

Abstract
Immune checkpoint blockades have revolutionized the treatment landscape for several cancer indications, yet they have not gained traction in a range of other tumors such as triple-negative breast cancer. Despite durable disease control by many patients, a third of cancer patients relapse due to acquired resistance. Combined immunotherapy has shown significant promise to overcome these grand challenges. In this report, we describe the synthesis and characterization of dual-action small-molecule PARP1/PD-L1 inhibitor conjugates as potential targeted anticancer agents. These conjugates display significant apoptosis and cytotoxic efficacy to approximately 2–20-fold better than their individual agents in a panel of cancer cell lines. This was underscored by derived combination indices, which was consistent with strong synergy when cells were treated with the individual agents, olaparib and BMS-001 using the Chou–Talalay method. Furthermore, we sought to unravel the mechanistic behavior of the conjugates and their implications on the PARP/PD-L1 axis. We used apoptosis, cell cycle, immunoblotting, and T-cell proliferation assays to establish the synergy imparted by these conjugates. These multifunctional compounds enable the discovery of small-molecule immunochemotherapeutic agents and chemical probes to elucidate the cross-talk between DNA repair and PD-L1 pathways.
Introduction
Immunotherapy involving immune checkpoint modulation, adoptive cell transfer, and cancer vaccine is the strategy du jour for cancer treatment.1 Programmed cell death receptor 1/programmed cell death-ligand 1 (PD-1/PD-L1) inhibition, a subset of immune checkpoint modulation, is one of the most clinically efficient strategies, which provide durable response rates, with accompanying low toxicity profile.1,2 Immunotherapeutics seek to suppress the various tumor immune evasion mechanisms, which in turn enable host immune responses to combat tumors.3 Immune evasion, as a tumor survival strategy, includes all mechanisms through which tumor avoids lethal adaptive immunologic responses, enhancing survival in its milieu as well as evolving to maximize its growth in the host.4 Implicit in these tumor evasion mechanisms are the alteration of specific co-inhibitory and co-stimulatory pathways,namely, immune checkpoints. The PD-1/PD-L1 axis is a co-inhibitory immune checkpoint. The engagement of the PD-1 receptor protein with its ligand triggers suppressive signals that play a key role in the maintenance of immune homeostasis and preventing immunopathogenesis and autoimmune diseases.5,6 Tumors can deploy the PD-1/PD-L1 pathway to institute an immune evasive roadblock, which compromises the cytotoxic abilities of activated T-cells and the immune system in general.7,8
To overcome resistance and improve potency associated with PD-1/PD-L1 therapy, several clinical combinations have emerged.9 One of such novel combinations strategy is PARP inhibitors (PARPi) with PD-1/PD-L1 inhibitors.10,11 For instance, the potency of a PD-L1 antibody, atezolizumab, was improved by conjugating it, through a PEG-linker, to doxorubicin.12 Combining the PARPi (niraparib) and the PD-1 antibody (pembrolizumab) has also shown to be synergistic and provides enhanced antitumor activities in both BRCA-proficient and BRCA-deficient tumors.13 Equally, PARPi and anti-PD-L1 mAb in combination therapies are also synergistic and improve antitumor responses.14−16
Few small-molecule PD-L1 inhibitors exist, and extensive research work has been done to decode their specific modes of interaction with the PD-L1 cell surface protein.17 Similar structural studies have also been done for PD-L1 antibodies, with the goal to develop potent small-molecule PD-L1 inhibitors that can get through to the clinics.18−20 Small molecule combinations for PARPi and PD-L1 are virtually nonexistent due to lack of clinically approved small-molecule anti-PD-L1 agents.
PARP inhibition is a first-line treatment regimen for tumors that harbor BRCA1/2 mutations due to synthetic lethality. DNA damage occurs in cells routinely and can be caused by several reasons including genetic insults, exposure to radiation, etc. Several cell-based mechanisms employ repair proteins to correct the damage according to type: single- or double-stranded breaks. BRCA1 and BRCA2 are classical examples of such repair proteins.21 These repair double-strand DNA deformations by a process called homologous recombination repair (HRR). Alteration in the genes that encode these repair proteins can ultimately affect this repair process, resulting in error-laden DNA repair, subsequently leading to cancer.22 PARP1 is a repair protein that corrects single-stranded nicks in the DNA. If unrepaired until replication, then these single-stranded breaks can degenerate into double-stranded breaks.23−25 PARP inhibitors demonstrate synthetic lethality in cells with impaired homologous recombination (HR)-mediated DNA repair function, particularly BRCA1/2-associated tumors. Thus, PARP inhibition is particularly effective against BRCA-1/2-mutated tumors via synthetic lethality.26,27 It is asserted that tumors with greater mutational burden are vulnerable to immune checkpoint blockade. For example, BRCA1-mutated tumors have high lymphocyte infiltration than non-BRCA1 tumors, resulting in improved survival and antitumor immune response.28
Several PARPi conjugates exist. Classical examples include the novel conjugate of SiR fluorochromes and olaparib derivatives as probes for cellular nuclear targeting.29 Related radiolabeled PARPi-conjugates have found applications as radiotracers.30−32 A PARPi conjugate prodrug, which targets PARP1 and releases nitric oxide, has also shown promise.25
Recent studies suggest the existence of a cross-talk between PARP and PD-1/PD-L1 axis. The inhibition of PARP results in the overexpression of PD-L1 via GSK3β inactivation.33 This enables tumor cells to evade immune assault via the immunosuppressive activity of the PD-1/PD-L1 pathway. Thus, the combined inhibition of PARP1 and PD-1/PD-L1 will be a more effective therapeutic solution than the monotherapy of each.10,33 To test this concept, we envisioned the use of small-molecule agents as dual inhibitors of both PARP1 and PD-L1 (Figure 1).
Figure 1.

Small-molecule drug candidates under investigation in this report.
Traditionally, combination therapy has been one of the ways used to circumvent drug toxicity/resistance associated with monotherapy in cancer treatment.34 From a clinical perspective, this strategy is used to either enhance the therapeutic effect obtained from monotherapy or to obtain a similar therapeutic effect but has less toxic side effects (dose-limiting toxicities).35 Combination therapy achieves the endpoint of mild toxicity, right dose, and efficient responses.36 Hybrid drug conjugates are comparable to combination therapies, and in some cases, exhibit relatively superior drug efficacy.37 Hybrid drug conjugates are designed based on the merging or linking of the active scaffolds of two or more different drugs with different and/or the same mechanism into one compound.38 Like combination therapies, hybrid compounds can be designed based on compounds that target the same protein interaction via different mechanisms, or it can be based on different interactions that produce the same therapeutic effect.39 Conjugates offer several added advantages than combination therapies. These include improved solubility, that is, different partner drugs may have different solubility; thus, the uptake rates in the blood may vary at a fixed dose. Additionally, with the freedom to vary linkers in the design of hybrid compounds, one can use a linker that contributes effectively to the solubility of the conjugate. The pharmacokinetics of a single conjugate is also relatively easier to predict, and this makes it amenable to modifications. Hybrid compounds also offer the added advantage to do structure–activity relationship studies more rapidly.38 High-throughput conjugation chemistries can be adopted to append functionalized monoagents to desired counterparts, and their biological effects can be evaluated. This approach is more effective, faster and reduces false positives/negatives that can be associated with combination therapy of individual agents.
We therefore sought to design and study the cytotoxicity as well as decipher the related biological profile of small-molecule conjugates that target both PARP and PD-L1. The constructs display improved potency in cancers cells. These compounds offer the ability to glean insights into the PARP/PD-L1 axis and are predictably amenable to structural modification/studies for the enhanced synergistic antitumor effect.
Results and Discussion
Design and Synthesis of Dual-Action Conjugates
The improved efficacy and information gleaned from using combination therapies provide the basis for the design of hybrid compounds. Inspiration from the clinically relevant combination therapy involving PARP inhibitors and mAb PD-1/PD-L1 inhibitors led us to design conjugates 1–3. These conjugates were developed by using the PD-L1 inhibitor, BMS-001, which is a potent small molecule developed by Bristol-Meyers Squibb (BMS), and a well-known PARP1 inhibitor, olaparib, in current clinical use. We conjectured that the validated pharmacokinetic and toxicity profile of olaparib could compensate for deficiencies associated with BMS-001. Thus, different analogs of these conjugates could be synthesized using different spacers. The piperazine ring of the olaparib derivative is amenable to modification at the N-terminus; and this does not alter its PARP inhibition properties. However, if the piperazine ring core in the olaparib framework is substantially modified or replaced with other bioisosteres, it significantly alters its cytotoxicity and DNA damaging properties.40 Therefore, we choose to effect modifications at the N-terminus of the piperazine ring core of the olaparib derivative.
The design principle was based on restriction and flexibility rules.41,42 Our choice of linkers were informed by preliminary docking studies, which gave us an insight into the potential of our compounds to outperform the reference compounds via interaction with the respective human PARP1 (PDB ID: 4R6E) and PD-L1 (PDB ID: 5N2F) protein structures.43,44 The in silico interaction of conjugates with PARP1 and PD-L1 proteins is shown in Figure 7 and 8. We envisioned a conjugate that is capable of targeting both PARP1 and PD-L1 without succumbing to hydrolysis to release the individual components, hence the design of conjugate 1. Then, we rationalized that establishing an ester bond via a linker to the PD-L1 small-molecule inhibitor will connect the two compounds, and after intracellular hydrolysis, the intact BMS-001 will be released. This led us to test two different spacers, namely, hydrocarbon and PEGylated-like linker toward conjugates 2 and 3, respectively. Generally, ethoxy-ethoxy-linker is supposed to improve cell permeability and the drug transport and kinetics of these conjugates. The PARPi and BMS-001 compounds were synthesized according to reported protocols and were purified and fully characterized by 1H-NMR and 13C-NMR spectroscopy. These compounds were subsequently used in the synthesis of all conjugates (Scheme 1). Based on our design strategy, we synthesized 1, direct coupling of the PD-L1 molecule to the olaparib derivative, PARPi, via an amide bond.25
Figure 7.
