

Abstract
Background
Spinal chordomas, a subtype of primary spinal column malignancies (PSCM), are rare tumors with poor prognosis, and we have limited understanding of the molecular drivers of neoplasia.
Methods
Study design was a retrospective review of prospectively collected data with cross-sectional survival. Archived paraffin embedded pathologic specimens were collected for 133 patients from 6 centers within Europe and North America between 1987 and 2012. Tumor DNA was extracted and the human telomerase reverse transcriptase (hTERT) promoter was sequenced. The hTERT mutational status was correlated with overall survival (OS) and time to first local recurrence.
Results
Ninety-two chordomas, 26 chondrosarcomas, 7 osteosarcomas, 3 Ewing’s sarcomas, and 5 other malignant spinal tumors were analyzed. Median OS following surgery was 5.8 years (95% CI: 4.6 to 6.9) and median time to first local recurrence was 3.9 years (95% CI: 2.5 to 6.7). Eight chordomas, 2 chondrosarcomas, 1 Ewing’s sarcoma, and 1 other malignant spinal tumor harbored either a C228T or C250T mutation in the hTERT promoter. In the overall cohort, all patients with hTERT mutation were alive at 10 years postoperative with a median OS of 5.1 years (95% CI: 4.5 to 6.6) (P = 0.03). hTERT promoter mutation was observed in 8.7% of spinal chordomas, and 100% of chordoma patients harboring the mutation were alive at 10 years postoperative compared with 67% patients without the mutation (P = 0.05).
Conclusions
We report for the first time that hTERT promoter mutations C228T and C250T are present in approximately 8.7% of spinal chordomas. The presence of hTERT mutations conferred a survival benefit and could potentially be a valuable positive prognostic molecular marker in spinal chordomas.
Keywords: chordoma, hTERT promoter mutation, primary spinal column malignancy, survival
Key Points.
1. hTERT promoter mutations C228T and C250T are present in 8.7% of spinal chordomas.
2. hTERT promoter mutations are associated with a positive overall survival benefit in spinal chordomas.
Importance of the Study.
Primary spinal column malignancies are rare tumors with poor prognosis and few systemic treatment options, and we have limited understanding of the molecular drivers of neoplasia. The current study consists of 133 patients from an international, multicenter database and represents the largest surgical cohort study on molecular genetics of these rare tumors. We report for the first time that hTERT promoter mutations C228T and C250T are present in 8.7% of spinal chordomas. All patients with hTERT mutation were alive at 10 years postoperative compared with those patients who lacked the mutations. hTERT could potentially be a valuable molecular biomarker in prognostication of spinal chordomas.
Primary spinal column malignancies (PSCMs) are a rare and heterogeneous group of oncologic lesions.1 Chordoma, chondrosarcoma, osteosarcoma, Ewing’s sarcoma, and other malignant soft tissue tumors of the spine are typically classified as PSCM. Clinical presentations are usually insidious and up to 33% of patients are found to have metastatic disease at initial evaluation.2 While chordoma and chondrosarcoma can be indolent but locally invasive, Ewing’s sarcoma and osteosarcoma are more aggressive and associated with higher rates of tumor progression and recurrence.3
Surgical, radiation, and systemic treatment options may be limited in the management of PSCM. En bloc resection is the evidence-based treatment, particularly for chordoma and chondrosarcoma.4,5 Unfortunately the spine’s anatomical restraints make achieving wide margins exceedingly difficult, frequently resulting in neurological sacrifice, high morbidity, and sometimes limited ability to achieve tumor-free resection margins. Even with adjuvant chemo and radiation therapy,6–11 PSCMs are known to have a poorer prognosis, even worse than similar tumors in the appendicular skeleton. Because of these high stakes, surgeons and oncologists need more precise prognostic variables to help guide treatment. Despite the evolving use of molecular markers in many cancers, there is currently limited understanding of the molecular drivers of carcinogenesis in PSCM.
