J Med. 1995;99(03):249–254. doi: 10.1016/s0002-9343(99)80156-9. [DOI] [PubMed] [Google Scholar]
- 28.Marcos E, Adnot S, Pham M H et al. Serotonin transporter inhibitors protect against hypoxic pulmonary hypertension. Am J Respir Crit Care Med. 2003;168(04):487–493. doi: 10.1164/rccm.200210-1212OC. [DOI] [PubMed] [Google Scholar]
- 29.Abenhaim L, Moride Y, Brenot F et al. Appetite-suppressant drugs and the risk of primary pulmonary hypertension. N Engl J Med. 1996;335(09):609–616. doi: 10.1056/NEJM199608293350901. [DOI] [PubMed] [Google Scholar]
- 30.Eddahibi S, Humbert M, Fadel E et al. Serotonin transporter overexpression is responsible for pulmonary artery smooth muscle hyperplasia in primary pulmonary hypertension. J Clin Invest. 2001;108(08):1141–1150. doi: 10.1172/JCI12805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Loscalzo J. Genetic clues to the cause of primary pulmonary hypertension. N Engl J Med. 2001;345(05):367–371. doi: 10.1056/NEJM200108023450511. [DOI] [PubMed] [Google Scholar]
- 32.Best D H, Austin E D, Chung W K, Elliott C G. Genetics of pulmonary hypertension. Curr Opin Cardiol. 2014;29(06):520–527. doi: 10.1097/HCO.0000000000000105. [DOI] [PubMed] [Google Scholar]
- 33.Battle R W, Davitt M A, Cooper S M et al. Prevalence of pulmonary hypertension in limited and diffuse scleroderma. Chest. 1996;110(06):1515–1519. doi: 10.1378/chest.110.6.1515. [DOI] [PubMed] [Google Scholar]
- 34.Minter K R, Gladwin M T. Pulmonary complications of sickle cell anemia. A need for increased recognition, treatment, and research. Am J Respir Crit Care Med. 2001;164(11):2016–2019. doi: 10.1164/ajrccm.164.11.2104101. [DOI] [PubMed] [Google Scholar]
- 35.Platt O S, Brambilla D J, Rosse W F et al. Mortality in sickle cell disease. Life expectancy and risk factors for early death. N Engl J Med. 1994;330(23):1639–1644. doi: 10.1056/NEJM199406093302303. [DOI] [PubMed] [Google Scholar]
- 36.Gabler N B, French B, Strom B L et al. Validation of 6-minute walk distance as a surrogate end point in pulmonary arterial hypertension trials. Circulation. 2012;126(03):349–356. doi: 10.1161/CIRCULATIONAHA.112.105890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wensel R, Francis D P, Meyer F J et al. Incremental prognostic value of cardiopulmonary exercise testing and resting haemodynamics in pulmonary arterial hypertension. Int J Cardiol. 2013;167(04):1193–1198. doi: 10.1016/j.ijcard.2012.03.135. [DOI] [PubMed] [Google Scholar]
- 38.Warwick G, Thomas P S, Yates D H. Biomarkers in pulmonary hypertension. Eur Respir J. 2008;32(02):503–512. doi: 10.1183/09031936.00160307. [DOI] [PubMed] [Google Scholar]
- 39.Jaïs X, Olsson K M, Barbera J A et al. Pregnancy outcomes in pulmonary arterial hypertension in the modern management era. Eur Respir J. 2012;40(04):881–885. doi: 10.1183/09031936.00141211. [DOI] [PubMed] [Google Scholar]
- 40.Thorne S, Nelson-Piercy C, MacGregor A et al. Pregnancy and contraception in heart disease and pulmonary arterial hypertension. J Fam Plann Reprod Health Care. 2006;32(02):75–81. doi: 10.1783/147118906776276486. [DOI] [PubMed] [Google Scholar]
- 41.Cohn J N. Optimal diuretic therapy for heart failure. Am J Med. 2001;111(07):577. doi: 10.1016/s0002-9343(01)00915-9. [DOI] [PubMed] [Google Scholar]