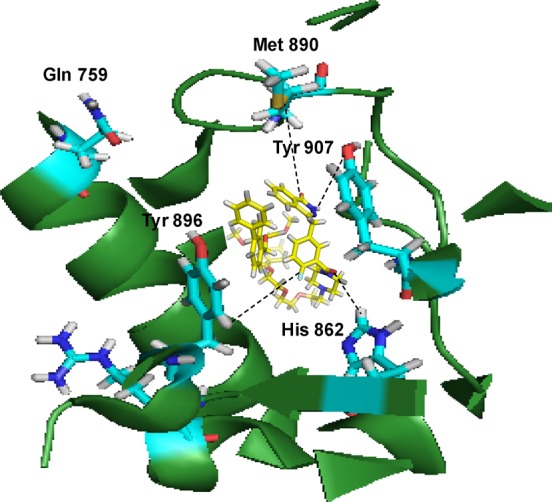
Conjugate 3 binds in the C-terminal catalytic domain of PARP1. Best pose of conjugate 3 (yellow sticks) forming hydrogen bond interactions (yellow lines) with key amino acid residues (cyan sticks) in NI, PH, and AD sites. PARP1 is shown as green ribbon.
Figure 8.
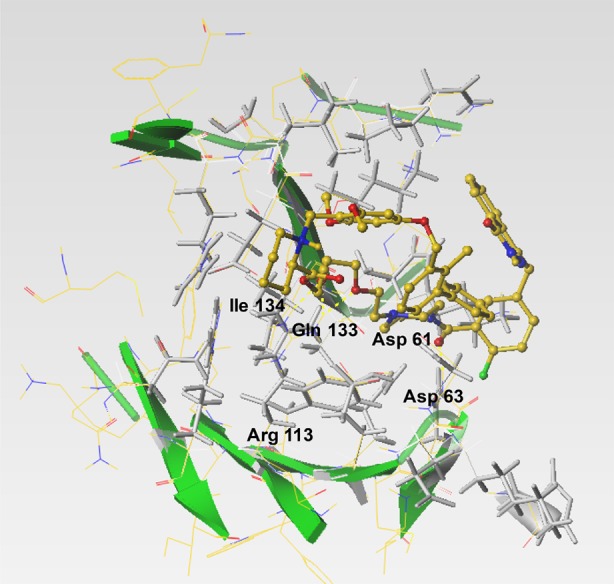
Conjugate 3 interacts with amino acid residues in the interfacial binding cleft of the PD-L1 dimer. Conjugate 3 (yellow sticks) interacts with hot spot aspartate, glutamine, isoleucine, and arginine amino acid residues (depicted as gray sticks)
Scheme 1. General Reaction Scheme toward the Synthesis of Small-Molecule PARP/PD-L1 Inhibitor Conjugates.

The use of HOBt in the coupling was to avoid unwanted racemization of the conjugate at the picolinic acid, which has (S) configuration. Unlike compound 1, different types of spacers were used as conduits in the hybrid inhibitor design of conjugates 2 and 3. For 2, the olaparib derivative, PARPi, was functionalized with bromoethanol by N-alkylation of the piperazine moiety. The alcohol handle was then used in an esterification with the carboxyl handle of BMS-001 using EDCl/HOBt. Similarly, in the synthesis of conjugate 3, the piperazine scaffold (of PARPi) was N-alkylated with chloroethoxyethoxy followed by esterification with BMS-001. All conjugates were purified by silica gel column chromatography, recrystallized, and fully characterized by 1H-NMR and 13C-NMR spectroscopy and high-resolution mass spectrometry (HRMS); the purity of the conjugates were ascertained by high-performance liquid chromatography (HPLC) (Supporting Information, Figures S1−S17). These conjugates were then used for biological assessment.
Photophysical and Stability Properties of Conjugates
The UV absorption profile of conjugates 1–3 were measured in a buffered system as well as Dulbecco’s modified Eagle medium (DMEM), which contains millimolar concentrations of amino acids and other biomolecules. The conjugates show a characteristic absorption peak at 260 nm. Stock solutions of 1–3 were prepared in DMSO and subsequently dissolved in PBS or DMEM. Furthermore, conditioned media, which contain metabolites, growth factors, and extracellular matrix proteins, were also used to evaluate the photophysical and stability properties of the conjugates. We monitored the peak at 260 nm over a period of 72 h (Figure 2, and Figures S35–S38). In PBS, the peaks were unaltered, while a minimal red-shift by 2 nm in the absorption maxima was observed for conjugate 1 in DMEM at 72 h. Compounds 2 and 3 follow a similar profile as in 1, indicative of relative stability in PBS and DMEM. Experiments in conditioned media did not deviate from the observation with DMEM. In addition to the characteristic peak of the conjugates at about 240 nm that remained unaltered during the 72 h study period was a peak at ∼300 nm, which is likely from an interaction of the compound with matrix proteins or metabolites from the medium. The studies suggest that our compounds are fairly stable in DMEM with characteristic absorption features of the compounds displayed over the 72 h duration of the experiment. Subsequently, conjugates 1–3 were found to be stable in bovine serum albumin (pH 5.2) over 72 h. In this experiment, we monitored the UV–vis absorption profile (Figures S39–S45) and at the end of the experiment subjected the solution to mass spectrometry (Figures S40–44). We found that the signature m/z corresponding to the exact mass of the conjugates was dominant. Given that human serum albumin is the most abundant protein in blood, this study affirms the stability of our conjugates under relevant biological conditions.
Figure 2.

Representative absorption profile of conjugates. (Left) 1 in PBS over 72 h, (middle) 1 in DMEM over 72 h, and (right) 1 in conditioned DMEM over 72 h. The compound concentration was 25 μM. Three replicates were performed for each experiment.
Microsomal Stability Assay
To understand the metabolism of our compounds, the synthesized conjugates were subjected to a microsomal stability assay. Liver microsomes are essentially subcellular fractions containing membrane-bound drug metabolizing enzymes. The study was conducted by incubating our compounds at a final concentration of 15 μM with pooled human liver microsomes (0.5 mg ml–1) and then analyzed with UV–vis (Figures S45–S47) and ESI-MS (Figure S48). The conjugates were incubated for 0, 5, 10, 15, 30, and 45 min. The UV profiles of the microsomal assay extracts suggest that conjugates 1–3 are relatively stable to metabolism. Particularly, following the study involving compound 3, the mass spectrometry showed the presence of the [M + H] peak at 956.4635 corresponding to the exact mass of 3. This indicates that after 45 min of incubation with liver microsomes, there is an unmodified compound. Overall, the compounds are fairly stable under relevant biological conditions.
Cellular Response to PARP and PD-L1 Inhibition
We examined the cytotoxicity of our PARP1/PD-L1 inhibitor conjugates in a panel of cancerous cell lines. The cell lines selected were from human ovary, lung, and breast origin. The cells were treated with conjugates 1–3, PARPi, BMS-001, or olaparib alone in varying concentrations after which cells were incubated for 7 days and assayed using crystal violet. The inhibitory concentration that resulted in 50% cell death was extrapolated from dose–response curves and is summarized in Table 1.
Table 1. IC50 Values (μM) of 1–3, BMS-001, PARPi, and Olaparib against a Panel of Cancer Cell Lines after 7 Days of Exposurea.
| cell line | 1 | 2 | 3 | BMS-001 | PARPi | olaparib |
|---|---|---|---|---|---|---|
| OVCAR8 | 13.62 ± 1.7 | 5.02 ± 0.3 | 6.29 ± 1.9 | 25.53 ± 0.15 | 25.5 ± 00.1 | 11.14 ± 5.4 |
| MDA-MB-231 | 10.99 ± 6.7 | 9.51 ± 7.5 | 5.89 ± 5.6 | 14.83 ± 7.7 | 26.61 ± 1.13 | 14.00 ± 7.1 |
| A2780 | 12.68 ± 8.6 | 5.82 ± 10.2 | 5.40 ± 3.42 | 24.26 ± 10.2 | 25.68 ± 8.4 | 9.32 ± 0.2 |
| SKOV3 | 3.8 ± 34.48 | 5.13 ± 6.1 | 12.17 ± 0.43 | 24.36 ± 2.31 | 50.0 ± 70.1 | |
| H460 | 13.6 ± 0.2 | 10.64 ± 0.48 | 5.91 ± 0.13 | 21.49 ± 0.19 | 28.57 ± 0.82 | 8.1 ± 1.2 |
| HCC1937 | 10.79 ± 0.4 | 10.67 ± 0.59 | 5.26 ± 0.05 | 13.54 ± 0.11 | 20.77 ± 0.17 | 18.44 ± 2.4 |
Compounds were freshly prepared in DMSO and used immediately. DMSO concentration was <1%.
The conjugates, 1–3, displayed a 2–20-fold improved anticancer efficacy against cancer cells over individual agents alone. Particularly, the triple-negative breast cancer (TNBC), MDA-MB-231 possess endogenous expression of PD-L145 and represent a recalcitrant tumor subtype for chemotherapeutic treatment. The cell-killing effect of 3 was five times more potent than the olaparib derivative and ∼9-fold efficacious by mixing the two individual components in MDA-MB-231 cells (Figure 3). Dose–response curves of the compounds under investigation are shown in (Figure 3 and Figures S18–S23). Additionally, the IC50 of olaparib and its derivative (aka PARPi) were similar for all cell lines studied.
Figure 3.
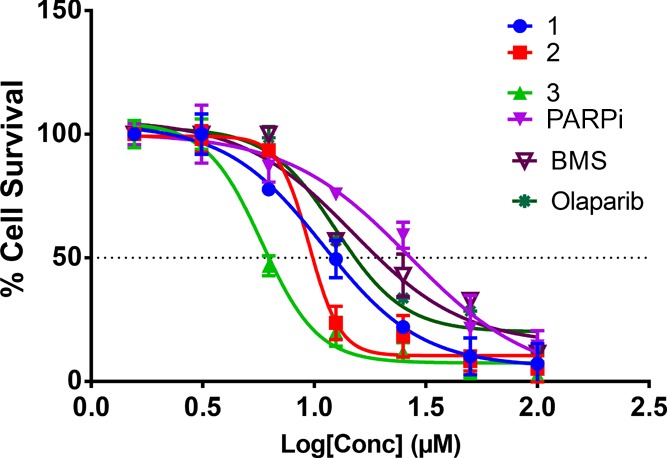
Dose–response curves of 1–3, BMS-001, PARPi, and olaparib in MDA-MB-231 cells for 7 days. The data were from three-independent replicates.