Human telomerase reverse transcriptase (hTERT) is an important catalytic subunit of the telomerase complex and contributes to telomere maintenance during tumorigenesis and cellular immortalization.12hTERT overexpression is observed in many human cancer types, including renal cell carcinoma,13 melanoma,14 urothelial carcinoma,15,16 oral squamous cell carcinoma,17 thyroid carcinomas,18 and gliomas.19,20 Two hotspot somatic mutations in the hTERT promoter region, 1,295,228 C > T (C228T) and 1,295,250 C > T (C250T), have been identified in a recurrent fashion, particularly in human melanoma and gliomas.21,22 The C228T and C250T promoter mutations generate a de novo binding motif for E-twenty-six transcription factors and can upregulate the transcriptional activity of hTERT by 2- to 6-fold in human melanomas.23,24 Although hTERT promoter mutations are frequently associated with worse survival outcomes in thyroid carcinoma, non–small cell lung cancer, and melanoma,25–27 the prognostic value of these mutations in central nervous system neoplasms is unclear. Recent research in glioma patients demonstrated a survival benefit in patients with hTERT promoter mutations in isocitrate dehydrogenase (IDH) wildtype/O6-methylguanine-DNA methyltransferase (MGMT) promoter methylated glioblastoma (GBM) and is strongly associated with IDH mutant‒1p/19q codeleted oligodendroglial neoplasm.28,29
Few studies have investigated the role of hTERT promoter region mutations in PSCM.30–32 The present study investigates the prognostic value of hTERT promoter mutations on overall survival (OS) and time to local recurrence in a large international cohort of PSCM patients, with a subgroup analysis focused on spinal chordoma.
Materials and Methods
Design
Study design was a retrospective review of prospectively collected data with cross-sectional survival in the AOSpine Knowledge Forum Tumor (AOSKFT) primary database. The AOSKFT model has been described in previous publications.33–35 An initial cohort of 1495 patients with primary spinal column tumors were treated at 13 centers within Europe, North America, and Australia between December 1985 and January 2013. Ethics approval was received at each center from appropriate institutional review boards; likewise, written informed consent was obtained from each subject. Excluded from the current study were 1322 patients because the pathologic specimens were not evaluated for hTERT promoter mutation or the specimens were not definitively identified as PSCM. Archived paraffin embedded pathologic specimens were available for 133 patients from 6 of the 13 centers; 92 spinal chordomas were then selected for subgroup analysis.
A secure web-based application (REDCap, Vanderbilt University) was used to gather demographic, clinical, diagnostic, therapeutic, local recurrence, perioperative morbidity, and survival data. Information regarding patient mortality and disease-free survival was acquired cross-sectionally.
Definitions and Staging
Histologic classification and tumor grading were performed by a musculoskeletal tumor pathologist at each individual center. Chordoma histologic subtypes included classic (notochordal cells with regions of chondroid), chondroid (containing chordoma and chondrosarcoma components), and de-differentiated (loss of the integrase interactor 1 gene, increased mitotic potential). Chondrosarcoma histologic subtypes included de-differentiated and myxoid. The validated Enneking classification was used to characterize surgical margins of resected tumors (intralesional, marginal, or wide). If wide or marginal margins were achieved, the specimen was classified as Enneking appropriate (EA) resection. If intralesional resection was performed, the specimen was classified as Enneking inappropriate (EI) resection. Tumor grade and stage were classified based on the Enneking staging system for malignant musculoskeletal tumors.36
Patient Follow-Up
Follow-up examinations were performed based on individual institutional protocols. Local recurrence was defined as time interval from tumor resection to radiographic tumor reappearance at or near the surgical resection cavity. OS was defined as the interval between time of surgery and death.
hTERT Promoter Mutation Genotyping
Tumor DNA was extracted from formalin-fixed and paraffin-embedded (FFPE) specimens using the DNA FFPE Tissue Kit (Qiagen, #56404). Genotyping of the hTERT promoter was performed using Sanger sequencing. The small amplicon, 163 base pair fragment was amplified using the following primers: hTERT-seq-for 5′- CAGCGCTGCCTGAAACTC-3′ and hTERT-seq-rev 5′-GTCCTGCCCCTTCACCTT-3′. The large amplicon, 193 base pair fragment of the hTERT promoter region spanning the hotspot mutations on chromosome 5 (C228T and C250T) was amplified using the following primers: hTERT-seq-for 5′- CACCCGTCCTGCCCCTTCACCTT-3′ and hTERT-seq-rev 5′- GGCTTCCCACGTGCGCAGCAGGA-3′. Primer sequences were tagged with T7 forward (caggaaacagctatgac) and M13 reverse tags (taatacgactcactataggg). DNA was first quantitated using NanoDrop and diluted to 10 ng/μL. PCR was performed on Tetrad (Bio-Rad) with the following conditions: 94°C for 2 minutes; 35 cycles with 94°C (30 seconds), 62°C (30 seconds), and 68°C (30 seconds); 68°C for 5 minutes; and 4°C for hold. Post-PCR products were treated with ExoSAP-IT (Thermo Fisher Scientific, #78201.1.ML). Finally, BigDye Terminator v3.1 (Thermo Fisher Scientific, #4337455) was utilized for cycle sequencing on the ABI capillary electrophoresis platform.
