

ABSTRACT
The role of inflammation response has been well documented in the development of acute lung injury (ALI). However, little is known about the functions of miRNAs in the regulation of inflammation in ALI. The aim of this study was to explore the effects of miRNAs in the regulation of inflammation in ALI and to elucidate the biomolecular mechanisms responsible for these effects. The expression profiles of miRNAs in lung tissues from lipopolysaccharide (LPS)-induced ALI mice model were analyzed using a microarray. It was observed that microRNA-221-3p (miR-221) was significantly increased in lung tissues in ALI mice. The inhibition of miR-221 attenuated lung injury including decreased lung W/D weight ratio and lung permeability and survival rates of ALI mice, as well as apoptosis, whereas its agomir-mediated upregulation exacerbated the lung injury. Concomitantly, miR-221 inhibition significantly reduced LPS-induced pulmonary inflammation, while LPS-induced pulmonary inflammation was aggravated by miR-221 upregulation. Of note, suppressor of cytokine signaling-1 (SOCS1), an effective suppressor of the NF-κB signaling pathway, was found to be a direct target of miR-221 in RAW264.7 cells. Overexpression of SOCS1 by pcDNA-SOCS1 plasmids markedly reversed the miR-221 inhibition-mediated inhibitory effects on inflammation and apoptosis in LPS-treated RAW264.7 cells. Finally, it was found that miR-221 inhibition suppressed LPS induced the activation of the NF-κB signaling pathway, as demonstrated by downregulation of phosphorylated-IκBα, p-p65 and upregulation of IκBα, whilst miR-221 overexpression had an opposite result in ALI mice. Our findings demonstrate that inhibition of miR-221 can alleviate LPS-induced inflammation via inactivation of SOCS1/NF-κB signaling pathway in ALI mice.
KEYWORDS: Acute lung injury, microRNA-221, inflammation, SOCS1, NF-κB signaling pathway
Introduction
Acute lung injury (ALI) is a common critical condition in patients with sepsis or infection, and it mainly characterized with an excessive-uncontrolled inflammatory response [1–5]. Although several available therapies have been applied to cure ALI such as nitric oxide, surfactant, glucocorticoids, the morbidity and mortality in critically ill patient remain high [6,7]. Therefore, it was an urgent need for the development of novel and effective treatments.
Figure 7.
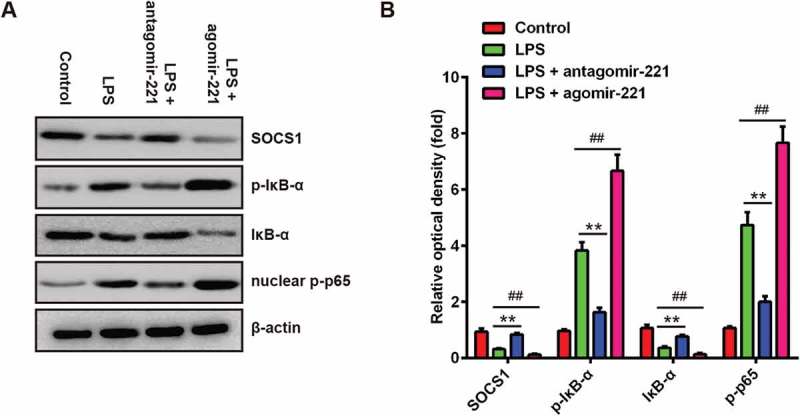
The effects of abnormally expressed miR-221 on SOCS1/NF-κB pathway in LPS-induced ALI mice. Mice were injected intravenously (tail vein) with agomir-221, antagomir-221, control antagomir or control agomir (2 mg/kg) 24 h prior to 2 mg/kg LPS. The mice were sacrificed at 24 h after LPS administration and then lung tissues were collected for analysis. (a) The levels of SOCS1, IκB-α, p-IκB-α and nuclear p-p65 were measured by Western Blot (n = 3). (b) The bands were semi-quantitatively analyzed by using Image J software, normalized to β-actin density. Data represent the mean ± SD of three independent experiments. **p < 0.01 vs. control group. ##p < 0.01 vs. LPS alone group.
Increasing evidence has confirmed that inflammatory response plays an important role in the pathogenesis of ALI [8]. Once activated, the inflammatory cell including neutrophils and macrophages in the lung tissue produces large amounts of pro-inflammatory cytokines such as tumor necrosis factor-α (TNF-α), interleukin (IL)-1β and IL-6 that increase the capillary permeability leading to tissue edema and exacerbation of tissue injury [9–12]. In ALI patients, the persistent elevation of pro-inflammatory cytokines always predicts a poor outcome [13]. Thus, reduction of inflammatory response could be a benefit for the treatment of ALI.
MicroRNAs (miRNAs) are a class of highly conserved, single-stranded, short noncoding RNAs that suppress the expression of protein-coding genes at the posttranscriptional level [14]. Emerging evidence reveals that miRNAs play a major role in the amelioration of various inflammatory diseases, including ALI [15–17]. Recent studies in mouse ALI model showed that miRNA expression profiles are altered after ALI, and modulation of miRNA expression can affect the development of ALI [16,18,19]. For example, Guo et al. showed that enforced expression of miR-125b was able to ameliorate the LPS-induced ALI [20]. Li et al. found that miR-181a inhibition protected mice against LPS-induced ALI by targeting Bcl-2 [16]. However, the regulations of the inflammatory response by miRNAs in ALI have rarely been reported.
In the present study, we conducted a miRNA microarray to define the miRNA pattern in ALI mice, and miR-221 attracted our attention. Subsequently, using a mouse model of ALI, the role of miR-221 in inflammation in ALI was examined, and the role of the SOCS1/NF-κB signaling pathway in the protective activity of miR-221 against ALI was further confirmed. These results suggest that miR-221 may serve as a potential therapeutic target for the treatment of ALI.
Materials and methods
Animals
Male BALB/c mice (18–22 g) were purchased from The Third Affiliated Hospital of Zhengzhou University (Zhengzhou, China). All surgical and care procedures were finished in accordance with the guide for the international guidelines on the ethical use of animals. All mice were housed in a specific pathogen-free condition at 22 ℃ ± 2 ℃ on 12 h light/dark cycle.
In miRNA microarray analysis, mice were randomly divided into two groups, namely control group (saline) and ALI group (n = 3/group). A control group with intra-tracheal instillation of 2 mg/kg normal saline (NS) and an ALI group with intra-tracheal instillation of 2 mg/kg LPS [21] (Escherichia coli 055:B5; Sigma). Mice were humanely killed at 24 h by an overdose of anesthesia after LPS treatment. Three pairs of lung tissue samples were subjected to miRNAs identification using miRNA microarray analysis.
miR-221 antagomir (antagomir-221) (5ʹ-GAAACCCAGCAGACAAUGUAGCU-3ʹ) and miR-221 agomir (agomir-221) (5ʹ-CAAACCCAGCAGACAAUGUAGCU-3ʹ) experiments. The mice were randomly divided into six groups (n = 16/group): (1) Control group (saline), (2) LPS group (3) LPS + agomir-221 group, (4) LPS + agomir-NC group, (5) LPS + antagomir-221 group, (6) LPS + antagomir-NC group. Mice were injected intravenously (tail vein) with agomir-221, antagomir-221, control antagomir or control agomir (2 mg/kg, Ribobio, Guangzhou, China) 24 h prior to 2 mg/kg LPS. Another 16 mice treated with saline were used as controls. Mice were humanely killed at 24 h by an overdose of anesthesia after LPS treatment except for survival experiment. Lung tissue samples, splenocytes and the bronchoalveolar lavage fluid (BALF) were collected for subsequent analysis. Whole blood from mice was collected in Heparin containing DNase/RNase-free tubes through the tail vein.