Importantly, cancer cells display unique phenotypes and genetic profiles that include mutated BRCA status and varying PD-L1 expression levels among others. The cell lines used in this study possess differentiated BRCA and PD-L1 status as shown in Table 2. The combination index (CI) of olaparib and BMS-001 was conducted to enable us to determine if there exists any form of synergy between olaparib and BMS-001.
Table 2. BRCA/PD-L1 Status of Panel of Cell Lines.
| cell line | BRCA status/expression | PD-L1 status |
|---|---|---|
| OVCAR8 | BRCA-hypermethylated/BRCA-deficient | N/Aa |
| H460 | BRCA1 wild-type | expressive |
| MDA-MB-231 | BRCA1 wild-type | expressive |
| HCC1937 | BRCA1-mutated | expressive |
| SKOV3 | BRCA1 wild-type | N/Aa |
| A2780 | BRCA1 wild-type | N/Aa |
N/A: not applicable.
Combination Effect of BMS-001 and Olaparib
We sought to determine the combination effect of olaparib, a first-in-class PARP inhibitor with a small-molecule PD-L1 inhibitor, BMS-001. The synergy of the combination of BMS-001 with olaparib was determined using the Chou–Talalay method.46 The combination index (CI) was calculated using Compusyn software, which computes the combination index based on the Chou–Talalay CI formula:
D1 and D2 represent the IC50 of olaparib and BMS-001, respectively, whereas Dm1 and Dm2 are the IC50 values of olaparib and BMS-001 alone, respectively. Based on the CI values, the combination is deemed synergistic, additive, or antagonistic. If CI < 1, the drug combination pair is synergistic; CI = 1, the pair is additive; or if CI > 1, the pair is antagonistic. Further classifications are as follows: (1) if CI <0.7, then the pair exhibits strong synergy, (2) 0.7–0.85 implies moderate synergism,(3) 0.85–0.9 implies slight synergism, (4) 1.0 implies an additive pair, and (5) if CI >1, it is antagonistic. Notably, olaparib–BMS-001 combinations that are cytotoxic to 90% of cells (Fa = 0.9) after 72 h were determined across all cell lines as displayed in Figures S24–S33. The combination of olaparib–BMS-001 was found to be synergistic across several molar ratio points in a panel of cell lines (Table 3). Most importantly, at 1:1 combination ratio, the olaparib–BMS-001 pair was found that to be synergistic across the selected cell lines. This implies that an equimolar presence of olaparib and for that matter PARPi with BMS-001 potentiates cytotoxicity in cancer cells. Consistent with this observation is the cytotoxicity results of the conjugates that show an improved antiproliferative effect. Taken together, coefficients of synergy were established for the treatment of cancer cells by BMS-001 and olaparib, which corroborates dual targeting of PD-L1 and PARP.
Table 3. Summary of the Combination Index (CI) Values for Olaparib–BMS-001 Combination at Fa = 0.9a.

CI value indicates the type of the combination effect: CI = 1, additive (gray); CI > 1, antagonistic (red); CI < 1, synergistic (strong synergy <0.7 (green), moderate synergy (pale green) ≤0.7–0.85, and 0.85–0.9, slight synergism (aqua)). Column headers indicate the olaparib–BMS-001 ratio.
Apoptosis
Furthermore, we sought to understand how the conjugates affected cell death, in the context of apoptosis. MDA-MB-231 cells were treated with 10 μM compounds—BMS-001, olaparib, PARPi, 1, 2, or 3. As depicted in Figure 4, 1–3 induced significant early- to late-stage apoptosis-mediated cell death: 26% for compound 1, 60% for 2, and 60% for 3. Comparatively, olaparib (25%) and PARPi (22%) exhibited a relatively lower early- to late-stage apoptosis-related cell death. BMS-001 also showed ∼19% early- to late-stage apoptosis-related cell death. Specifically, we observed a large cell population occurring at late-stage apoptosis for compounds 2 and 3 as shown in Figure 4. The conjugates showed enhanced in vitro potency via late-stage apoptosis in comparison to PARP and PD-L1 inhibitor moieties.
Figure 4.
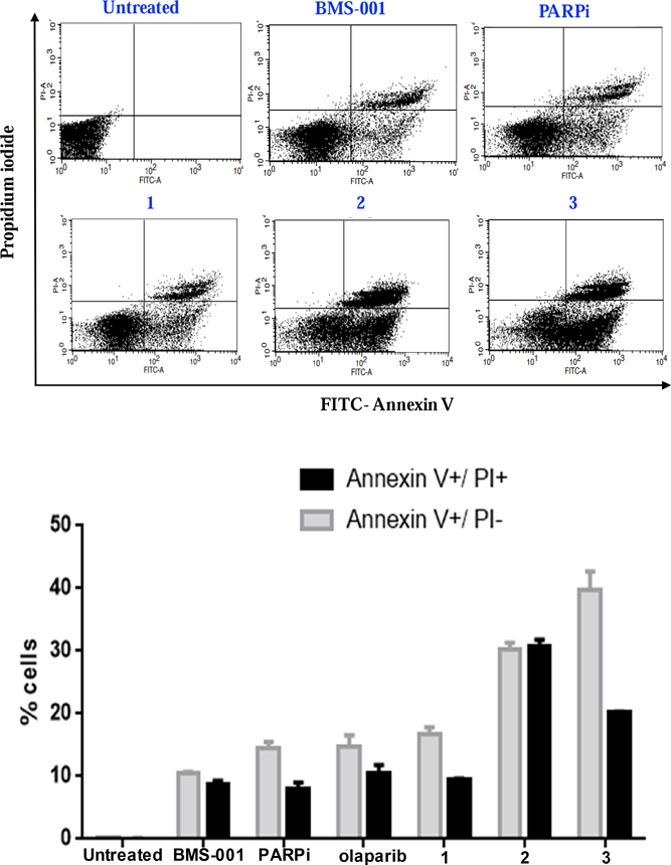
FITC Annexin V/PI apoptosis dead cell assay detect apoptosis in MDA-MB-231 cells. The percentages of early apoptotic (Annexin V+/PI–, gray) and late apoptotic (Annexin V+/PI+, black) cells were quantified. Data are presented as mean ± SD and were representative of three independent biological experiments.
Additionally, these data imply that the potency of conjugates (1–3) is not additive but rather synergistic. Taken together, these results are consistent with studies that show tumor growth inhibition by combining olaparib and anti-PD-L1 mAb. It is worth noting that several clinical trials are currently on going to evaluate the combined effect of FDA approved PARP1 inhibitors and immune checkpoint antibodies. Effective small-molecule alternatives could be a game-changer while reducing adverse immunologic effects.
PARP1 and PD-L1 Potentiation by Conjugates
Effect on PARP
Intracellular effects of the compounds under investigation in this report were examined using immunoblotting. Modulation of PARP by 1–3 in cells was evaluated by immunoblotting, using cancer cells that endogenously expressed PD-L1 (Figure 5a). Consequently, H460 (lung) cells were treated with 10 or 20 μM of various compounds for 24 h and analyzed by Western blotting. In addition, MDA-MB-231 (breast) cells were treated with 10 μM, and lysates were collected for 24, 48, and 72 h for Western blotting. It is evident that H460 cells treated with the conjugates inhibited PARP in a dose-dependent manner. The conjugates equally inhibit PARP in MDA-MB-231 cells (Figure 5b), implying that the various conjugates potentiate PARP expression. It can be inferred from our design principle that all the conjugates (1–3) possess a PARP inhibiting scaffold whose functional activity remains uncompromised, at least at the cellular level. This is consistent with our docking studies where all our conjugates interacted specifically with amino acid residues in the catalytic domain of the PARP1 protein (vide infra).
Figure 5.

(a) H460 or (b) MDA-MB-231 cells were treated with compounds 1–3 for indicated time points (24–72 h) and concentration (10 or 20 μM). Lysates were generated and subjected to immunoblotting. Densitometric quantification of blots are shown on the right of each blot. Data were analyzed as mean ± SD and were representative of three independent biological experiments.
PARP Inhibition Upregulates PD-L1
The inhibition of PARP leads to the upregulation of PD-L1 in some cancer cells.14 The PD-1/PD-L1-mediated anticancer suppression requires the inhibition of the coupling of PD-1 to its endogenous peptidic partner, PD-L1, implying that if either PD-L1 or PD-1 can be upregulated, then the inhibition of this pathway becomes relatively easier. Of note, MDA-MB-231 cells endogenously express the PD-L1 protein, and the PD-1/PD-L1 ligation occurs at the cell surface, leading to immunosuppressive effects.47 To determine if our conjugates as well as the reference PARP inhibitors can downregulate cell surface PD-L1, MDA-MB-231 cells were treated with 10 μM conjugates, and reference compounds for 24 h, and subsequently incubated with a fluorophore-labeled PD-L1 antibody and then subjected to FACS (Figure 6). Cell surface PD-L1 was upregulated in cells, after treatment with olaparib and PARPi. However, they were downregulated after treatment with BMS-001 and the conjugates 1–3. Importantly, 3 was found to significantly downregulate cell surface PD-L1 than the reference PD-L1 inhibitor, BMS-001, establishing it as our best candidate for PD-L1 inhibition.
Figure 6.
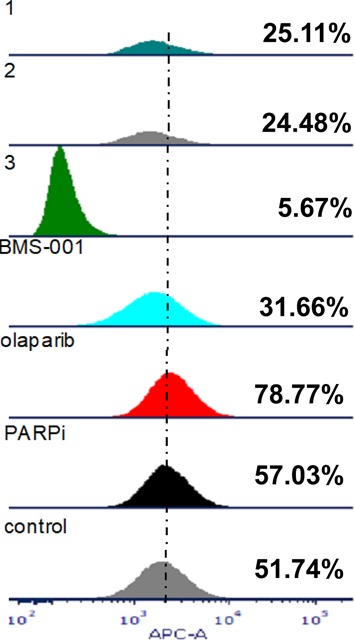
PARP inhibition upregulates PD-L1 in MDA-MB-231 cells and can be attenuated by small-molecule inhibitors of PD-L1. Treated cells were subjected to FACS after 24 h of treatment with compounds. Quadruplicate experiments were performed.