Statistical Analysis
Data were described using descriptive statistics (mean ± standard deviation or median/interquartile range for continuous variables; absolute number/percentage for categorical variables). Comparisons between wildtype and mutational subgroups were done in a supervised manner. Further statistical subgroup analysis was performed in the spinal chordoma group. The chi-square test (Pearson’s or Fisher’s exact) and Student’s t-tests or Wilcoxon Mann–Whitney tests were used for comparison between cohorts. Kaplan–Meier survival analyses were performed over a 10-year period and the Mantel–Cox log-rank test was used to evaluate factors associated with OS and overall time to first local recurrence. Statistical significance was set at P ≤ 0.05. All statistical analyses were performed using Stata v12.0.
Results
Patient Population and Baseline Characteristics
Of the 133 patients included in the study, approximately 64% were male and 84% Caucasian. Patients had a mean age of 56 ± 16 years at the time of surgery. Although over 90% of the patients had pain as a presenting symptom at diagnosis, only 7% of the patients had a diagnosis of pathological fracture. Of the 133 patients, 15 (11%) had undergone previous resection, with intralesional resection achieved in 10/15 (67%) of the cases. Tissue diagnosis of PSCM was obtained by CT-trocar guided biopsy in 59% of the cases and intraoperative biopsy in 17%. Lesions of the mobile spine were seen in 36% of the patients, while 64% had lesions of the fixed spine (Table 1). The 133-patient cohort comprised 92 chordomas, 26 chondrosarcomas, 7 osteosarcomas, 3 Ewing’s sarcomas, and 5 other malignant spinal tumors. Patient follow-up ranged from 2 days to 22.5 years postoperatively.
Table 1.
Patient and tumor characteristics
| Variables | |
|---|---|
| Sex (n = 133) | |
| Male | 85 (63.9) |
| Female | 48 (36.1) |
| Ethnicity (n = 115) | |
| African | 3 (2.6) |
| Asian/Pacific Islander | 4 (3.5) |
| Caucasian | 96 (83.5) |
| East Indian | 2 (1.7) |
| Hispanic | 8 (7.0) |
| Other | 2 (1.7) |
| Age at time of surgery (years) (n = 133) | 55.8 ± 16.4 |
| Pain at Diagnosis (n = 126) | |
| No | 11 (8.7) |
| Yes | 115 (91.3) |
| Pathologic Fracture at Diagnosis (n = 126) | |
| No | 117 (92.9) |
| Yes | 9 (7.1) |
| Previous Spine Tumor Operation (n = 133) | |
| No | 118 (88.7) |
| Yes | 15 (11.3) |
| Intralesional | 10 (66.7) |
| Marginal | 0 (0.0) |
| Wide | 1 (6.7) |
| Unknown | 4 (26.7) |
| How the Diagnosis Was Performed (n = 123) | |
| Open biopsy | 17 (13.8) |
| CT-trocar biopsy | 72 (58.5) |
| Intraoperative biopsy | 21 (17.1) |
| Other | 13 (10.6) |
| Spinal Level (n = 132) | |
| Mobile | 47 (35.6) |
| Fixed | 85 (64.4) |
| Diagnosis (n = 133) | |
| Chordoma | 92 (69.2) |
| Chondrosarcoma | 26 (19.5) |
| Ewing’s sarcoma | 3 (2.3) |
| Osteosarcoma | 7 (5.3) |
| Other malignant soft tissue tumors | 5 (3.8) |
| Tumor Grade (n = 133) | |
| Low (Ia/Ib) | 83 (62.4) |
| High (IIa/IIb) | 50 (37.6) |
| TERT Promoter Mutation (n = 133) | |
| No | 121 (91.0) |
| Yes | 12 (9.0) |
| TERT Status (n = 133) | |
| Wildtype | 121 (91.0) |
| C228T | 11 (8.3) |
| C250T | 1 (0.7) |
Data are presented as N (%), mean ± standard deviation or median (p25, p75).
Treatment
All patients underwent surgical resection of their lesion. In the overall cohort, 125/133 had Enneking appropriateness information available. Surgery was EA in 83/125 patients (66%) and EI in 42/125 patients (34%). The 15 patients with prior tumor resection were regraded on the Enneking resection scale after the last surgical resection. Of 130 patients, 111 (85%) of patients did not receive any chemotherapy and 96/131 (73%) did not receive any radiation therapy.