Microarray analysis
Total RNA was isolated from LPS-infected lung tissues collected from the sacrificed mice using the miRNeasy mini kit (QIAGEN). After passing the RNA quantity measurement using the NanoDrop 1000, the samples were labeled using the miRCURY™ Hy3™/Hy5™ power labeling kit and hybridized on the miRCURY™ LNA Array (v18.0). The slides were scanned using the Axon GenePix 4000B microarray scanner. After grid alignment and data extraction using GenePix Pro 6.0 software (Axon), differentially expressed miRNAs were then identified through Fold Change filtering (Fold Change >2.0). Finally, hierarchical clustering was performed to show distinguishable miRNA expression profiling among samples.
Quantitative real-time PCR analysis
Total RNA was isolated from tissues or cells using the miRNeasy mini kit (QIAGEN) following the manufacturer’s protocol. For microRNA analysis, cDNA conversion was carried using miRNA specific stem-loop primers (Applied Biosystems, Foster City, CA, USA). For mRNA, 1.0 μg total RNA was reverse transcribed into cDNA using a PrimeScript RT Reagent Kit (Takara Bio, China) according to the manufacturer’s protocol. qPCR was performed using a standard protocol from the SYBR Green PCR kit (Toyobo, Osaka, Japan) on an ABI PRISM 7500 Real-time PCR system (Life Technologies, USA). U6 and GAPDH were used as references for miRNAs and mRNAs, respectively. The primer sequences were as follows: miR-211, forward 5ʹ-GTTCGTGGGAGCTACATTGTCTGC-3ʹ and re-verse 5ʹ-GTGCAGGGTCCGAGGT-3ʹ; U6 forward: 5ʹ-GCTTCGGCAGCACATATACTAAAAT-3ʹ, re-verse: 5ʹCGCTTCACGAATTTGCGTGTCAT-3ʹ; SOCS1 forward: 5ʹ-CTGGGATGCCGTGTTATTT-3ʹ, reverse: 5ʹ-TAGGAGGTGCGAGTTCAGGT-3ʹ GAPDH forward: 5ʹ-AGGTCGGTGTGAACGGATTTG-3ʹ, reverse: 5ʹ-TGTAGACCATGTAGTTGAGGTCA-3ʹ. Data were calculated using the 2−ΔΔCT method [22].
Evaluation of lung permeability
Lung permeability was assessed using the Evans blue dye extravasation method, as described in a previous study [23]. In brief, using the tail vein, 4 mL/kg of 2% Evans blue (Sigma-Aldrich, St. Louis, MO, USA) in normal saline was injected 10 h before the animals were euthanized. After adequate perfusion with normal saline, Evans blue dye was extracted from the lung using formamide for 18 h at 60°C and measured as the absorbance of the supernatant at 620 nm on a microplate reader (BioTek, Winooski, Vermont, United States) and was reported as the amount of EB per wet tissue weight (μg/g).
Lung wet/dry (W/D) ratio
Mice were sacrificed, the right lungs were excised, and the lung tissues were placed in an incubator at 80°C for 48 h to obtain the dry weight. The ratio of the wet lung to dry lung was calculated to assess tissue edema.
Histopathologic evaluation of lung tissues
Upper and lower lobe lung samples were excised 24 h after LPS challenge, fixed with 10% formalin and microsectioned at 5 μm. Then, the tissues embedded in paraffin and stained with hematoxylin and eosin. Finally, lung injury score was assessed in a blinded fashion using a semi-quantitative light microscopy evaluation as previously described [24]. The images were captured by Nikon E100 microscope at ×400 magnification.
Terminal deoxynucleotidyl transferase (TdT)-mediated dUTP nickend labeling (TUNEL) assay
For TUNEL staining, fluorescence staining was performed using a commercially available in situ Cell Death Detection Kit (Roche Diagnostics, Indianapolis, IN) according to the manufacturer’s instructions. Results are expressed as the average number of TUNEL-positive staining cells per 400 × magnification field. Fluorescence images were obtained on a Nikon E100 microscope. Quantification of TUNEL-positive cells was done by determining the percentage of TUNEL-positive (green) cells in multiple high-power fields (n = 3 sections per mouse strain and treatment group). Each sample was measured in triplicate.
Bronchoalveolar lavage fluid (BALF) collection and analysis
BALF was collected 24 h after LPS treatment from all groups as described in a previous study [25]. The BALF was centrifuged 1500 rpm for 10 min at 4°C, and the supernatant aliquots were separated and preserved at −80°C for further cytokine quantification. The sedimented cells were re-suspended in PBS for analyzing the number of cells and neutrophils with the help of a light microscope, using Wright–Giemsa method employing hemocytometer.
Measurement of pro-inflammatory cytokines and chemokines
The levels of TNF-α, IL-6, and IL-1β in BALF were measured using sandwich ELISA kits according to the manufacturer’s instructions protocol (BioLegend, CA, USA). Chemokine levels including KC (the murine homolog of human IL-8) in BALF were measured using cytokine-specific bead kits (R&D Systems).
Cell culture and treatment
The RAW 264.7 macrophages are widely used in a model of LPS-induced ALI, and macrophages play an essential role in the regulation of inflammation responses [26,27]. RAW 264.7 cells were purchased from the American Type Culture Collection (Manassas, VA, USA) and cultured in DMEM/F12 (Gibco BRL, Grand Island, NY, USA) supplemented with 10% FBS (Invitrogen, Carlsbad, CA, USA) at 37℃ in a humidified atmosphere with 5% CO2. For LPS treatments, cells were plated in 12-well plates (1 × 106 cells/well) and were cultured at 37°C for 24 h. Then, cells were exposed to 0, 0.1, 1, 2 and 10 μg/ml LPS for 5 h.
Cell transfection
miR-221 mimics (5ʹ-AGCUACAUUGUCUGCUGGGUUUC-3ʹ), miR-221 inhibitor (5ʹ-GAAACCCAGCAGACAAGUAGCU-3ʹ), and the respective negative controls (NCs) i.e. mimics NC and inhibitors NC, si-SOCS1, were chemically synthesized from GenePharma Co., Ltd. (Shanghai, China). RAW 264.7 cells in logarithmic phase were transfected with miRNAs, si-SOCS1 and their controls using Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s instructions. Post-transfection, the cells were stimulated with LPS (1 μg/mL) for 24 h and then utilized in subsequent experiments.
Cell viability assay
RAW 264.7 cells were seeded in 96-well plate (5000 cells/well) and treated with LPS for 5 h. Then, viability was assessed by Cell Counting Kit-8 assay (CCK-8, Dojindo Molecular Technologies, Gaithersburg, MD, USA). Briefly, after treatment, CCK-8 solution (10 μl) was added into the culture medium followed by incubation at 37°C in humidified 5% CO2 for 1 h. After that, the optical density of each sample at 450 nm was measured using a Microplate Reader (Bio-Rad, Hercules, CA, USA).
Cell apoptosis assay
After 48 h transfection in RAW 264.7 cells, apoptosis assay was performed with Annexin V-FITC Apoptosis Detection Kit (Abcam, Cambridge, UK) according to the manufacturer’s instructions. The cells were harvested and washed twice with PBS, then stained with Annexin V and PI. After incubation for 15 min at room temperature in the dark, cell apoptosis was analyzed on a FACScan flow cytometer (FCM; Bechman Coulter, CA) and then analyzed by FlowJo 8.7.1 software (Ashland, OR).
Luciferase assay
The 3´-UTR of SOCS1, with wild-type or mutant (Mut) binding sites for miR-221, was amplified and cloned into the pGL3 vector (Promega, Madison, WI, USA) to generate the plasmid pGL3-WT-SOCS1-3´-UTR or pGL3-Mut-SOCS1-3´-UTR, respectively. For the luciferase reporter assay, RAW 264.7 cells were co-transfected with the luciferase reporter vectors and miR-221 mimics, miR-221 inhibitor or corresponding negative control (100 nM, Genecopoeia, Guangzhou) using Lipofectamine 2000 reagent. The pRL-TK plasmid (5 ng, Promega, Madison, USA) was used as a normalizing control. After 24 h of incubation, luciferase activity was analyzed using the Dual-Luciferase Reporter Assay System (Promega) according to the manufacturer’s protocol.