Molecular Docking Insights
Insight into the interaction of these dual-action conjugates will be beneficial for further optimization and discovery. Therefore, we sought to study the interaction of our conjugates with the various proteins (herein, PARP and PD-L1) under study. To unravel, with cheminformatic finesse, the possibility of our conjugates to interrogate both PARP and PD-L1 proteins, we prepared and docked our conjugates into the crystal structures of the respective protein structures. SYBYL-X molecular modeling and simulation suite (version 2.1) was used for the generation of the relevant protonated and tautomeric molecular forms of PARPi and the conjugates. The preparation of the crystal structures of the proteins and ligand files can be found elsewhere (see Experimental Section). PARP has three functional domains: the automodification domain, N-terminal DNA-binding, and C-terminal catalytic domain.48,49 The PARP1 catalytic domain has a major donor site (ART domain), which possesses a β-α-loop-β-α signature NAD+-specific binding motif.50 Conjugate 3 and PARPi share a similar scaffold in that their phthalazinone moiety is a nicotinamide mimic. Therefore, it is expected that both will bind to the nicotinamide-specific binding pocket (NI site). Unsurprisingly, 3 and reference compound, PARPi, both bind to the PARP protein in the NI site of its C-terminal catalytic domain. Conjugate 3 anchors itself into this nicotinamide-specific binding pocket (NI site) via several hydrogen bond networks and hydrophobic interactions (Figure 7a). More precisely, the carboxamide portion of the phthalizinone functional group of conjugate 3 forms two hydrogen bonds with Arg865 (αJ) and Tyr896 (βd) in the NI site of the PARP1 catalytic domain. But PARPi interacts via the carbonyl of its phthalizinone with Met890 (D-loop) in the NI site. Additional π–π interactions in Tyr907 also stabilize conjugate 3 in the NI region. Both PARPi and conjugate 3 interact with the glutamine backbone (Gln759) within the phosphate binding site (PH site). However, conjugate 3 also extends into other parts of the donor site of PARP1’s catalytic domain. Conjugate 3 forms hydrogen bond networks with histidine and arginine residues (His862 and Arg865, respectively), which are in the adenine ribose-binding (AD) pocket of the catalytic domain. It can be inferred from previous research that potency and selectivity can be conferred to inhibitors simply by extending reactive side groups from the NI site into other binding pockets of the catalytic domain.50 We propose that as conjugate 3 sprawls into the AD site, it is expected that enhanced PARP inhibition will be achieved.
PD-L1 hot spots are characterized by key amino acid residues such as Phe19, Tyr56, Glu58, Gln66, Arg113, Met115, and Tyr123.17,51 Docking results show that interaction of conjugate 3 and BMS-001 within the binding cleft of the PD-L1 dimer via a miscellany of interactions: hydrogen bonding, salt bridge interactions, and hydrophobic interactions (Figure 8). In addition to the two hydrogen bonds formed between the O atom on the ethoxy-ethoxy-linker of conjugate 3 with the Arg113 residues in the binding pocket, there exists further hydrogen bond stabilization between the carbonyl in proxy to the piperazine ring (of 3) with the Asp63 side chain (Figure 8b). Other noncovalent interactions, π–π interactions with tyrosine residues, in the binding cleft further stabilize conjugate 3. These modeling results suggest that there is no loss of functionality, in the context of PARP and PD-L1 inhibition profiles of 3. Rather, these properties are predicted to be enhanced in the conjugates.
Cell Cycle Studies
To gain insight into the DNA content response to the dual-action agents, cell cycle studies were conducted in the presence or absence of compound 3, olaparib, and BMS-001 (Figure 9a,b). Cells were treated with 5 μM of various compounds, at 24 and 48 h incubation points, and then subjected to flow cytometric propidium iodide analysis. After 24 h of treatment with compound 3, an increase in the fraction of cells in the G1 phase was observed, that is, 71.07% in untreated cells to 75.56% in cells incubated with 3. Consistent with 3, olaparib and BMS-001 induced G1 phase accumulation after 24 h (Figure 9c). Changes in the number of cells in the S phase for 3-treated cells peaked at 48 h and returned to initial levels by 72 h, whereas negligible changes were observed for olaparib and BMS-001 under the same duration. Recent studies on the combination of cyclic-dependent kinase inhibitor, dinaciclib and PARP inhibition in TNBCs demonstrate increased sub-G1 cell cycle population.52 This is quite consistent with our observation of 3 in MDA-MB-231 cells.
Figure 9.

(a) Histograms of the DNA content of each phase of the cell cycle in MDA-MB-231 cells following treatment with compounds. (b) Bar chart extrapolated from the DNA histograms in panel (a). (c) Comparison of G1 phase accumulation. Data were analyzed by means ± SD and were representative of three independent biological experiments.
T-Cell Proliferation
We investigated the functional activity of PD-L1 modulation by studying the proliferation of T-cells co-cultured with PD-L1 expressing MDA-MB-231 cells. Following the hypothesis that PD-L1 upregulation in cancer cells renders the cells resistant to T-cell killing, we studied the effect of PD-L1 inhibition by our conjugates on T-cell proliferation. Briefly, human peripheral blood mononuclear cells (PBMCs) were activated with 100 ng/mL CD3 antibody, 100 ng/mL CD28 antibody, and 10 ng/mL r-IL-2. The activated PBMCs were labeled with carboxyfluoresceine succinimidyl ester (CFSE) and co-cultured with conjugate-treated or untreated MDA-MB-231cells. The labeled PBMCs were subjected to FACS analysis on day 0 and day 5 (Figure 10). After the 5 day treatment, co-cultured T-cells with MDA-MB-231 cells treated with 1–3 proliferated exponentially with complete reduction of the fluorescent signal corresponding to original T-cell population and the consequent formation of multiple peaks arising from dye dilution due to cell division. This implies that PD-L1 inhibition induced by our conjugates promotes proliferation and may restore immunity by blocking the PD-1/PD-L1.
Figure 10.

T-cell proliferation assay using CFSE stained anti-CD3/CD28-activated PBMCs co-cultured with treated or untreated MDA-MB-231 cells. Three independent experiments with different PBMCs were used.
Conclusions
In summary, we have developed small-molecule hybrid inhibitors of PARP and PD-L1. The compounds were synthesized and well-characterized, and their purity was confirmed. In all our biological characterization, our conjugates, 1–3, offered superior synergistic cytotoxicity, apoptosis, and PD-L1 inhibition than the individual inhibitors, BMS-001 and PARPi alone. Additionally, these compounds act as proof-of-concept tools to delineate the PARP1/PD-L1 axis to better understand the cross-talk that exist between these pathways. Using molecular docking, we gleaned insights into the interaction of small-molecule conjugates with the respective proteins, offering the opportunity for further optimization and discovery of more efficacious dual-action compounds. Future studies outlined include intracellular kinetic rates of interaction of the compounds with its respective targets, in vivo studies using syngeneic breast cancer mouse models and detailed PARP1/PD-L1 characterization in vivo.
Experimental Section
Chemistry
General Materials and Methods
Reagents and solvents used were purchased from commercial vendors (Acros, MilliporeSigma, USA) and used without further purification unless otherwise stated. Acetonitrile (CH3CN) and dimethylformamide (DMF) were used from AcroSeal anhydrous bottle containing 3 Å molecular sieves. Dry dichloromethane (CH2Cl2) was distilled over CaH2 and stored over 4 Å molecular sieves in an anhydrous solvent bottle. Tetrahydrofuran (THF) was distilled from a mixture of sodium metal and benzophenone ketyl under nitrogen.
Physical and Spectroscopic Measurements
Coupling constant, J, was reported in Hertz unit (Hz). Nuclear magnetic resonance spectroscopy (1H, 13C NMR) was recorded on a Varian Unity 400/500 NMR with a Spectro Spin superconducting magnet in the University of Kentucky NMR Facility. Chemical shifts were reported in ppm, 1H NMR spectra were internally referenced to solvent signals (1H NMR: DMSO at δ = 2.50 ppm, CD3CN at δ = 1.94 ppm; 13C NMR: DMSO δ = 39.52 ppm). Column chromatography was done on a CombiFlash from Teledyne ISCO. Mass spectra were obtained on a high-resolution mass spectrometer using the electrospray ionization (ESI) method. The HPLC data were obtained on an Agilent 1100 series HPLC using a normal-phase column. GC–MS data were obtained on an Agilent 6890N Network GC system.
Synthesis of 2,6-Dimethoxy-4-[(2-methyl[1,1′-biphenyl]-3-yl)methoxy]benzaldehyde (c)
Diisopropyl azodicarboxylate (1.027 mL, 5.04 mmol) in dry THF was added dropwise to a cooled (0 °C) solution of 4-hydroxy-2,6-dimethoxybenzaldehyde (918.68 mg, 5.04 mmol), 3-hydroxymethyl-2-methylbiphenyl (1000 mg, 5.04 mmol), and triphenylphosphine (1322.73 mg, 5.04 mmol) in dry THF, and the resulting solution was stirred overnight under nitrogen at room temperature. THF was removed in vacuo, and the crude was then dissolved in diethyl ether. This solution was cooled on ice for 3 h and filtered to remove the triphenylphosphine oxide. The filtrate was concentrated in vacuo to give the crude residue, which was purified by flash column chromatography (solid phase: silica gel and mobile phase: 1:1 ethyl acetate–hexane) to afford the white crystals of the product, 2,6-dimethoxy-4-[(2-methyl[1,1′-biphenyl]-3-yl)methoxy]benzaldehyde. Yield: (1698.4 mg, 92%); Rf = 0.55 (solvent system: 1:1 ethyl acetate–hexane). 1H NMR (400 MHz, CDCl3): δ 10.40 (s, 1H), 7.45 (q, J = 5.5, 3.8 Hz, 3H), 7.40 (d, J = 7.1 Hz, 1H), 7.36–7.30 (m, 4H), 6.23 (s, 2H), 5.18 (s, 2H), 3.92 (s, 6H), 2.30 (s, 3H). 13C NMR (101 MHz, CDCl3): δ 187.67, 165.44, 164.14, 143.27, 141.75, 134.54, 134.17, 130.66, 129.34, 128.15, 127.00, 125.75, 109.12, 91.08, 76.69, 69.52, 56.06, 16.26. HRMS ESI/Q-TOF for C23H22O4 calcd mass, 362.1518; found [M]+, 362.1520 m/z.