- 42.Roberts D H, Lepore J J, Maroo A, Semigran M J, Ginns L C. Oxygen therapy improves cardiac index and pulmonary vascular resistance in patients with pulmonary hypertension. Chest. 2001;120(05):1547–1555. doi: 10.1378/chest.120.5.1547. [DOI] [PubMed] [Google Scholar]
- 43.Fuster V, Steele P M, Edwards W D, Gersh B J, McGoon M D, Frye R L. Primary pulmonary hypertension: natural history and the importance of thrombosis. Circulation. 1984;70(04):580–587. doi: 10.1161/01.cir.70.4.580. [DOI] [PubMed] [Google Scholar]
- 44.Sitbon O, Humbert M, Jaïs X et al. Long-term response to calcium channel blockers in idiopathic pulmonary arterial hypertension. Circulation. 2005;111(23):3105–3111. doi: 10.1161/CIRCULATIONAHA.104.488486. [DOI] [PubMed] [Google Scholar]
- 45.Galiè N, Ussia G, Passarelli P, Parlangeli R, Branzi A, Magnani B. Role of pharmacologic tests in the treatment of primary pulmonary hypertension. Am J Cardiol. 1995;75(03):55A–62A. doi: 10.1016/s0002-9149(99)80384-1. [DOI] [PubMed] [Google Scholar]
- 46.Montani D, Savale L, Natali D et al. Long-term response to calcium-channel blockers in non-idiopathic pulmonary arterial hypertension. Eur Heart J. 2010;31(15):1898–1907. doi: 10.1093/eurheartj/ehq170. [DOI] [PubMed] [Google Scholar]
- 47.Rich S, Kaufmann E, Levy P S. The effect of high doses of calcium-channel blockers on survival in primary pulmonary hypertension. N Engl J Med. 1992;327(02):76–81. doi: 10.1056/NEJM199207093270203. [DOI] [PubMed] [Google Scholar]
- 48.Hoeper M M, Apitz C, Grünig E et al. Targeted therapy of pulmonary arterial hypertension: Updated recommendations from the Cologne Consensus Conference 2018. Int J Cardiol. 2018;272S:37–45. doi: 10.1016/j.ijcard.2018.08.082. [DOI] [PubMed] [Google Scholar]
- 49.Valerio C J, Coghlan J G. Bosentan in the treatment of pulmonary arterial hypertension with the focus on the mildly symptomatic patient. Vasc Health Risk Manag. 2009;5:607–619. doi: 10.2147/vhrm.s4713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Channick R N, Simonneau G, Sitbon Oet al. Effects of the dual endothelin-receptor antagonist bosentan in patients with pulmonary hypertension: a randomised placebo-controlled study Lancet 2001358(9288):1119–1123. [DOI] [PubMed] [Google Scholar]
- 51.Rubin L J, Badesch D B, Barst R J et al. Bosentan therapy for pulmonary arterial hypertension. N Engl J Med. 2002;346(12):896–903. doi: 10.1056/NEJMoa012212. [DOI] [PubMed] [Google Scholar]
- 52.Humbert M, Barst R J, Robbins I M et al. Combination of bosentan with epoprostenol in pulmonary arterial hypertension: BREATHE-2. Eur Respir J. 2004;24(03):353–359. doi: 10.1183/09031936.04.00028404. [DOI] [PubMed] [Google Scholar]
- 53.Galiè N, Beghetti M, Gatzoulis M A et al. Bosentan therapy in patients with Eisenmenger syndrome: a multicenter, double-blind, randomized, placebo-controlled study. Circulation. 2006;114(01):48–54. doi: 10.1161/CIRCULATIONAHA.106.630715. [DOI] [PubMed] [Google Scholar]
- 54.Galiè N, Rubin Lj, Hoeper Met al. Treatment of patients with mildly symptomatic pulmonary arterial hypertension with bosentan (EARLY study): a double-blind, randomised controlled trial Lancet 2008371(9630):2093–2100. [DOI] [PubMed] [Google Scholar]
- 55.McLaughlin V, Channick R N, Ghofrani H-A et al. Bosentan added to sildenafil therapy in patients with pulmonary arterial hypertension. Eur Respir J. 2015;46(02):405–413. doi: 10.1183/13993003.02044-2014. [DOI] [PubMed] [Google Scholar]