Western blot
Lung tissue samples or cells were lysed in a RIPA buffer (Cell Signaling Technology TDanvers, MA, USA) with protease and phosphatase inhibitors (Roche, Indianapolis, IN, USA) for 30 min. Nuclear protein from cells was extracted using Cytoplasmic and Nuclear Protein Extraction Kit (cat no. 78,833, ThermoFisher Scientific, USA). The protein concentrations were measured using a BCA protein assay kit (Beyotime, China), and 40 μg of proteins was electrophoretically transferred onto a PVDF membrane, followed by blocking with 5% non-fat milk for 1 h. Membranes were incubated with primary antibodies against SOCS1 (cat no.ab9870, 1:2000), nuclear-p65 (cat no.ab86299, 1:2000), p-IκBα (cat no.ab133462, 1:5000), IκBα (cat no.ab32518, 1:5000) and β-actin (cat no.ab8226, 1:2000) (all from Abcam, Cambridge) at 4°C overnight. After washing, the membrane was incubated with appropriate horseradish peroxidase (HRP)-conjugated secondary antibodies including goat anti-rabbit IgG (cat no.ab6721, 1:5000) and goat anti-mouse IgG (cat no.ab6789, 1:5000) for 1 h at room temperature. A chemiluminescence detection system (Millipore, Billerica, MA, USA) was used for visualization of the results and quantification of the bands was performed using Quantity One software (Bio-Rad, Hercules, CA, USA).
Statistical analysis
Statistical analysis was performed using GraphPad Prism 5.0 (GraphPad Software, Inc., San Diego, CA, USA). Data are presented as the means ± standard deviation (SD). Statistical differences were analyzed using the Student’s t-test or one-way analysis of variance (ANOVA) with the Tukey’s test. P values < 0.05 were considered significant.
Results
The expression of pulmonary miRNA in ALI mice
In order to identify miRNAs associated with ALI, we conducted a miRNA expression microarray to identify the types of miRNA in a mice model of ALI induced by LPS. The miRNA expression microarray revealed that the number of miRNAs with over a twofold change in expression levels was 57, including 35 upregulated and 22 downregulated miRNAs (Figure 1(a)). Among these aberrant miRNAs, miR-221 (miR-221-3p) was selected for further investigation as its expression level was identified as the highest in the ALI group. Previous studies have reported that miR-221 protects macrophages against the inflammatory response, and are critically involved in the development of inflammatory disorders, including those affecting the lungs [28,29]. However, whether miR-221 has a protective effect on inflammatory response in ALI remains unknown.
Figure 1.
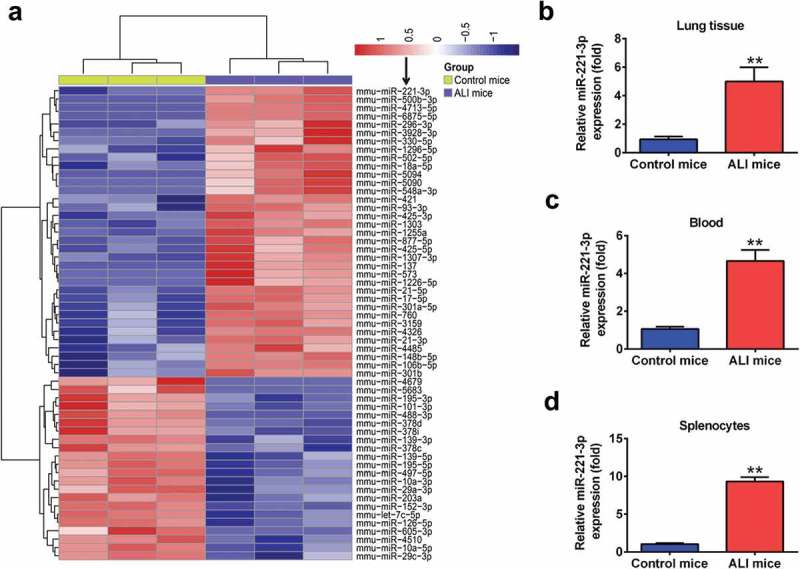
miR-221 was upregulated in LPS-induced ALI in mice. Mice (n = 3 each) were treated with a single dose of LPS (2 mg/kg) or normal saline, and total RNAs were isolated from their lungs at 24 h after the treatment. (a) Heat map of miRNA profiles represented the significantly regulated miRNAs (n = 3/group). (b-d) miR-221 expression was validated by qRT-PCR in lung tissues, peripheral blood and splenocytes of mice challenged with LPS (n = 6/group). Data represent the mean ± SD of three independent experiments. **p < 0.01 vs. control group.
To further validate the results of the microarray study, we determined the expression level of miR-221 in lung tissues of LPS-induced ALI mice at 24 h after LPS challenge. We found that the expression level of miR-221 in lung tissues was significantly increased compared with the control group (Figure 1(b)). Similarly, the expression of miR-221 was also significantly upregulated in peripheral blood and splenocytes in the ALI group, compared with that in control group (Figure 1(c,d)). These results indicated that miR-221 might be involved in the development of ALI.
The effects of miR-221 on LPS-induced injury in vivo
To further investigate the role of miR-221 in ALI, agomir-221 and antagomir-221 were injected into ALI mice. The qRT-PCR analysis showed that the expression of miR-221 in lung tissues markedly increased or decreased by the agomir-221 and antagomir-221, compared with that in the LPS group (Figure 2(a)). Then, the survival rates of mice in different groups were calculated. As shown in Figure 2(b), the survival rate in antagomir-221 group was significantly high, compared with that in LPS group, while the survival rate in the agomir-221 group was even lower than LPS group. The capillary permeability was evaluated by Evans Blue assay and the results showed that the increased capillary permeability induced by LPS was dramatically reduced by antagomir-221, but it was enhanced by agomir-221 (Figure 2(c)). Figure 2(d) shows that LPS treatment led to a significant increase in lung wet to dry (W/D) weight ratio, compared to control mice and this promoting effect by LPS was attenuated by antagomir-221, but was enhanced by agomir-221. Moreover, histopathological results in LPS-induced ALI mice showed excessive cellular infiltration in the alveolar and interstitial spaces of the tissue. However, antagomir-221 injection significantly reduced this LPS-driven cellular infiltration into the lungs, closely resembling lungs from control mice, but agomir-221 aggravated LPS-induced cellular infiltration into the lung (Figure 2(e)). Further, the severity of lung injury was also scored using a semi-quantitative histopathology score system, and we found that antagomir-221 could significantly reduce the lung injury score of LPS-induced ALI mice, whereas agomir-221 could enhance the lung injury score. Overall, these results demonstrated that miR-221 inhibition exerted a protective effect against LPS-induced injuries in mice.
Figure 2.
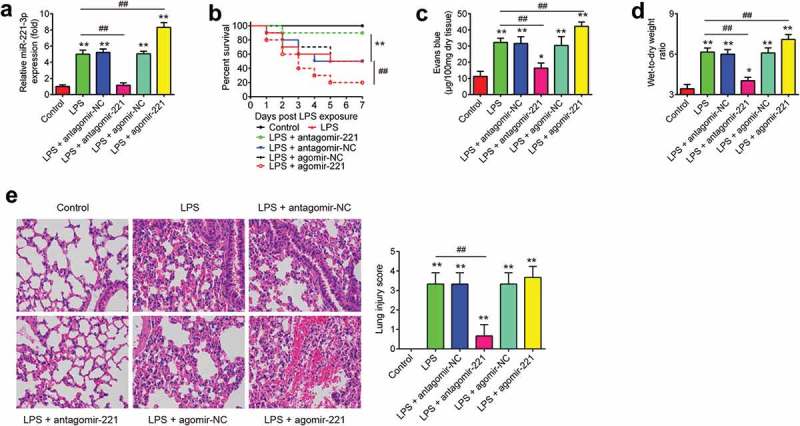
The effects of abnormally expressed miR-221 on LPS-induced injury. Mice were injected intravenously (tail vein) with agomir-221, antagomir-221, control antagomir or control agomir (2 mg/kg) 24 h prior to 2 mg/kg LPS treatment. The mice were sacrificed after LPS administration for 24 h and then lung tissues were collected for analysis. (a) The efficiency of agomir-221 or antagomiR-221 was determined by qRT-PCR. (b) The Kaplan–Meier method was used to compare the survival rates among different groups (n = 10/group). (c) Lung permeability was assessed using the Evans blue dye extravasation method (n = 6/group). (d) The pulmonary edema was evaluated by the lung wet/dry weight ratio (n = 6/group). (e) The degree of injury of lung samples was assessed with H&E staining at ×400 magnification (n = 6/group). *p < 0.05, **p < 0.01 vs. control group. ##p < 0.01 vs. LPS alone group.