Synthesis of (S)-1-(2,6-Dimethoxy-4-((2-methyl-[1,1′-biphenyl]-3-yl)methoxy)benzyl)piperidine-2-carboxylic Acid (BMS-001)
A 250 mL well-dried Schlenk flask was charged with 2,6-dimethoxy-4-((2-methylbiphenyl-3-yl)methoxy)benzaldehyde (1000 mg, 2.76 mmol), (S)-piperidine-2-carboxylic acid (356.1 mg, 2.76 mmol), and sodium triacetoxyborohydride (1755 mg, 8.28 mmol). The flask was purged and back-filled with nitrogen in three cycles. Dry dichloromethane was added to the mixture and stirred at 85 °C for 2 h after which it was allowed to cool to room temperature and then diluted with 10 mL of DCM and 10 mL of distilled water. The aqueous layer was extracted with 3:1 DCM–IPA (10 mL × 2). The combined organic layer was dried over anhydrous magnesium sulfate and filtered. The filtrate was concentrated in vacuo to give the crude residue, which was purified by flash column chromatography using silica gel with a mobile phase of 5% methanol in DCM to afford the product, (S)-1-(2,6-dimethoxy-4-((2-methyl-[1,1′-biphenyl]-3-yl)methoxy)benzyl)piperidine-2-carboxylic acid as a white crystalline solid. Yield: (711.2 mg, 54%); Rf = 0.31 (solvent system: 5% methanol–DCM). 1H NMR (400 MHz, DMSO-d6): δ 7.50–7.40 (m, 3H), 7.36 (d, J = 7.3 Hz, 1H), 7.27 (dd, J = 13.6, 7.9 Hz, 3H), 7.18 (d, J = 7.6 Hz, 1H), 6.40 (s, 2H), 5.16 (s, 2H), 4.10 (s, 2H), 3.77 (s, 6H), 3.07 (d, J = 38.2 Hz, 2H), 2.66 (s, 1H), 2.19 (s, 3H), 1.80 (s, 2H), 1.54 (s, 2H), 1.37 (s, 2H). 13C NMR (101 MHz, DMSO): δ 176.21, 161.56, 161.55, 160.33, 142.71, 141.86, 135.83, 134.60, 130.26, 129.64, 127.45, 126.05, 118.10, 92.06, 69.24, 56.42, 48.65, 46.17, 39.38, 29.52, 25.84, 21.13, 16.42. HRMS ESI/Q-TOF for C29H33NO5 calcd mass, 475.2359; found [M + H]+, 476.2360 m/z.
Synthesis of 4-[[4-Fluoro-3-[[4-(2-hydroxyethyl)-1-piperazinyl]carbonyl]phenyl]methyl]-1(2H)-phthalazinone (e)
A mixture of 4-(4-fluoro-3-(piperazine-1-carbonyl)benzyl)phthalazin-1(2H)-one (1500 mg, 4.09 mmol), potassium carbonate (1 mg, 12.27 mmol), 2-bromoethanol (1380 mg, 8.19 mmol), and 12 mL of DMF was stirred at 65 °C for 24 h. The reaction was cooled to room temperature, and 20 mL of distilled water was added. The aqueous layer was extracted with DCM (10 mL × 2), and the resulting organic layer was dried over anhydrous magnesium sulfate and filtered. The filtrate was concentrated in vacuo to give the crude residue, which was purified by flash column chromatography using silica gel with a mobile phase of 5% methanol in DCM to afford the product, e. Yield: (1310.2 mg, 78%). Rf = 0.27 (solvent system: 5% methanol–DCM). 1H NMR (400 MHz, Chloroform-d): δ 10.36 (s, 1H), 8.44 (s, 1H), 7.75 (d, J = 17.5 Hz, 3H), 7.33 (s, 2H), 7.05 (d, J = 8.5 Hz, 1H), 4.27 (s, 2H), 3.98 (s, 2H), 3.79 (s, 2H), 3.50 (s, 2H), 3.10 (d, J = 7.4 Hz, 2H), 2.78 (d, J = 26.1 Hz, 4H). 13C NMR (101 MHz, CDCl3): δ 164.86, 160.39, 158.29, 155.83, 145.56, 134.26, 133.72, 131.65, 129.55, 129.27, 128.33, 127.18, 125.02, 116.29, 116.07, 59.64, 57.45, 53.77, 53.08, 52.59, 46.32, 42.01, 41.21, 37.64. LRMS ESI for C22H23FN4O3 calcd mass, 410.17; found [M + H]+, 411.18 m/z.
Synthesis of 4-(4-Fluoro-3-(4-(2-(2-(2-hydroxyethoxy)ethoxy)ethyl)piperazine-1-carbonyl)benzyl)phthalazin-1(2H)-One (f)
A mixture of 4-(4-fluoro-3-(piperazine-1-carbonyl)benzyl)phthalazin-1(2H)-one (1500 mg, 4.09 mmol), potassium carbonate (1695.78 mg, 12.27 mmol), 2-(2-(2-chloroethoxy)ethoxy)ethanol (1380 mg, 8.19 mmol), and 12 mL of DMF was stirred at 85 °C for 24 h. The reaction was allowed to cool, and 20 mL of distilled water was added. The aqueous layer was extracted with DCM (10 mL × 2). Then, the resulting organic layer was dried over anhydrous magnesium sulfate and filtered. The filtrate was concentrated in vacuo to give the crude residue, which was purified by flash column chromatography using silica gel with a mobile phase of 5% methanol in DCM to afford the product, f. Yield: (1753.9 mg, 86%). Rf = 0.19 (solvent system: 5% methanol–DCM). 1H NMR (400 MHz, Chloroform-d): δ 10.62 (s, 1H), 8.49–8.44 (m, 1H), 7.74 (dd, J = 15.2, 7.4 Hz, 3H), 7.30 (d, J = 6.3 Hz, 2H), 7.01 (t, J = 8.7 Hz, 1H), 4.27 (s, 2H), 3.82 (s, 2H), 3.74–3.70 (m, 2H), 3.66 (s, 3H), 3.64–3.60 (m, 5H), 3.33 (s, 2H), 2.68–2.59 (m, 4H), 2.49 (s, 1H). 13C NMR (101 MHz, CDCl3): δ 164.78, 160.32, 155.89, 145.57, 133.68, 131.62, 129.57, 128.37, 127.19, 125.05, 116.26, 116.05, 76.71, 72.59, 70.21, 68.46, 61.62, 57.58, 53.59, 53.04, 46.79, 41.69, 37.64. HRMS ESI/Q-TOF for C26H31FN4O5 calcd mass, 498.2278; found [M + H]+, 499.2280 m/z.
Synthesis of (S)-4-(3-(4-(1-(2,6-Dimethoxy-4-((2-methyl-[1,1′-biphenyl]-3-yl)methoxy)benzyl)piperidine-2-carbonyl)piperazine-1-carbonyl)-4-fluorobenzyl)phthalazin-1(2H)-one (1)
To (2,6-dimethoxy-4-((2-methyl-[1,1′-biphenyl]-3-yl)methoxy)benzyl)piperidine-2-carboxylic acid (359.54 mg, 0.756 mmol) was added 4-(4-fluoro-3-(piperazine-1-carbonyl)benzyl)phthala-zin-1(2H)-one (277 mg, 0.756 mmol) in EDCl (434.78 mg,2.268 mmol), HOBt (306.56 mg, 2.268 mmol) in dry DCM, and DIPEA (0.4 mL, 2.268 mmol). The reaction mixture was then stirred at room temperature for 24 h under an inert atmosphere. The mixture was diluted with 20 mL of distilled water. The aqueous layer was extracted with DCM (10 mL × 2), and the combined organic layers were dried over anhydrous magnesium sulfate and filtered. The filtrate was concentrated in vacuo to give the crude residue, which was purified by flash column chromatography using silica gel with a mobile phase of 5% methanol in DCM to afford the product as a brownish powder. Yield: (220.6 mg, 70%); Rf = 0.43 (solvent system: 6% methanol in DCM). 1H NMR (400 MHz, DMSO-d6): δ 12.55 (s, 0H), 8.05–7.65 (m, 2H), 7.41 (d, J = 7.2 Hz, 2H), 7.38 (s, 5H), 7.26 (d, J = 7.5 Hz, 2H), 7.16 (s, 2H), 6.36 (d, J = 39.0 Hz, 2H), 5.11 (t, J = 14.8 Hz, 2H), 4.27 (s, 1H), 3.73 (d, J = 27.9 Hz, 7H), 3.61 (d, J = 9.6 Hz, 2H), 3.31–3.25 (m, 70H), 3.12 (s, 1H), 2.94 (s, 1H), 2.16 (s, 2H), 1.64 (s, 2H), 1.29 (s, 1H). HRMS ESI/Q-TOF for C49H50FN5O6 calcd mass, 823.3745; found [M + H]+, 824.3899 m/z.
Synthesis of 2-(4-(2-Fluoro-5-((4-oxo-3,4-dihydrophthalazin-1-yl)methyl)benzoyl)piperazin-1-yl)ethyl (S)-1-(2,6-Dimethoxy-4-((2-methyl-[1,1′-biphenyl]-3-yl)methoxy)benzyl)piperidine-2-Carboxylate (2)
To (S)-1-(2,6-dimethoxy-4-((2-methyl-[1,1′-biphenyl]-3-yl)methoxy)benzyl)piperidine-2-carboxylic acid (550 mg, 1.16 mmol), EDCl (222.4 mg,1.16 mmol), and HOBt (156.8 mg,1.16 mmol) in dry DCM were added DIPEA (0.2 mL, 1.16 mmol) and compound e (474.7 mg, 1.16 mmol). The reaction mixture was then stirred at room temperature for 24 h under inert conditions. The mixture was diluted with 20 mL of distilled water. The aqueous layer was extracted with DCM (10 mL × 2), and the combined organic layers were dried over anhydrous magnesium sulfate and filtered. The filtrate was concentrated in vacuo to give the crude residue, which was purified by flash column chromatography using silica gel with a mobile phase of 5% methanol in DCM to afford the product as a yellowish powder. Yield: (783.3 mg, 78%); Rf = 0.36 (solvent system: 5% methanol in DCM).