- 56.Galié N, Badesch D, Oudiz R et al. Ambrisentan therapy for pulmonary arterial hypertension. J Am Coll Cardiol. 2005;46(03):529–535. doi: 10.1016/j.jacc.2005.04.050. [DOI] [PubMed] [Google Scholar]
- 57.Galiè N, Olschewski H, Oudiz R J et al. Ambrisentan for the treatment of pulmonary arterial hypertension: results of the ambrisentan in pulmonary arterial hypertension, randomized, double-blind, placebo-controlled, multicenter, efficacy (ARIES) study 1 and 2. Circulation. 2008;117(23):3010–3019. doi: 10.1161/CIRCULATIONAHA.107.742510. [DOI] [PubMed] [Google Scholar]
- 58.Pulido T, Adzerikho I, Channick R N et al. Macitentan and morbidity and mortality in pulmonary arterial hypertension. N Engl J Med. 2013;369(09):809–818. doi: 10.1056/NEJMoa1213917. [DOI] [PubMed] [Google Scholar]
- 59.Galiè N, Ghofrani H A, Torbicki A et al. Sildenafil citrate therapy for pulmonary arterial hypertension. N Engl J Med. 2005;353(20):2148–2157. doi: 10.1056/NEJMoa050010. [DOI] [PubMed] [Google Scholar]
- 60.Rubin L J, Badesch D B, Fleming T R et al. Long-term treatment with sildenafil citrate in pulmonary arterial hypertension: the SUPER-2 study. Chest. 2011;140(05):1274–1283. doi: 10.1378/chest.10-0969. [DOI] [PubMed] [Google Scholar]
- 61.Iversen K, Jensen A S, Jensen T V, Vejlstrup N G, Søndergaard L. Combination therapy with bosentan and sildenafil in Eisenmenger syndrome: a randomized, placebo-controlled, double-blinded trial. Eur Heart J. 2010;31(09):1124–1131. doi: 10.1093/eurheartj/ehq011. [DOI] [PubMed] [Google Scholar]
- 62.Galiè N, Brundage B H, Ghofrani H A et al. Tadalafil therapy for pulmonary arterial hypertension. Circulation. 2009;119(22):2894–2903. doi: 10.1161/CIRCULATIONAHA.108.839274. [DOI] [PubMed] [Google Scholar]
- 63.Oudiz R J, Brundage B H, Galiè N et al. Tadalafil for the treatment of pulmonary arterial hypertension: a double-blind 52-week uncontrolled extension study. J Am Coll Cardiol. 2012;60(08):768–774. doi: 10.1016/j.jacc.2012.05.004. [DOI] [PubMed] [Google Scholar]
- 64.Jing Z-C, Yu Z-X, Shen J-Y et al. Vardenafil in pulmonary arterial hypertension: a randomized, double-blind, placebo-controlled study. Am J Respir Crit Care Med. 2011;183(12):1723–1729. doi: 10.1164/rccm.201101-0093OC. [DOI] [PubMed] [Google Scholar]
- 65.Ghofrani H-A, Galiè N, Grimminger F et al. Riociguat for the treatment of pulmonary arterial hypertension. N Engl J Med. 2013;369(04):330–340. doi: 10.1056/NEJMoa1209655. [DOI] [PubMed] [Google Scholar]
- 66.Rubin L J, Galiè N, Grimminger F et al. Riociguat for the treatment of pulmonary arterial hypertension: a long-term extension study (PATENT-2) Eur Respir J. 2015;45(05):1303–1313. doi: 10.1183/09031936.00090614. [DOI] [PubMed] [Google Scholar]
- 67.Sitbon O, Delcroix M, Bergot E et al. EPITOME-2: An open-label study assessing the transition to a new formulation of intravenous epoprostenol in patients with pulmonary arterial hypertension. Am Heart J. 2014;167(02):210–217. doi: 10.1016/j.ahj.2013.08.007. [DOI] [PubMed] [Google Scholar]
- 68.Barst R J, Rubin L J, Long W A et al. A comparison of continuous intravenous epoprostenol (prostacyclin) with conventional therapy for primary pulmonary hypertension. N Engl J Med. 1996;334(05):296–301. doi: 10.1056/NEJM199602013340504. [DOI] [PubMed] [Google Scholar]