The effects of miR-221 on apoptosis in LPS-induced ALI mice
Apoptosis is also one of the critical pathologies that contribute to ALI [30]. To determine the effect of miR-221 on apoptosis in LPS-induced ALI mice, we quantified the number of apoptotic events in the lungs of ALI mice using TUNEL assay. As shown in Figure 3(a), B, LPS treatment showed a significant increase in the number of TUNEL-positive cells. However, antagomir-221 injection resulted in a significant reduction in the number of TUNEL-positive cells, compared to the LPS group, while agomir-221 dramatically enhanced the number of TUNEL-positive cells induced by LPS. These data indicate that inhibition of miR-221 suppressed the apoptosis in LPS-induced ALI mice.
Figure 3.
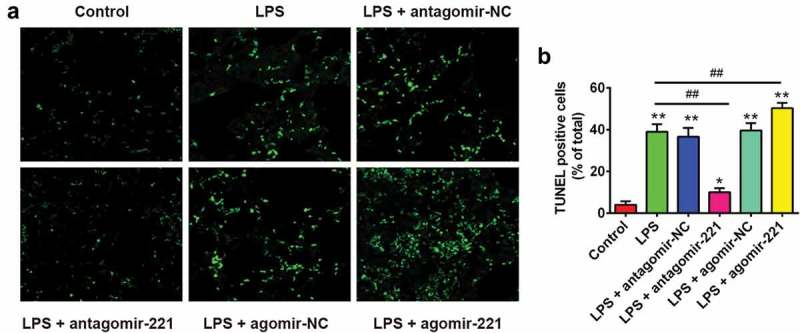
The effects of altered miR-221 expression on apoptosis in LPS-induced ALI mice. Mice were injected intravenously (tail vein) with agomir-221, antagomir-221, control antagomir or control agomir (2 mg/kg) at 24 h prior to 2 mg/kg LPS treatment. The mice were sacrificed after LPS administration for 24 h and then lung tissues were collected for analysis. (a) Apoptotic cells in the tissues were observed using TUNEL assay at ×400 magnification (n = 6/group). (b) The quantity of TUNEL-positive cells was elevated in ALI mice (n = 6/group). Data represent the mean ± SD of three independent experiments. *p < 0.05, **p < 0.01 vs. control group. ##p < 0.01 vs. LPS alone group.
The effects of miR-221 on pulmonary inflammation in LPS-induced ALI mice
Next, we further investigated the effects of miR-221 on the pulmonary inflammation in LPS-induced ALI mice. First, the total cell and neutrophil counts in BALF were detected. As shown in Figure 4(a), B, LPS treatment resulted in an elevation of the total cell and neutrophil numbers. However, antagomir-221 injection significantly reduced the total cell counts and neutrophils compared to the LPS group, while agomir-221 dramatically enhanced the total cell and neutrophil counts (Figure 4(a,b)). In order to further confirm that miR-221 could attenuate LPS-caused lung injury via inflammation improvement, pro-inflammatory cytokines (IL-1β, TNF-α and IL-6) and chemokines (KC) levels in BALF were measured using ELISA. As expected, LPS-induced increase in protein levels of IL-1β, TNF-α, IL-6 and KC was obviously attenuated by antagomir-221, while the ability of LPS to increase these inflammatory mediator expressions was dramatically enhanced by agomir-221 (Figure 4(c–f)). The data above indicated that inhibition of miR-221 may attenuate the lung injury induced by LPS through suppression of pulmonary inflammation.
Figure 4.
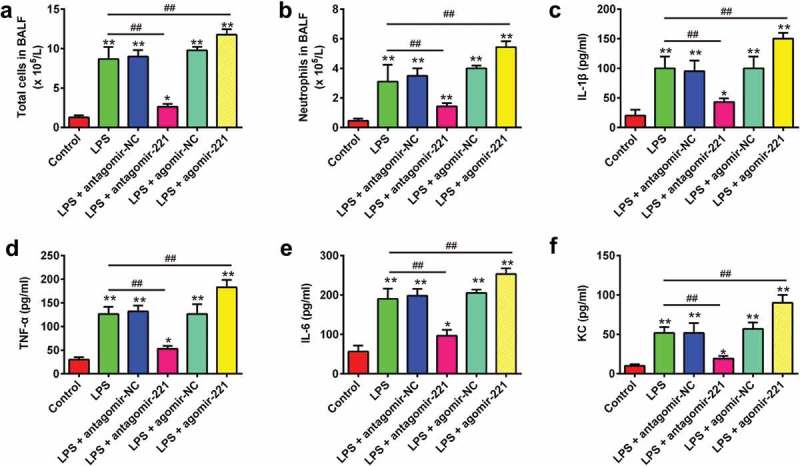
The effects of altered miR-221 expression on pulmonary inflammation in LPS-induced ALI mice. Mice were injected intravenously (tail vein) with agomir-221, antagomir-221, control antagomir or control agomir (2 mg/kg) at 24 h prior to 2 mg/kg LPS treatment. The mice were sacrificed after LPS administration for 24 h and then BALF were collected for analysis. (a, b) The total counts of cells and neutrophils from the BALF were counted using a hemocytometer (n = 6/group). (C-F) IL-1β, TNF-α, IL-6 and chemokines levels in BALF were measured using commercial ELISA kits (n = 6/group). Data represent the mean ± SD of three independent experiments. *p < 0.05, **p < 0.01 vs. control group. ##p < 0.01 vs. LPS alone group.
SOCS1 was a direct target of miR-221
In search of potential target genes for miR-221, the webservers Targetscan, miRanda and PicTar were used, and found that suppressor of cytokine signaling-1 (SOCS1) was a target of miR-221, with the target site located in the 3´-UTR (Figure 5(a)). It has previously been reported that SOCS1 is a negative feedback inhibitor of cytokine signaling and plays an important role in the amelioration of inflammation response [31–33]. As SOCS1 has been proven to be the target of miR-221 [34,35], we sought to determine whether miR-221 regulates the inflammatory response through targeting SOCS1 in ALI. To confirm that SOCS1 was negatively regulated by miR-221, the levels of SOCS1 mRNA and protein expression were analyzed in RAW 264.7 cells by qRT-PCR and Western Blot analysis. It was found that SOCS1 levels were significantly downregulated after miR-221 mimics transfection, while upregulated after transfection with miR-221 inhibitor (Figure 5(b,c)). To further validate that miR-221 directly binds to SOCS1, a luciferase reporter assay was performed. We observed that overexpression of miR-221 decreased relative luciferase activity of RAW 264.7 cells in the presence of the wild-type 3´-UTR, whereas knockdown of miR-221 increased the relative luciferase activity (Figure 5(d)). Similarly, we observed that the luciferase activity did not change significantly when the targeted sequence of SOCS1 was mutated in the miR-221-binding site. In addition, we also measured the protein expression of SOCS1 in lung tissues from ALI mice injected with agomir-221 or antagomir-221. The results of Western Blot showed that LPS treatment significantly decreased the expression of SOCS1 compared to control group. However, antagomir-221 reversed the inhibitory effect of LPS on the expression of SOCS1, whereas agomir-221 resulted in a more pronounced reduction in the expression of SOCS1 (Figure 5(e)). These results above show that miR-221/SOCS1 may play an important role in the inhibition of inflammation in ALI.