1H NMR (400 MHz, Chloroform-d): δ 10.02 (s, 1H), 8.41–8.35 (m, 1H), 7.66 (dd, J = 20.7, 6.9 Hz, 3H), 7.35 (d, J = 7.1 Hz, 3H), 7.30 (d, J = 6.9 Hz, 1H), 7.24 (d, J = 7.6 Hz, 4H), 6.94 (s, 1H), 6.17 (s, 2H), 5.03 (s, 2H), 4.19 (s, 2H), 3.80–3.51 (m, 10H), 3.19 (s, 3H), 2.60–2.45 (m, 3H), 2.38 (d, J = 12.4 Hz, 2H), 2.33–2.24 (m, 1H), 2.20 (s, 3H), 1.98 (dd, J = 21.9, 7.3 Hz, 2H), 1.63–1.33 (m, 6H). 13C NMR (101 MHz, CDCl3): δ 170.37, 162.32, 160.40, 143.12, 141.74, 134.51, 134.43, 130.51, 129.30, 128.51, 128.32, 128.08, 126.90, 125.67, 111.22, 110.18, 110.07, 110.04, 110.01, 110.00, 109.95, 109.92, 109.90, 109.85, 109.82, 98.45, 91.13, 77.32, 77.21, 77.00, 76.69, 69.46, 64.75, 55.87, 55.82, 49.16, 46.33, 25.41, 21.93, 21.22, 21.01, 16.24, 14.17. HRMS ESI/Q-TOF for C51H54FN5O7 calcd mass, 867.4008; found [M + H]+, 868.4140 m/z.
Synthesis of 2-(2-(2-(4-(2-Fluoro-5-((4-oxo-3,4-dihydrophthalazin-1-yl)methyl)benzoyl)piperazin-1-yl)ethoxy)ethoxy)ethyl (S)-1-(2,6-Dimethoxy-4-((2-methyl-[1,1′-biphenyl]-3-yl)methoxy)benzyl)piperidine-2-carboxylate (3)
To a stirring solution of (S)-1-(2,6-dimethoxy-4-((2-methyl-[1,1′-biphenyl]-3-yl)methoxy)benzyl)piperidine-2-carboxylic acid (476.96 mg, 1.00 mmol) in dry DCM were added EDCl (192.3 mg, 1.00 mmol), HOBt (135.58 mg, 1.00 mmol), DIPEA (0.174 mL, 1.00 mmol), and f (500 mg, 1.00 mmol). The reaction mixture was then stirred at room temperature for 24 h under inert conditions. The mixture was diluted with 20 mL of distilled water. The aqueous layer was extracted with DCM (10 mL × 2), and the combined organic layer was dried over anhydrous magnesium sulfate and filtered. The filtrate was concentrated in vacuo to give the crude residue, which was purified by flash column chromatography using silica gel with a mobile phase of 6% methanol in DCM to afford the product as a brownish powder. Yield: (675 mg, 70%); Rf = 0.33 (solvent system: 6% methanol in DCM). 1H NMR (400 MHz, Chloroform-d): δ 10.41 (s, 1H), 8.44 (s, 1H), 7.82–7.68 (m, 3H), 7.42 (t, J = 7.1 Hz, 3H), 7.36 (d, J = 7.1 Hz, 1H), 7.31 (d, J = 7.4 Hz, 4H), 7.00 (t, J = 8.8 Hz, 1H), 6.23 (s, 2H), 5.08 (s, 2H), 4.43 (s, 1H), 4.26 (s, 3H), 3.76 (s, 6H), 3.68–3.57 (m, 6H), 3.32 (d, J = 19.9 Hz, 3H), 2.56 (s, 4H), 2.43 (s, 2H), 2.26 (s, 3H), 2.15–1.44 (m, 6H). 13C NMR (101 MHz, CDCl3): δ 164.71, 160.39, 160.23, 158.22, 155.81, 155.76, 145.48, 143.03, 141.81, 134.82, 134.48, 134.06, 133.61, 131.53, 131.17, 130.36, 129.51, 129.32, 129.07, 128.31, 128.07, 127.11, 126.87, 125.63, 125.05, 124.28, 124.10, 116.18, 115.96, 91.11, 77.31, 70.35, 69.29, 68.75, 57.51, 55.61, 53.59, 52.99, 46.89, 41.80, 37.67, 30.29, 29.67, 16.23. HRMS ESI/Q-TOF for C55H62FN5O9 calcd mass, 955.4532; found [M + H]+, 956.4657 m/z.
Biology
FACS and UV–vis Characterization
UV–vis data were obtained using a Shimadzu UV-1280 UV–vis spectrophotometer. Flow data were obtained from the University of Kentucky Flow Cytometry and Immune Function Core Facility.
Cell Culture
All cells were obtained from ATCC and routinely grown in a humidified incubator at 37 °C with 5% CO2. MDA-MB-231 cells were grown in DMEM containing 10% FBS, 1% penicillin/streptomycin, and 4 mM glutamine. A2780, OVCAR8, HCC1937, H460 and SKOV3 cells were grown in RPMI containing 10% FBS, 1% penicillin/streptomycin, and 4 mM glutamine. All media and supplements were obtained from VWR, USA.
Cell Viability Studies
The various established human cancer cell lines were seeded in a 96-well plate (2 × 103 cells/well) and were incubated with RPMI or DMEM supplemented with 10% FBS (150 μL) for 24 h and at 37 °C. Cells were treated with the BMS-001, PARPi, olaparib or conjugates 1–3 at increasing concentrations for 7 days. The various stock solutions were prepared in DMSO. Thereafter, cellular viability was assessed via the crystal violet assay. Measurements of absorbance were subsequently performed using a GENios plate reader at 570 nm (peak absorbance). All experiments were conducted in triplicates. Also, cell viability experiments were done to study the synergy of olaparib and BMS001, at increasing concentrations for 72 hours.
Apoptosis Studies
MDA-MB-231 cells (500,000 cells per well) were plated into six-well plates for 24 h. After complete adhesion, the cells were treated with olaparib, BMS-001, or 1–3 at a fixed concentration (10 μM) for 48 h and harvested by trypsinization. The Annexin V-FITC Apoptosis Detection Kit (BD Biosciences), according to the manufacturer protocol, was used to determine the fraction of cells that underwent apoptosis, using fluorescence-activated cell sorting (FACS) analysis (BD Biosciences, USA) and by following the manufacturer protocol. The data were analyzed by FlowJo software.
Cell Cycle Analysis
MDA-MB-231 cells (500,000 cells per well) were plated into six-well plates for 24 h. After complete adhesion, the cells were treated with a fixed concentration (5 μM) of olaparib, BMS-001, 1, 2, or 3, along with the various untreated controls for 24, 48, and 72 h. The cells were harvested by trypsinization and pelleted. The collected pellets were washed twice with 1 mL of PBS. Fifty microliters of 100 μg/mL RNase solution was added and incubated on ice for 20 min. Two hundred microliters of 50 μg/mL propidium iodide solution was added to each sample, resuspended, and filtered through the cell strainer cap of a 5 mL Corning Falcon test tube. Then, the samples were analyzed with flow cytometry, and the data were analyzed using Modfit.
Immunoblotting
MDA-MB-231 cells in a fresh complete medium were plated (500,000 cells per well) into six-well plates for 24 h. After complete adhesion, cells were treated with olaparib or the conjugates, 1–3 at concentration of 10 μM for 24 h, 48, and 72 h, scraped into SDS-PAGE loading buffer (64 mM Tris-HCl (pH 6.8)/9.6% glycerol/2% SDS/5% β-mercaptoethanol/0.01% bromophenol blue), and incubated at 95 °C on a heat block for 10 min. The cell samples were cooled and stored at −20 °C until ready for use. Whole cell lysates were resolved by 4–20% sodium dodecylsulfate polyacrylamide gel electrophoresis (SDS-PAGE, 200 V for 25 min) followed by electrotransfer to the polyvinylidene difluoride membrane, PVDF (350 mA for 1 h). Membranes were incubated in blocking buffer containing using 5% (w/v) bovine serum albumin (BSA) in TBST (TBS/0.1% Tween 20) for 1 h at room temperature. Subsequently, the membranes were probed separately with PARP or β-actin, primary antibodies (Cell Signaling, USA) in blocking buffer overnight at 4 °C (Cell Signaling Technology and Santa Cruz Biotechnology, USA). After washing three times, over a period of 15 min, with TBST (3 × 5 mL), the membrane was incubated with the related alkaline phosphatase-conjugated secondary antibodies (Cell Signaling Technology) in fresh BSA blocking solution. Protein–antibody conjugates were visualized using one-step NBT–BCIP (nitro blue tetrazolium chloride (NBT) and 5-bromo-4-chloro-3′-indoyl phosphate p-toluidine (BCIP)) visualization solution (Thermo Fisher, USA), according to the manufacturer instruction. Images of the blots were then digitally obtained.
Detection of Cell Surface PD-L1
MDA-MB-231 cells (500,000 cells per well) were plated in a six-well plate and treated with the various compounds—PARPi, olaparib, BMS-001, compounds 1, 2, or 3. For the detection of cell surface PD-L1, MDA-MB-231 cells were suspended in 100 μL of cell staining buffer (#420201, BioLegend) and incubated with the APC-conjugated anti-human PD-L1 antibody (#329708, BioLegend) on ice for 30 min. After washing in the staining buffer, the stained cells were analyzed by FACS.
Stability Studies of Conjugates
A DMSO stock solution of 1, 2, or 3 was diluted to 25 μM solutions with either PBS or DMEM. Three milliliters of each sample was aliquoted into a cuvette, and the absorbance was measured on a Shimadzu UV-1280 UV–Vis spectrophotometer. The data were plotted using GraphPad Prism.
Molecular Modeling
Compound Database
The structure of olaparib (in .mol2) was downloaded from the ZINC database. The structures of 1, 2, 3, PARPi, and BMS-001 were drawn with Chemdraw software and prepared into a 3D .sdf/.mol file by using the ligand preparation tool in SYBYL-X
Protein Preparation
The crystal structure of the PARP1 protein bound with niraparib (PDB: 4R6E) and PD-L1 protein in complex with small molecule, 4-[[4-[[3-(2,3-dihydro-1,4-benzodioxin-6-yl)-2-methyl-phenyl]methoxy]-2,5-bis(fluoranyl)phenyl]methylamino]-3-oxidanylidene-butanoic acid, (PDB: 5N2F) was retrieved from the RSCB Protein Data Bank was downloaded from the protein data as .pdb file. This file was imported into the SYBYL working panel and converted into a .mol2 file. The small molecule, 4-[[4-[[3-(2,3-dihydro-1,4-benzodioxin-6-yl)-2-methyl-phenyl]methoxy]-2,5-bis(fluoranyl)phenyl]methylamino]-3-oxidanylidene-butanoic acid, was selected and removed from the 5N2F protein. Polar hydrogens were added, as water molecules were deleted. The termini/amide chains as well as B-factors were corrected. Gasteiger–Marsili charges were then added, and the protonation states were set to 7.4 (physiological). Equally, similar protocol was followed in preparing the human PARP protein, 4R6E; after the ligand, niraparib have been removed
Ligand Screening
To initiate the docking process, protomols were generated from the interfacial amino acid residues within the active sites of both the PARP1 and PD-L1 proteins. Then, the generated 3D structure set was docked into the protomol, scored, and ranked. To validate the docking method, the removed ligands were docked back in the prepared protein active site. The corresponding pose of the ligands were similar to its original form in the crystal structures, implying that our docking method is reliable.