- 69.Sitbon O, Humbert M, Nunes H et al. Long-term intravenous epoprostenol infusion in primary pulmonary hypertension: prognostic factors and survival. J Am Coll Cardiol. 2002;40(04):780–788. doi: 10.1016/s0735-1097(02)02012-0. [DOI] [PubMed] [Google Scholar]
- 70.Rubin L J, Mendoza J, Hood M et al. Treatment of primary pulmonary hypertension with continuous intravenous prostacyclin (epoprostenol). Results of a randomized trial. Ann Intern Med. 1990;112(07):485–491. doi: 10.7326/0003-4819-112-7-485. [DOI] [PubMed] [Google Scholar]
- 71.Badesch D B, Tapson V F, McGoon M D et al. Continuous intravenous epoprostenol for pulmonary hypertension due to the scleroderma spectrum of disease. A randomized, controlled trial. Ann Intern Med. 2000;132(06):425–434. doi: 10.7326/0003-4819-132-6-200003210-00002. [DOI] [PubMed] [Google Scholar]
- 72.Doran A K, Ivy D D, Barst R J, Hill N, Murali S, Benza R L; Scientific Leadership Council of the Pulmonary Hypertension Association.Guidelines for the prevention of central venous catheter-related blood stream infections with prostanoid therapy for pulmonary arterial hypertension Int J Clin Pract Suppl 20081605–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Barst R J, McGoon M, McLaughlin V et al. Beraprost therapy for pulmonary arterial hypertension. J Am Coll Cardiol. 2003;41(12):2119–2125. doi: 10.1016/s0735-1097(03)00463-7. [DOI] [PubMed] [Google Scholar]
- 74.Hoeper M M, Gall H, Seyfarth H J et al. Long-term outcome with intravenous iloprost in pulmonary arterial hypertension. Eur Respir J. 2009;34(01):132–137. doi: 10.1183/09031936.00130408. [DOI] [PubMed] [Google Scholar]
- 75.Olschewski H, Simonneau G, Galiè N et al. Inhaled iloprost for severe pulmonary hypertension. N Engl J Med. 2002;347(05):322–329. doi: 10.1056/NEJMoa020204. [DOI] [PubMed] [Google Scholar]
- 76.Simonneau G, Barst R J, Galie N et al. Continuous subcutaneous infusion of treprostinil, a prostacyclin analogue, in patients with pulmonary arterial hypertension: a double-blind, randomized, placebo-controlled trial. Am J Respir Crit Care Med. 2002;165(06):800–804. doi: 10.1164/ajrccm.165.6.2106079. [DOI] [PubMed] [Google Scholar]
- 77.White R J, Levin Y, Wessman K, Heininger A, Frutiger K. Subcutaneous treprostinil is well tolerated with infrequent site changes and analgesics. Pulm Circ. 2013;3(03):611–621. doi: 10.1086/674304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Hiremath J, Thanikachalam S, Parikh K et al. Exercise improvement and plasma biomarker changes with intravenous treprostinil therapy for pulmonary arterial hypertension: a placebo-controlled trial. J Heart Lung Transplant. 2010;29(02):137–149. doi: 10.1016/j.healun.2009.09.005. [DOI] [PubMed] [Google Scholar]
- 79.McLaughlin V V, Benza R L, Rubin L J et al. Addition of inhaled treprostinil to oral therapy for pulmonary arterial hypertension: a randomized controlled clinical trial. J Am Coll Cardiol. 2010;55(18):1915–1922. doi: 10.1016/j.jacc.2010.01.027. [DOI] [PubMed] [Google Scholar]
- 80.Jing Z-C, Parikh K, Pulido T et al. Efficacy and safety of oral treprostinil monotherapy for the treatment of pulmonary arterial hypertension: a randomized, controlled trial. Circulation. 2013;127(05):624–633. doi: 10.1161/CIRCULATIONAHA.112.124388. [DOI] [PubMed] [Google Scholar]