Figure 5.
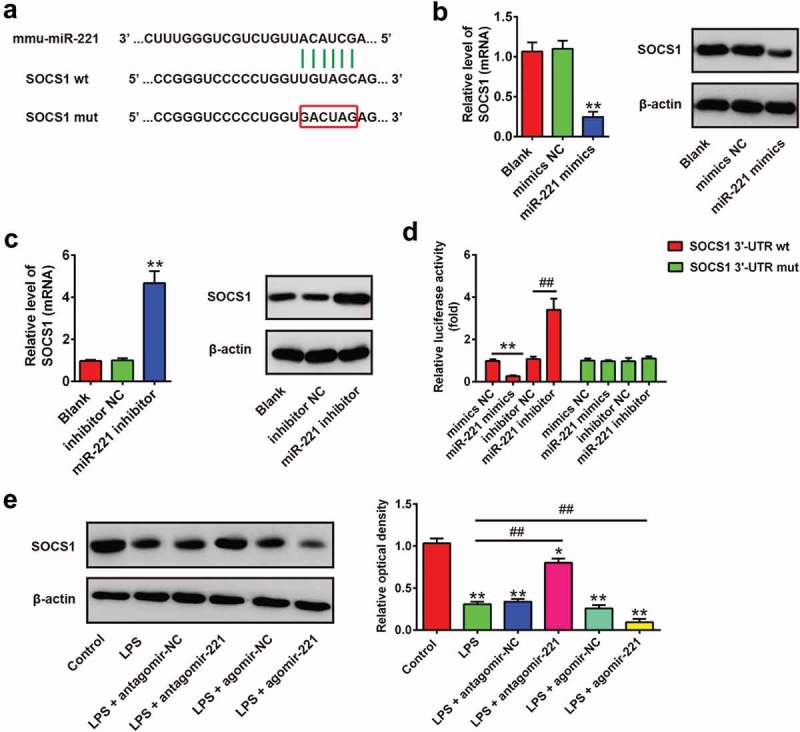
SOCS1 was a direct target of miR-221. (a) The putative binding site of miR-221 and SOCS1 is shown. (b, c) The expressions of SOCS1 mRNA and protein after transfection with miR-221 mimics or miR-221 inhibitor were measured by qRT-PCR and Western Blot. Data represent the mean ± SD of three independent experiments. **p < 0.01 vs. Blank group. (d) Luciferase assay of RAW264.7 cells co-transfected with firefly luciferase constructs containing the SCOS1 wild-type or mutated 3´-UTRs and miR-221 mimics, mimics NC, miR-221 inhibitor or inhibitor NC, as indicated (n = 3). Data represent the mean ± SD of three independent experiments. **p < 0.01 vs mimics NC, ## p < 0.01 vs inhibitor NC. (e) Expression of SOCS1 in lung tissues was measured using Western Blot in ALI mice (n = 6/group). Data represent the mean ± SD of three independent experiments. *p < 0.05, **p < 0.01 vs. control group. ##p < 0.01 vs. LPS alone group.
MiR-221 regulates the inflammatory response in LPS-treated RAW 264.7 cells through targeting SOCS1
As mentioned above, SOCS1 was a direct target of miR-221 in RAW 264.7 cells; therefore, the present study further investigated whether miR-221 protected RAW 264.7 cells from LPS-induced inflammatory response by regulating SOCS1. RAW 264.7 cells were treated with different concentrations of LPS (0.1, 1, 2 or 10 μg/ml) for 6 h. The results showed that the viability of RAW 264.7 cells was obviously decreased in a high dose of LPS (2 μg/ml and 10 μg/ml). However, no remarkable change in cell viability was observed when cells received LPS at the doses of 0.1 and 1.0 μg/ml (Figure 6(a)). Therefore, LPS in a dose of 1 μg/ml was used for subsequent experiments, which is consistent with a previous study [36]. To confirm the role of SOCS1 in the observed protective effects of miR-221 inhibition on LPS-induced inflammatory response, RAW 264.7 cells were co-transfected with miR-221 inhibitor and si-SOCS1 plasmids, and then treated with LPS for 6 h, followed by the assessment of cell apoptosis and pro-inflammatory cytokines (IL-1β, TNF-α and IL-6). First, we measured the expression of miR-221 in LPS-treated RAW 264.7 cells after miR-221 inhibitor transfection by qRT-PCR. The results showed that miR-221 inhibitor transfection markedly reduced the expression of miR-221 compared to the LPS group (Figure 6(b)). In order to verify the effect of miR-221 on the apoptosis in LPS-stimulated cell model, flow cytometer was performed. As shown in Figure 6(c), LPS-induced cell apoptosis was reduced by miR-221 inhibitor, while the inhibitory effect was attenuated when SOCS1 was knocked down. Moreover, we found that the expression of IL-1β, TNF-α and IL-6 was remarkably decreased in LPS-induced RAW264.7 cells after the treatment with miR-221 inhibitor. However, these inhibitory effects of miR-221 knockdown were reversed by si-SOCS1. (Figure 6(d–f)). Above all, these results revealed that miR-221 inhibition attenuated LPS-induced cell apoptosis and inflammation through upregulating SOCS1 expression.
Figure 6.
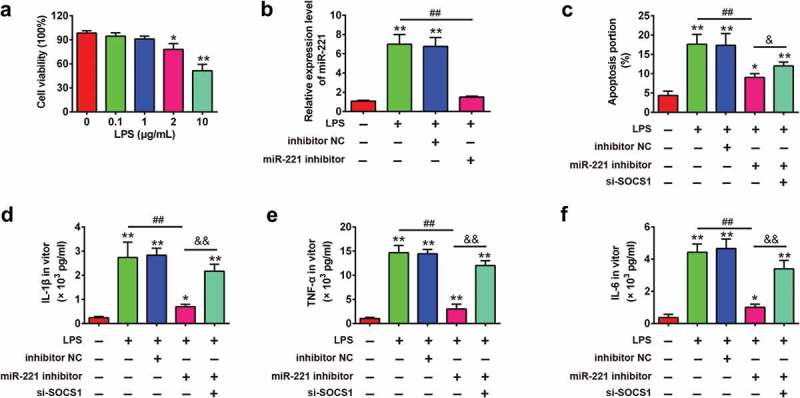
miR-221 regulates the inflammatory response in LPS-treated RAW 264.7 cells through targeting SOCS1. (a) LPS with different doses (0.1, 1, 2 or 10 μg/ml) was used to treat RAW 264.7 cells for 6 h, then cell viability was measured by CCK-8 assay. Data represent the mean ± SD of three independent experiments. *p < 0.05, **p < 0.01 vs. non-treated group. (b) The expression of miR-221 was detected by qRT-PCR. Then, RAW 264.7 cells were co-transfected with miR-221 inhibitor (20 nM) and si-SOCS1, and then treated with LPS (1 μg/ml) for 6 h, followed by the assessment of cell apoptosis and pro-inflammatory cytokines. (c) The apoptosis was determined by flow cytometer. (d-f) IL-1β, TNF-α and IL-6 levels in BALF were measured using commercial ELISA kits. Data represent the mean ± SD of three independent experiments. *p < 0.05, **p < 0.01 vs. control group. ##p < 0.01 vs. LPS alone group. && p < 0.01 vs. LPS + miR-221 inhibitor group.