Acknowledgments
We are grateful to the staff of the University of Kentucky, which supported this work.
Glossary
Abbreviations
- PARP-1
Poly(ADP-ribose) polymerase-1
- PARPi
Poly(ADP-ribose) polymerase-1 inhibitor
- PD-L1
Programmed death-ligand 1
- PD-1
programmed cell death protein 1
- DIPEA
N,N-Diisopropylethylamine
- HOBt
Hydroxybenzotriazole
- EDCl
1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride
- DCM
Dichloromethane
- DMEM
Dulbecco’s modified Eagle medium
- PBS
Phosphate-buffered saline
- TBS
Tris-buffered saline
- TBST
Tris-buffered saline and Tween 20
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsomega.9b01106.
List of characterization data including NMR spectroscopy, dose–response curves, UV–Vis spectra, and HPLC trace of compounds (PDF)
Author Present Address
† 505 Rose Street, Department of Chemistry, University of Kentucky, Lexington KY 40506.
Financial support was provided by the University of Kentucky (UK). This study made use of the UK NMR facility, with funds from the MRI Program (grants CHE-0319176 and CHE-1625732) as well as UK Flow Cytometry & Immune Function core facility is supported in part by the Office of the Vice President for Research, the Markey Cancer Center, and an NCI Center Core Support Grant (P30 CA 177558) to the University of Kentucky Markey Cancer Center.
The authors declare no competing financial interest.
Supplementary Material
References
- Bu X.; Yao Y.; Li X.. Immune Checkpoint Blockade in Breast Cancer Therapy. In Translational Research in Breast Cancer: Biomarker Diagnosis; Targeted Therapies and Approaches to Precision Medicine, Song E.; Hu H., Eds.; Springer Singapore: Singapore, 2017; pp 383–402. [DOI] [PubMed] [Google Scholar]
- Marshall H. T.; Djamgoz M. B. A. Immuno-Oncology: Emerging Targets and Combination Therapies. Front. Oncol. 2018, 8, 315–315. 10.3389/fonc.2018.00315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farkona S.; Diamandis E. P.; Blasutig I. M. Cancer immunotherapy: the beginning of the end of cancer?. BMC Med. 2016, 14, 73–73. 10.1186/s12916-016-0623-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vinay D. S.; Ryan E. P.; Pawelec G.; Talib W. H.; Stagg J.; Elkord E.; Lichtor T.; Decker W. K.; Whelan R. L.; Kumara H. M. C. S.; Signori E.; Honoki K.; Georgakilas A. G.; Amin A.; Helferich W. G.; Boosani C. S.; Guha G.; Ciriolo M. R.; Chen S.; Mohammed S. I.; Azmi A. S.; Keith W. N.; Bilsland A.; Bhakta D.; Fujii H.; Aquilano K.; Ashraf S. S.; Nowsheen S.; Yang X.; Choi B. K.; Kwon B. Immune evasion in cancer: Mechanistic basis and therapeutic strategies. Semin. Cancer Biol. 2015, 35, S185–S198. 10.1016/j.semcancer.2015.03.004. [DOI] [PubMed] [Google Scholar]
- Riella L. V.; Paterson A. M.; Sharpe A. H.; Chandraker A. Role of the PD-1 pathway in the immune response. Am. J. Transplant. 2012, 12, 2575–87. 10.1111/j.1600-6143.2012.04224.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen X.; Zhao B. Efficacy of PD-1 or PD-L1 inhibitors and PD-L1 expression status in cancer: meta-analysis. Br. Med. J. 2018, 362, k3529. 10.1136/bmj.k3529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webb E. S.; Liu P.; Baleeiro R.; Lemoine N. R.; Yuan M.; Wang Y. Immune checkpoint inhibitors in cancer therapy. J. Biomed. Res. 2018, 32, 317–326. 10.7555/JBR.31.20160168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng J.; Hamanishi J.; Matsumura N.; Abiko K.; Murat K.; Baba T.; Yamaguchi K.; Horikawa N.; Hosoe Y.; Murphy S. K.; Konishi I.; Mandai M. Chemotherapy Induces Programmed Cell Death-Ligand 1 Overexpression via the Nuclear Factor- B to Foster an Immunosuppressive Tumor Microenvironment in Ovarian Cancer. Cancer Res. 2015, 75, 5034. 10.1158/0008-5472.CAN-14-3098. [DOI] [PubMed] [Google Scholar]
- Song M.; Chen X.; Wang L.; Zhang Y. Future of anti-PD-1/PD-L1 applications: Combinations with other therapeutic regimens. Chin. J. Cancer Res. 2018, 30, 157–172. 10.21147/j.issn.1000-9604.2018.02.01. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu-Lieskovan S.; Ribas A. New Combination Strategies Using Programmed Cell Death 1/Programmed Cell Death Ligand 1 Checkpoint Inhibitors as a Backbone. Cancer J. 2017, 23, 10–22. 10.1097/PPO.0000000000000246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Topalian S. L.; Drake C. G.; Pardoll D. M. Targeting the PD-1/B7-H1(PD-L1) pathway to activate anti-tumor immunity. Curr. Opin. Immunol. 2012, 24, 207–212. 10.1016/j.coi.2011.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sau S.; Petrovici A.; Alsaab H.; Iyer A. K. Abstract 3707: PD-L1 antibody drug conjugate for cancer immune-chemo combination therapy. Cancer Res. 2018, 78, 3707. 10.1158/1538-7445.AM2018-3707. [DOI] [Google Scholar]
- Wang Z.; Sun K.; Xiao Y.; Feng B.; Mikule K.; Ma X.; Feng N.; Vellano C. P.; Federico L.; Marszalek J. R.; Mills G. B.; Hanke J.; Ramaswamy S.; Wang J. Niraparib activates interferon signaling and potentiates anti-PD-1 antibody efficacy in tumor models. Sci. Rep. 2019, 9, 1853. 10.1038/s41598-019-38534-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart R. A.; Pilié P. G.; Yap T. A. Development of PARP and Immune-Checkpoint Inhibitor Combinations. Cancer Res. 2018, 6717. 10.1158/0008-5472.CAN-18-2652. [DOI] [PubMed] [Google Scholar]
- Criscuolo D.; Morra F.; Giannella R.; Visconti R.; Cerrato A.; Celetti A. New combinatorial strategies to improve the PARP inhibitors efficacy in the urothelial bladder Cancer treatment. J. Exp. Clin. Cancer Res. 2019, 38, 91. 10.1186/s13046-019-1089-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedlander M.; Meniawy T.; Markman B.; Mileshkin L. R.; Harnett P. R.; Millward M.; Lundy J.; Freimund A. E.; Norris C.; Mu S.; Wu J.; Paton V. E.; Wang L.; Gao B. A phase 1b study of the anti-PD-1 monoclonal antibody BGB-A317 (A317) in combination with the PARP inhibitor BGB-290 (290) in advanced solid tumors. J. Clin. Oncol. 2017, 35, 3013. 10.1200/JCO.2017.35.15_suppl.3013. [DOI] [Google Scholar]
- Zak K. M.; Grudnik P.; Guzik K.; Zieba B. J.; Musielak B.; Dömling A.; Dubin G.; Holak T. A. Structural basis for small molecule targeting of the programmed death ligand 1 (PD-L1). Oncotarget 2016, 7, 30323–30335. 10.18632/oncotarget.8730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang F.; Qi X.; Wang X.; Wei D.; Wu J.; Feng L.; Cai H.; Wang Y.; Zeng N.; Xu T.; Zhou A.; Zheng Y. Structural basis of the therapeutic anti-PD-L1 antibody atezolizumab. Oncotarget 2017, 8, 90215–90224. 10.18632/oncotarget.21652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee H. T.; Lee J. Y.; Lim H.; Lee S. H.; Moon Y. J.; Pyo H. J.; Ryu S. E.; Shin W.; Heo Y. S. Molecular mechanism of PD-1/PD-L1 blockade via anti-PD-L1 antibodies atezolizumab and durvalumab. Sci. Rep. 2017, 7, 5532. 10.1038/s41598-017-06002-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J. Y.; Lee H. T.; Shin W.; Chae J.; Choi J.; Kim S. H.; Lim H.; Heo T. W.; Park K. Y.; Lee Y. J.; Ryu S. E.; Son J. Y.; Lee J. U.; Heo Y.-S. Structural basis of checkpoint blockade by monoclonal antibodies in cancer immunotherapy. Nat. Comm. 2016, 7, 13354. 10.1038/ncomms13354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okuma H. S.; Yonemori K.. BRCA Gene Mutations and Poly(ADP-Ribose) Polymerase Inhibitors in Triple-Negative Breast Cancer. In Translational Research in Breast Cancer: Biomarker Diagnosis; Targeted Therapies and Approaches to Precision Medicine, Song E.; Hu H., Eds. Springer Singapore: Singapore, 2017; pp 271–286. [DOI] [PubMed] [Google Scholar]