- 81.Tapson V F, Torres F, Kermeen F et al. Oral treprostinil for the treatment of pulmonary arterial hypertension in patients on background endothelin receptor antagonist and/or phosphodiesterase type 5 inhibitor therapy (the FREEDOM-C study): a randomized controlled trial. Chest. 2012;142(06):1383–1390. doi: 10.1378/chest.11-2212. [DOI] [PubMed] [Google Scholar]
- 82.Tapson V F, Jing Z-C, Xu K-F et al. Oral treprostinil for the treatment of pulmonary arterial hypertension in patients receiving background endothelin receptor antagonist and phosphodiesterase type 5 inhibitor therapy (the FREEDOM-C2 study): a randomized controlled trial. Chest. 2013;144(03):952–958. doi: 10.1378/chest.12-2875. [DOI] [PubMed] [Google Scholar]
- 83.Sitbon O, Channick R, Chin K M et al. Selexipag for the treatment of pulmonary arterial hypertension. N Engl J Med. 2015;373(26):2522–2533. doi: 10.1056/NEJMoa1503184. [DOI] [PubMed] [Google Scholar]
- 84.Galiè N, Palazzini M, Manes A. Pulmonary arterial hypertension: from the kingdom of the near-dead to multiple clinical trial meta-analyses. Eur Heart J. 2010;31(17):2080–2086. doi: 10.1093/eurheartj/ehq152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Galiè N, Barberà J A, Frost A E et al. Initial use of Ambrisentan plus Tadalafil in pulmonary arterial hypertension. N Engl J Med. 2015;373(09):834–844. doi: 10.1056/NEJMoa1413687. [DOI] [PubMed] [Google Scholar]
- 86.Prior D L, Adams H, Williams T J. Update on pharmacotherapy for pulmonary hypertension. Med J Aust. 2016;205(06):271–276. doi: 10.5694/mja16.00468. [DOI] [PubMed] [Google Scholar]
- 87.Yoon K L. New therapeutic target for pulmonary arterial hypertension. Korean Circ J. 2018;48(12):1145–1147. doi: 10.4070/kcj.2018.0250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Marcu R, Kotha S, Zhi Z et al. The mitochondrial permeability transition pore regulates endothelial bioenergetics and angiogenesis. Circ Res. 2015;116(08):1336–1345. doi: 10.1161/CIRCRESAHA.116.304881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Pullamsetti S S, Savai R, Seeger W, Goncharova E A. Translational advances in the field of pulmonary hypertension. from cancer biology to new pulmonary arterial hypertension therapeutics. targeting cell growth and proliferation signaling hubs. Am J Respir Crit Care Med. 2017;195(04):425–437. doi: 10.1164/rccm.201606-1226PP. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Satoh K, Kikuchi N, Satoh T et al. Identification of novel therapeutic targets for pulmonary arterial hypertension. Int J Mol Sci. 2018;19(12):4081. doi: 10.3390/ijms19124081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Noda K, Nakajima S, Godo S et al. Rho-kinase inhibition ameliorates metabolic disorders through activation of AMPK pathway in mice. PLoS One. 2014;9(11):e110446. doi: 10.1371/journal.pone.0110446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Satoh K, Satoh T, Kikuchi N et al. Basigin mediates pulmonary hypertension by promoting inflammation and vascular smooth muscle cell proliferation. Circ Res. 2014;115(08):738–750. doi: 10.1161/CIRCRESAHA.115.304563. [DOI] [PubMed] [Google Scholar]
- 93.Chang S A, Kim H K, Chang H J, Kim D K. Acute hemodynamic changes after single administration of udenafil in pulmonary arterial hypertension: a phase IIa study. Korean Circ J. 2019;49(04):353–360. doi: 10.4070/kcj.2018.0281. [DOI] [PMC free article] [PubMed] [Google Scholar]