MiR-221 inhibition suppressed NF-κB pathway through upregulation of SOCS1
It has previously been reported that NF-κB signaling pathway plays important roles in mediating inflammatory response [37]. As NF-κB acts as a downstream pathway of SOCS1, we sought to determine whether inhibition of miR-221 exerts its anti-inflammatory role through SOCS1-mediated inhibition of the NF-κB signaling pathway. Thus, we investigated the effect of miR-221 on the expressions of the key proteins of the NF-κB signaling pathway, including p-p65, IκBα and p-IκBα. It was observed that the expression levels of p-p65 and p-IκBα were both enhanced by LPS exposure in ALI mice, whereas the expression of SOCS1 and IκBα was significantly reduced, which suggest that LPS promoted the activation of SOCS1/NF-κB pathway. However, the activation of SOCS1/NF-κB signaling pathway induced by LPS was substantially suppressed by antagomir-221, as a result of the reduction of p-p65 and p-IκBα and promotion of SOCS1 and IκBα (Figure7(a,b)). In contrast, the activation of NF-κB signaling pathway induced by LPS was enhanced by agomir-221. These results indicated that miR-221 inhibition inactivated NF-κB pathway via upregulating SOCS1.
Discussion
In the present study, we found that miR-221 was significantly upregulated in lung tissues of ALI mice. miR-221 inhibition could attenuate LPS-induced lung injury through its anti-inflammatory activity in mice. Moreover, it was demonstrated that miR-221 suppressed LPS-induced inflammatory response through SOCS1/NF-κB pathway. These results suggest that inhibition of miR-221 may be a potential therapeutic target for the treatment of ALI.
Recently, miRNAs have been reported to be involved in the inflammatory response in many injury models [38,39]. For example, Ying et al. found that miR-127 modulated macrophage polarization and promoted lung inflammation and injury by activating the JNK pathway [21]. Liu et al. demonstrated that miR-147 attenuated the inflammatory response of macrophages through decreasing LPS-induced TNF-α and IL-6 production [39]. In ALI, Yang et al. showed that miR-16 protected against ALI in mice via attenuating inflammatory response through regulating the TLR4/NF-κB pathway [40]. Huang et al. found that downregulated miR-27b attenuated LPS-induced ALI via activation of NF-E2-related factor 2 (Nrf2) and inhibition of the NF-κB signaling pathway. Fu et al. reported that miR-92a antagonism attenuated LPS-induced pulmonary inflammation and injury in mice through suppressing the PTEN/AKT/NF-κB signaling pathway [41,42]. In this study, using microarray, we found that miR-221 was significantly upregulated in ALI mice and its high expression was also observed in peripheral blood and splenocytes from ALI mice. Previous studies revealed that miR-221 was closely associated with inflammatory disorders. For example, Zhu et al. showed that overexpression of miR-221 attenuated angiotensin II-induced endothelial inflammation in human umbilical vein endothelial cells [43]. Similarly, miR-221 was reported to inhibit nitric oxide (NO) production and activate NF-κB signaling in human endothelial cells, thus leading to exaggerated inflammation [44]. Zhao et al. reported that intratracheal instillation of miR-221 in ALI mice caused exaggerated lung injury and inflammation, suggesting that inhibition of miR-221 may improve lung injury induced by LPS [45]. In our study, we investigated the influence of miR-221 on LPS-induced ALI. Results have demonstrated that miR-221 inhibition could effectively attenuate LPS-mediated lung injury and mortality in mice, and overexpression of miR-221 had opposite effects. We further showed that the protective effect of miR-221 inhibition might be mediated through its ability to inhibit LPS-induced inflammatory cytokines release (TNF-α, IL-1β and IL-6) and to ameliorate of LPS-stimulated apoptosis. All these data support that miR-221 inhibition can improve LPS-induced ALI in mice.
As we know, miRNAs usually exert their functions by regulating the expression of target genes. The target genes of miR-221 have been widely investigated, such as Axis inhibition protein 2 (AXIN2) and RECK [46,47]. Thus, we further explored the possible target genes of miR-221 in LPS-treated RAW 264.7 cells. Through bioinformatic analysis and dual-luciferase reporter assays, we identified SOCS1, an important regulator in the regulation of inflammation [48], as a target of miR-221. Studies have reported that SOCS1 plays an important role in various lung inflammatory diseases including ALI. For example, Galam et al. showed that overexpression of SOCS-1 protected against ALI by inhibiting apoptosis signal-regulating kinase 1 (ASK1) and suppressing IL-1β levels [49]. Kolliputi N et al. revealed that IL-6 cytoprotection against hyperoxic acute lung injury (HALI) is associated with enhanced SOCS-1 expression [50]. Rao et al. found that miR-155 promoted SEB-mediated inflammation and lung injury through SOCS1 suppression [51]. Recently, Xia et al. demonstrated that inhibition of miR-221 alleviated neuropathic pain and neuroinflammation through increasing SOCS1 [52]. Combined with the above observations, we hypothesized that miR-221 inhibition protected against LPS-mediated inflammation through the modulation of SOCS1. As expected, our results showed that SOCS1 knockdown reversed the inhibitory effects of miR-221 inhibition on LPS-induced inflammation and apoptosis in RAW 264.7 cells. Therefore, it will be intriguing to examine whether and how SOCS1 signaling affects the protection of miR-221 inhibition against LPS-induced ALI.
Activation of the NF-κB signaling pathway has been implicated in various inflammatory diseases, which can induce a large number of inflammatory cytokine expressions [53]. In ALI mice model, inhibition of the NF-κB signaling pathway had the ability to protect against LPS-induced ALI [54–56]. Previous studies have reported that miRNAs are important in the regulation of inflammatory response through activating the NF-κB signaling pathway. For example, Yan et al. found that miR-223 attenuated LPS-induced inflammation in an acute lung injury model via the NF-κB signaling pathway [57]. Suo et al. showed that miRNA-1246 suppresses acute lung injury-induced inflammation and apoptosis via the NF-κB pathway [58]. Notably, miR-221 stimulated IL-4 secretion in mast cells through NF-κB pathway in a murine asthma model [59]. In order to determine the protective mechanism of miR-221, the effects of aberrantly expressed miR-221 on key kinases in the NF-κB pathway were examined in the present study. The data showed that the NF-κB pathway was activated in LPS-induced ALI mice, and the inhibition of miR-221 suppressed the activity of this signaling pathway, suggesting that the protective effect of miR-221 on LPS-induced ALI may mediate by the NF-κB pathway.
However, ALI is a much complex pathological condition, in which the apoptosis also serves an important role, in addition to the inflammatory response [60,61]. Interestingly, there are still 361 other genes which could also be the potential target genes of miR-221, including some apoptosis facilitator such as programmed cell death 10 (PDCD10); DNA-damage-inducible transcript 4 (DDIT4); BCL2 binding component 3 (BBC3); BCL2-like 11 IGSF1 (BCL2L11) and so on. Additionally, many key factors of cell apoptosis have proved to be the targets of miR-221-3p in other studies, such as BCL2-like 11 (BCL2L11) [62], DNA-damage-inducible transcript 4(DDIT4) [63] and BCL2 binding component 3 (BBC3) [64]. In the future, we would also explore the roles/functions between miR-221-3p and apoptosis-related targets during ALI. For more details, please refer to http://www.targetscan.org/cgi-bin/targetscan/vert_72/targetscan.cgi?species=Mouse&mir_sc=miR-221-3p/222-3p.
In summary, our findings revealed that miR-221 inhibition may exert lung protection in LPS-induced acute lung injury both in vitro and in vivo via anti-inflammatory by targeting SOCS1/NF-κB signaling pathway. These results suggest that miR-221 inhibition might hold promise as a potential therapeutic target for the treatment of ALI.
Funding Statement
Henan Academy of Medical Science and Technology Project [201602135]
Authors contribution
Tao Wang and Lihua Jiang were responsible for the main conceive of the study and the draft of the manuscript. Xiaoyong Wei, Zhenghua Dong, Xuena Wang, Bo Liu, Junbo Zhao, Lijuan Wang, Shangyou Zhou and Yuxia Wang helped to design the study and performed the statistical analysis. Tao Wang and Lihua Jiang helped to revise the manuscript and participated in its design. All authors have read and approved the final manuscript.