- Broustas C. G.; Lieberman H. B. DNA damage response genes and the development of cancer metastasis. Radiat. Res. 2014, 181, 111–130. 10.1667/RR13515.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sizemore S. T.; Mohammad R.; Sizemore G. M.; Nowsheen S.; Yu H.; Ostrowski M. C.; Chakravarti A.; Xia F. Synthetic Lethality of PARP Inhibition and Ionizing Radiation is p53-dependent. Mol. Cancer Res. 2018, 16, 1092–1102. 10.1158/1541-7786.MCR-18-0106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher A. E. O.; Hochegger H.; Takeda S.; Caldecott K. W. Poly(ADP-ribose) polymerase 1 accelerates single-strand break repair in concert with poly(ADP-ribose) glycohydrolase. Mol. Cell. Biol. 2007, 27, 5597–5605. 10.1128/MCB.02248-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maciag A. E.; Holland R. J.; Kim Y.; Kumari V.; Luthers C. E.; Sehareen W. S.; Biswas D.; Morris N. L.; Ji X.; Anderson L. M.; Saavedra J. E.; Keefer L. K. Nitric Oxide (NO) Releasing Poly ADP-ribose Polymerase 1 (PARP-1) Inhibitors Targeted to Glutathione S-Transferase P1-Overexpressing Cancer Cells. J. Med. Chem. 2014, 57, 2292–2302. 10.1021/jm401550d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashworth A.; Lord C. J. Synthetic lethal therapies for cancer: what’s next after PARP inhibitors?. Nat. Rev. Clin. Oncol. 2018, 15, 564–576. 10.1038/s41571-018-0055-6. [DOI] [PubMed] [Google Scholar]
- Mittica G.; Ghisoni E.; Giannone G.; Genta S.; Aglietta M.; Sapino A.; Valabrega G. PARP Inhibitors in Ovarian Cancer. Recent Pat. Anti-Cancer Drug Disc. 2018, 13, 392–410. 10.2174/1574892813666180305165256. [DOI] [PubMed] [Google Scholar]
- Savas P.; Salgado R.; Denkert C.; Sotiriou C.; Darcy P. K.; Smyth M. J.; Loi S. Clinical relevance of host immunity in breast cancer: from TILs to the clinic. Nat. Rev. Clin. Oncol. 2016, 13, 228. 10.1038/nrclinonc.2015.215. [DOI] [PubMed] [Google Scholar]
- Kim E.; Yang K. S.; Giedt R. J.; Weissleder R. Red Si-rhodamine drug conjugates enable imaging in GFP cells. Chem. Commun. 2014, 50, 4504–4507. 10.1039/c4cc00144c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zmuda F.; Malviya G.; Blair A.; Boyd M.; Chalmers A. J.; Sutherland A.; Pimlott S. L. Synthesis and Evaluation of a Radioiodinated Tracer with Specificity for Poly(ADP-ribose) Poymerase-1 (PARP-1) in Vivo. J. Med. Chem. 2015, 58, 8683–8693. 10.1021/acs.jmedchem.5b01324. [DOI] [PubMed] [Google Scholar]
- Wilson T. C.; Xavier M.-A.; Knight J.; Verhoog S.; Torres J. B.; Mosley M.; Hopkins S. L.; Wallington S.; Allen P. D.; Kersemans V.; Hueting R.; Smart S.; Gouverneur V.; Cornelissen B. PET Imaging of PARP Expression Using 18F-Olaparib. J. Nucl. Med. 2019, 60, 504–510. 10.2967/jnumed.118.213223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zmuda F.; Blair A.; Liuzzi M. C.; Malviya G.; Chalmers A. J.; Lewis D.; Sutherland A.; Pimlott S. L. An18F-Labeled Poly(ADP-ribose) Polymerase Positron Emission Tomography Imaging Agent. J. Med. Chem. 2018, 61, 4103–4114. 10.1021/acs.jmedchem.8b00138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiao S.; Xia W.; Yamaguchi H.; Wei Y.; Chen M.-K.; Hsu J.-M.; Hsu J. L.; Yu W.-H.; Du Y.; Lee H.-H.; Li C.-W.; Chou C.-K.; Lim S.-O.; Chang S.-S.; Litton J.; Arun B.; Hortobagyi G. N.; Hung M.-C. PARP Inhibitor Upregulates PD-L1 Expression and Enhances Cancer-Associated Immunosuppression. Clin. Cancer Res. 2017, 23, 3711–3720. 10.1158/1078-0432.CCR-16-3215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leary M.; Heerboth S.; Lapinska K.; Sarkar S. Sensitization of Drug Resistant Cancer Cells: A Matter of Combination Therapy. Cancers 2018, 10, 483. 10.3390/cancers10120483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mokhtari R. B.; Homayouni T. S.; Baluch N.; Morgatskaya E.; Kumar S.; Das B.; Yeger H. Combination therapy in combating cancer. Oncotarget 2017, 8, 38022–38043. 10.18632/oncotarget.16723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foucquier J.; Guedj M. Analysis of drug combinations: current methodological landscape. Pharmacol. Res. Perspect. 2015, 3, e00149. 10.1002/prp2.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aminake M. N.; Mahajan A.; Kumar V.; Hans R.; Wiesner L.; Taylor D.; de Kock C.; Grobler A.; Smith P. J.; Kirschner M.; Rethwilm A.; Pradel G.; Chibale K. Synthesis and evaluation of hybrid drugs for a potential HIV/AIDS-malaria combination therapy. Bioorg. Med. Chem. 2012, 20, 5277–5289. 10.1016/j.bmc.2012.06.038. [DOI] [PubMed] [Google Scholar]
- Agarwal D.; Gupta R. D.; Awasthi S. K. Are Antimalarial Hybrid Molecules a Close Reality or a Distant Dream?. Antimicrob. Agents Chemother. 2017, 61, e00249-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beckers T.; Mahboobi S.; Sellmer A.; Winkler M.; Eichhorn E.; Pongratz H.; Maier T.; Ciossek T.; Baer T.; Kelter G.; Fiebig H.-H.; Schmidt M. Chimerically designed HDAC- and tyrosine kinase inhibitors. A series of erlotinib hybrids as dual-selective inhibitors of EGFR, HER2 and histone deacetylases. Med. Chem. Commun. 2012, 3, 829–835. 10.1039/c2md00317a. [DOI] [Google Scholar]
- Reilly S. W.; Puentes L. N.; Wilson K.; Hsieh C.-J.; Weng C.-C.; Makvandi M.; Mach R. H. Examination of Diazaspiro Cores as Piperazine Bioisosteres in the Olaparib Framework Shows Reduced DNA Damage and Cytotoxicity. J. Med. Chem. 2018, 61, 5367–5379. 10.1021/acs.jmedchem.8b00576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCombs J. R.; Owen S. C. Antibody drug conjugates: design and selection of linker, payload and conjugation chemistry. AAPS J. 2015, 17, 339–351. 10.1208/s12248-014-9710-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu J.; Jiang F.; Lu A.; Zhang G. Linkers Having a Crucial Role in Antibody-Drug Conjugates. Int. J. Mol. Sci. 2016, 17, 561. 10.3390/ijms17040561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorsell A. G.; Ekblad T.; Karlberg T.; Löw M.; Pinto A. F.; Trésaugues L.; Moche M.; Cohen M. S.; Schüler H. Structural Basis for Potency and Promiscuity in Poly(ADP-ribose) Polymerase (PARP) and Tankyrase Inhibitors. J. Med. Chem. 2017, 60, 1262–1271. 10.1021/acs.jmedchem.6b00990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guzik K.; Zak K. M.; Grudnik P.; Magiera K.; Musielak B.; Törner R.; Skalniak L.; Dömling A.; Dubin G.; Holak T. A. Small-Molecule Inhibitors of the Programmed Cell Death-1/Programmed Death-Ligand 1 (PD-1/PD-L1) Interaction via Transiently Induced Protein States and Dimerization of PD-L1. J. Med. Chem. 2017, 60, 5857–5867. 10.1021/acs.jmedchem.7b00293. [DOI] [PubMed] [Google Scholar]
- Mittendorf E. A.; Philips A. V.; Meric-Bernstam F.; Qiao N.; Wu Y.; Harrington S.; Su X.; Wang Y.; Gonzalez-Angulo A. M.; Akcakanat A.; Chawla A.; Curran M.; Hwu P.; Sharma P.; Litton J. K.; Molldrem J. J.; Alatrash G. PD-L1 expression in triple-negative breast cancer. Cancer Immunol. Res. 2014, 2, 361–370. 10.1158/2326-6066.CIR-13-0127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou T.-C. Theoretical Basis, Experimental Design, and Computerized Simulation of Synergism and Antagonism in Drug Combination Studies. Pharmacol. Rev. 2006, 58, 621–681. 10.1124/pr.58.3.10. [DOI] [PubMed] [Google Scholar]
- Ikebuchi R.; Konnai S.; Okagawa T.; Yokoyama K.; Nakajima C.; Suzuki Y.; Murata S.; Ohashi K. Influence of PD-L1 cross-linking on cell death in PD-L1-expressing cell lines and bovine lymphocytes. Immunology 2014, 142, 551–561. 10.1111/imm.12243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J.; Hu L. Immunomodulators targeting the PD-1/PD-L1 protein-protein interaction: From antibodies to small molecules. Med. Res. Rev. 2019, 39, 265–301. 10.1002/med.21530. [DOI] [PubMed] [Google Scholar]
- Kinoshita T.; Nakanishi I.; Warizaya M.; Iwashita A.; Kido Y.; Hattori K.; Fujii T. Inhibitor-induced structural change of the active site of human poly(ADP-ribose) polymerase. FEBS Lett. 2004, 556, 43–46. 10.1016/S0014-5793(03)01362-0. [DOI] [PubMed] [Google Scholar]
- Steffen J. D.; Brody J. R.; Armen R. S.; Pascal J. M. Structural Implications for Selective Targeting of PARPs. Front. Oncol. 2013, 3, 301. 10.3389/fonc.2013.00301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Acúrcio R. C.; Scomparin A.; Conniot J.; Salvador J. A. R.; Satchi-Fainaro R.; Florindo H. F.; Guedes R. C. Structure–Function Analysis of Immune Checkpoint Receptors to Guide Emerging Anticancer Immunotherapy. J. Med. Chem. 2018, 61, 10957–10975. 10.1021/acs.jmedchem.8b00541. [DOI] [PubMed] [Google Scholar]
- Carey J. P. W.; Karakas C.; Bui T.; Chen X.; Vijayaraghavan S.; Zhao Y.; Wang J.; Mikule K.; Litton J. K.; Hunt K. K.; Keyomarsi K. Synthetic Lethality of PARP Inhibitors in Combination with MYC Blockade Is Independent of BRCA Status in Triple-Negative Breast Cancer. Cancer Res. 2018, 78, 742–757. 10.1158/0008-5472.CAN-17-1494. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.