Disclosure statement
No potential conflict of interest was reported by the authors.
References
- [1].Zhou T, Garcia JG, Zhang W.. Integrating microRNAs into a system biology approach to acute lung injury. Transl Res. 2011;157:180–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Herridge MS, Tansey CM, Matte A, et al. Functional disability 5 years after acute respiratory distress syndrome. N Engl J Med. 2011;364:1293–1304. [DOI] [PubMed] [Google Scholar]
- [3].Mendez JL, Hubmayr RD.. New insights into the pathology of acute respiratory failure. Curr Opin Crit Care. 2005;11:29–36. [DOI] [PubMed] [Google Scholar]
- [4].Rubenfeld GD, Caldwell E, Peabody E, et al. Incidence and outcomes of acute lung injury. N Engl J Med. 2005;353:1685–1693. [DOI] [PubMed] [Google Scholar]
- [5].Mei SH, McCarter SD, Deng Y, et al. Prevention of LPS-induced acute lung injury in mice by mesenchymal stem cells overexpressing angiopoietin 1. PLoS Med. 2007;4:e269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Stapleton RD, Wang BM, Hudson LD, et al. Causes and timing of death in patients with ARDS. Chest. 2005;128:525–532. [DOI] [PubMed] [Google Scholar]
- [7].Ji M, Chen M, Hong X, et al. The effect of diabetes on the risk and mortality of acute lung injury/acute respiratory distress syndrome: a meta-analysis. Medicine (Baltimore). 2019;98:e15095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Chen Z, Zhang X, Chu X, et al. Preventive effects of valnemulin on lipopolysaccharide-induced acute lung injury in mice. Inflammation. 2010;33:306–314. [DOI] [PubMed] [Google Scholar]
- [9].Grommes J, Soehnlein O. Contribution of neutrophils to acute lung injury. Mol Med. 2011;17:293–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Brigham KL. Lower tidal volume ventilation and plasma cytokine markers of inflammation in patients with acute lung injury. Curr Infect Dis Rep. 2005;7:327–328. [DOI] [PubMed] [Google Scholar]
- [11].Bhatia M, Moochhala S. Role of inflammatory mediators in the pathophysiology of acute respiratory distress syndrome. J Pathol. 2004;202:145–156. [DOI] [PubMed] [Google Scholar]
- [12].Takashima K, Matsushima M, Hashimoto K, et al. Protective effects of intratracheally administered quercetin on lipopolysaccharide-induced acute lung injury. Respir Res. 2014;15:150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Agouridakis P, Kyriakou D, Alexandrakis MG, et al. The predictive role of serum and bronchoalveolar lavage cytokines and adhesion molecules for acute respiratory distress syndrome development and outcome. Respir Res. 2002;3:25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Djuranovic S, Nahvi A, Green R. A parsimonious model for gene regulation by miRNAs. Science. 2011;331:550–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Xu Z, Zhang C, Cheng L, et al. The microRNA miR-17 regulates lung FoxA1 expression during lipopolysaccharide-induced acute lung injury. Biochem Biophys Res Commun. 2014;445:48–53. [DOI] [PubMed] [Google Scholar]
- [16].Li W, Qiu X, Jiang H, et al. Downregulation of miR-181a protects mice from LPS-induced acute lung injury by targeting Bcl-2. Biomed Pharmacothe. 2016;84:1375–1382. [DOI] [PubMed] [Google Scholar]
- [17].Luo J, Zhan J, You H, et al. MicroRNA146a/Tolllike receptor 4 signaling protects against severe burninduced remote acute lung injury in rats via antiinflammation. Mol Med Rep. 2018;17:8377–8384. [DOI] [PubMed] [Google Scholar]
- [18].Vaporidi K, Vergadi E, Kaniaris E, et al. Pulmonary microRNA profiling in a mouse model of ventilator-induced lung injury. Am J Physiol Lung Cell Mol Physiol. 2012;303:L199–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Ke XF, Fang J, Wu XN, et al. MicroRNA-203 accelerates apoptosis in LPS-stimulated alveolar epithelial cells by targeting PIK3CA. Biochem Biophys Res Commun. 2014;450:1297–1303. [DOI] [PubMed] [Google Scholar]
- [20].Guo Z, Gu Y, Wang C, et al. Enforced expression of miR-125b attenuates LPS-induced acute lung injury. Immunol Lett. 2014;162:18–26. [DOI] [PubMed] [Google Scholar]
- [21].Ying H, Kang Y, Zhang H, et al. MiR-127 modulates macrophage polarization and promotes lung inflammation and injury by activating the JNK pathway. J Iimmunol. 2015;194:1239–1251. [DOI] [PubMed] [Google Scholar]
- [22].Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods. 2001;25:402–408. [DOI] [PubMed] [Google Scholar]
- [23].Duan Y, Learoyd J, Meliton AY, et al. Inhibition of Pyk2 blocks lung inflammation and injury in a mouse model of acute lung injury. Respir Res. 2012;13:4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Barreto TR, Costola-de-Souza C, Margatho RO, et al. Repeated Domperidone treatment modulates pulmonary cytokines in LPS-induced acute lung injury in mice. Int Immunopharmacol. 2018;56:43–50. [DOI] [PubMed] [Google Scholar]
- [25].Li H, Qiang Y, Wang L, et al. Repair of lipopolysaccharide-induced acute lung injury in mice by endothelial progenitor cells, alone and in combination with simvastatin. Chest. 2013;144:876–886. [DOI] [PubMed] [Google Scholar]
- [26].Li C, Yang D, Cao X, et al. LFG-500, a newly synthesized flavonoid, attenuates lipopolysaccharide-induced acute lung injury and inflammation in mice. Biochem Pharmacol. 2016;113:57–69. [DOI] [PubMed] [Google Scholar]
- [27].Lei C, Jiao Y, He B, et al. RIP140 down-regulation alleviates acute lung injury via the inhibition of LPS-induced PPARgamma promoter methylation. Pulm Pharmacol Ther. 2016;37:57–64. [DOI] [PubMed] [Google Scholar]
- [28].Lee H, Zhang D, Zhu Z, et al. Epithelial cell-derived microvesicles activate macrophages and promote inflammation via microvesicle-containing microRNAs. Sci Rep. 2016;6:35250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Ortega FJ, Moreno M, Mercader JM, et al. Inflammation triggers specific microRNA profiles in human adipocytes and macrophages and in their supernatants. Clin Epigenetics. 2015;7:49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Yanling Q, Xiaoning C, Fei B, et al. Inhibition of NLRP9b attenuates acute lung injury through suppressing inflammation, apoptosis and oxidative stress in murine and cell models. Biochem Biophys Res Commun. 2018;503:436–443. [DOI] [PubMed] [Google Scholar]
- [31].Dragone T, Cianciulli A, Calvello R, et al. Resveratrol counteracts lipopolysaccharide-mediated microglial inflammation by modulating a SOCS-1 dependent signaling pathway. Toxicol In Vitro. 2014;28:1126–1135. [DOI] [PubMed] [Google Scholar]
- [32].Liu X, Li J, Peng X, et al. Geraniin inhibits LPS-induced THP-1 macrophages switching to M1 phenotype via SOCS1/NF-kappaB pathway. Inflammation. 2016;39:1421–1433. [DOI] [PubMed] [Google Scholar]
- [33].Ortiz-Munoz G, Martin-Ventura JL, Hernandez-Vargas P, et al. Suppressors of cytokine signaling modulate JAK/STAT-mediated cell responses during atherosclerosis. Arterioscler Thromb Vasc Biol. 2009;29:525–531. [DOI] [PubMed] [Google Scholar]
- [34].Xu G, Yang F, Ding CL, et al. MiR-221 accentuates IFNs anti-HCV effect by downregulating SOCS1 and SOCS3. Virology. 2014;462–463:343–350. [DOI] [PubMed] [Google Scholar]
- [35].Lu C, Huang X, Zhang X, et al. miR-221 and miR-155 regulate human dendritic cell development, apoptosis, and IL-12 production through targeting of p27kip1, KPC1, and SOCS-1. Blood. 2011;117:4293–4303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Huang C, Yang Y, Li WX, et al. Hyperin attenuates inflammation by activating PPAR-gamma in mice with acute liver injury (ALI) and LPS-induced RAW264.7 cells. Int Immunopharmacol. 2015;29:440–447. [DOI] [PubMed] [Google Scholar]
- [37].Yuan H, Ma J, Li T, et al. MiR-29b aggravates lipopolysaccharide-induced endothelial cells inflammatory damage by regulation of NF-kappaB and JNK signaling pathways. Biomed Pharmacothe. 2018;99:451–461. [DOI] [PubMed] [Google Scholar]
- [38].Bhaumik D, Scott GK, Schokrpur S, et al. MicroRNAs miR-146a/b negatively modulate the senescence-associated inflammatory mediators IL-6 and IL-8. Aging (Albany NY). 2009;1:402–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Liu G, Friggeri A, Yang Y, et al. miR-147, a microRNA that is induced upon Toll-like receptor stimulation, regulates murine macrophage inflammatory responses. Proc Natl Acad Sci U S A. 2009;106:15819–15824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Yang Y, Yang F, Yu X, et al. miR-16 inhibits NLRP3 inflammasome activation by directly targeting TLR4 in acute lung injury. Biomed Pharmacothe. 2019;112:108664. [DOI] [PubMed] [Google Scholar]
- [41].Fu L, Zhu P, Qi S, et al. MicroRNA-92a antagonism attenuates lipopolysaccharide (LPS)-induced pulmonary inflammation and injury in mice through suppressing the PTEN/AKT/NF-kappaB signaling pathway. Biomed Pharmacothe. 2018;107:703–711. [DOI] [PubMed] [Google Scholar]
- [42].Huang Y, Huang L, Zhu G, et al. Downregulated microRNA-27b attenuates lipopolysaccharide-induced acute lung injury via activation of NF-E2-related factor 2 and inhibition of nuclear factor kappaB signaling pathway. J Cell Physiol. 2019;234:6023–6032. [DOI] [PubMed] [Google Scholar]
- [43].Zhu N, Zhang D, Chen S, et al. Endothelial enriched microRNAs regulate angiotensin II-induced endothelial inflammation and migration. Atherosclerosis. 2011;215:286–293. [DOI] [PubMed] [Google Scholar]
- [44].Chen CF, Huang J, Li H, et al. MicroRNA-221 regulates endothelial nitric oxide production and inflammatory response by targeting adiponectin receptor 1. Gene. 2015;565:246–251. [DOI] [PubMed] [Google Scholar]
- [45].Zhao D, Zhuang N, Ding Y, et al. MiR-221 activates the NF-kappaB pathway by targeting A20. Biochem Biophys Res Commun. 2016;472:11–18. [DOI] [PubMed] [Google Scholar]
- [46].Nie X, Chen Y, Tan J, et al. MicroRNA-221-3p promotes pulmonary artery smooth muscle cells proliferation by targeting AXIN2 during pulmonary arterial hypertension. Vascul Pharmacol. 2017;116:24–35. [DOI] [PubMed] [Google Scholar]
- [47].Qin J, Luo M. MicroRNA-221 promotes colorectal cancer cell invasion and metastasis by targeting RECK. FEBS Lett. 2014;588:99–104. [DOI] [PubMed] [Google Scholar]
- [48].Nakagawa R, Naka T, Tsutsui H, et al. SOCS-1 participates in negative regulation of LPS responses. Immunity. 2002;17:677–687. [DOI] [PubMed] [Google Scholar]
- [49].Galam L, Soundararajan R, Breitzig M, et al. SOCS-1 rescues IL-1beta-mediated suppression of epithelial sodium channel in mouse lung epithelial cells via ASK-1. Oncotarget. 2016;7:29081–29091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Kolliputi N, Waxman AB. IL-6 cytoprotection in hyperoxic acute lung injury occurs via suppressor of cytokine signaling-1-induced apoptosis signal-regulating kinase-1 degradation. Am J Respir Cell Mol Biol. 2009;40:314–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Rao R, Rieder SA, Nagarkatti P, et al. Staphylococcal enterotoxin B-induced microRNA-155 targets SOCS1 to promote acute inflammatory lung injury. Infect Immun. 2014;82:2971–2979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Xia L, Zhang Y, Dong T. Inhibition of microRNA-221 alleviates neuropathic pain through targeting suppressor of cytokine signaling 1. J Mol Neurosci. 2016;59:411–420. [DOI] [PubMed] [Google Scholar]
- [53].Ni H, Ergin M, Huang Q, et al. Analysis of expression of nuclear factor kappa B (NF-kappa B) in multiple myeloma: downregulation of NF-kappa B induces apoptosis. Br J Haematol. 2001;115:279–286. [DOI] [PubMed] [Google Scholar]
- [54].Zhang A, Wang S, Zhang J, et al. Genipin alleviates LPS-induced acute lung injury by inhibiting NF-kappaB and NLRP3 signaling pathways. Int Immunopharmacol. 2016;38:115–119. [DOI] [PubMed] [Google Scholar]
- [55].Deng G, He H, Chen Z, et al. Lianqinjiedu decoction attenuates LPS-induced inflammation and acute lung injury in rats via TLR4/NF-kappaB pathway. Biomed Pharmacothe. 2017;96:148–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Tian L, Li W, Wang T. Therapeutic effects of silibinin on LPS-induced acute lung injury by inhibiting NLRP3 and NF-kappaB signaling pathways. Microb Pathog. 2017;108:104–108. [DOI] [PubMed] [Google Scholar]
- [57].Yan Y, Lu K, Ye T, et al. MicroRNA223 attenuates LPSinduced inflammation in an acute lung injury model via the NLRP3 inflammasome and TLR4/NFkappaB signaling pathway via RHOB. Int J Mol Med. 2019;43:1467–1477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Suo T, Chen GZ, Huang Y, et al. miRNA-1246 suppresses acute lung injury-induced inflammation and apoptosis via the NF-kappaB and Wnt/beta-catenin signal pathways. Biomed Pharmacothe. 2018;108:783–791. [DOI] [PubMed] [Google Scholar]
- [59].Zhou Y, Yang Q, Xu H, et al. miRNA-221-3p enhances the secretion of interleukin-4 in mast cells through the phosphatase and tensin homolog/p38/nuclear factor-kappaB pathway. PloS One. 2016;11:e0148821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Li W, Ma K, Zhang S, et al. Pulmonary microRNA expression profiling in an immature piglet model of cardiopulmonary bypass-induced acute lung injury. Artif Organs. 2015;39:327–335. [DOI] [PubMed] [Google Scholar]
- [61].Chen Y, Xian PF, Yang L, et al. MicroRNA-21 promotes proliferation of fibroblast-like synoviocytes through mediation of NF-kappaB nuclear translocation in a rat model of collagen-induced rheumatoid arthritis. Biomed Res Int. 2016;2016:9279078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Garofalo M, Romano G, Di Leva G, et al. EGFR and MET receptor tyrosine kinase-altered microRNA expression induces tumorigenesis and gefitinib resistance in lung cancers. Nat Med. 2011;18:74–82. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- [63].Pineau P, Volinia S, McJunkin K, et al. miR-221 overexpression contributes to liver tumorigenesis. Proc Natl Acad Sci U S A. 2010;107:264–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Zhang J, Sha J, Zhou Y, et al. Bufalin inhibits proliferation and induces apoptosis in osteosarcoma cells by downregulating microRNA-221. Evid Based Complement Alternat Med. 2016;2016:7319464. [DOI] [PMC free article] [PubMed] [Google Scholar]