Comparison of hTERT Mutant to Wildtype in the Overall Cohort
Of 133 patients, 121 (91%) were identified with hTERT wildtype, and 12/133 (9%) harbored a promoter mutation (Table 2). Eleven of 12 of the mutations were C228T with only 1 C250T. The hTERT mutations were identified in 8 chordomas, 2 chondrosarcomas, 1 Ewing’s sarcoma, and 1 other malignant spinal tumor. The wildtype and C228T/C250T cohorts were evenly matched in terms of sex distribution (P = 0.76), age at time of surgery (P = 0.52), ethnicity (P = 0.39), and location of lesion in mobile versus fixed spine (P = 0.10). There was no statistical difference between the 2 cohorts in use of adjuvant therapy (P = 0.34), timing of chemotherapy (P = 1.00), and timing of radiation therapy (P = 0.28). Tumor grade was low (Ia/Ib) in 62% of the wildtype cohort and 67% of the hTERT mutation cohort (P = 1.00). The adequacy of surgical resection was not statistically different between the 2 cohorts (EA 65% in wildtype vs 80% in mutation, P = 0.49).
Table 2.
Comparison of cohorts
| Variables | TERT promoter mutation | P-value | |
|---|---|---|---|
| No (n = 121) | Yes (n = 12) | ||
| Sex (n = 133) | 0.76* | ||
| Male | 78 (64.5) | 7 (58.3) | |
| Female | 43 (35.5) | 5 (41.7) | |
| Age at time of surgery (y) (n = 133) | 56.1 ± 16.5 | 52.9 ± 15.8 | 0.52† |
| Spinal Level (n = 132) | 0.10* | ||
| Mobile | 46 (38.0) | 1 (9.1) | |
| Fixed | 75 (62.0) | 10 (90.9) | |
| Ethnicity (n = 115) | 0.39* | ||
| Non-caucasian | 16 (15.4) | 3 (27.3) | |
| Caucasian | 88 (84.6) | 8 (72.7) | |
| Adjuvant Therapy Given (n = 131) | 0.34* | ||
| No | 77 (64.7) | 10 (83.3) | |
| Yes | 42 (35.3) | 2 (16.7) | |
| Timing of Chemotherapy (n = 130) | 1.00* | ||
| Preop | 8 (6.8) | 1 (8.3) | |
| Postop | 4 (3.4) | 0 (0.0) | |
| Both | 6 (5.1) | 0 (0.0) | |
| Neither (no chemo) | 100 (84.8) | 11 (91.7) | |
| Timing of Radiation Therapy (n = 131) | 0.28* | ||
| Preop | 9 (7.6) | 2 (16.7) | |
| Postop | 20 (16.8) | 0 (0.0) | |
| Both | 4 (3.4) | 0 (0.0) | |
| Neither (no radiation) | 86 (72.3) | 10 (83.3) | |
| Tumor Grade (n = 133) | 1.00* | ||
| Low (I) | 75 (62.0) | 8 (66.7) | |
| High (II) | 46 (38.0) | 4 (33.3) | |
| Enneking Appropriateness (n = 125) | 0.49* | ||
| EA | 75 (65.2) | 8 (80.0) | |
| EI | 40 (34.8) | 2 (20.0) | |
| Local Recurrence at 10 Years Postoperative (n = 130) | 0.75* | ||
| No | 77 (65.3) | 9 (75.0) | |
| Yes | 41 (34.7) | 3 (25.0) | |
| Survival at 10 Years Postoperative (n = 133) | 0.01* | ||
| Alive | 74 (61.2) | 12 (100.0) | |
| Dead | 47 (38.8) | 0 (0.0) | |
Data are presented as N (%) or mean ± standard deviation; EA: Enneking appropriate, EI: Enneking inappropriate.
*Fisher’s exact test,†Student’s t-test
Subgroup Analysis in Spinal Chordoma
The histologic subtype was known in 46 chordoma patients, with 35 (76%) with of subtype, 4 (9%) chondroid, 2 (4%) de-differentiated, and 5 (11%) classified as other. Eighty-four (91.3%) chordoma patients were wildtype compared with 8 (8.7%) patients with the hTERT mutation. The wildtype and C228T/C250T chordoma cohorts were evenly matched in terms of sex distribution (P = 0.43), age at time of surgery (P = 0.27), ethnicity (P = 0.34), and Enneking appropriateness (P = 1.00). All 8 patients with hTERT mutations received adjuvant therapy compared with 56 (68%) of the wildtype patients. No patients in the mutation group received chemotherapy compared with 6 (7.3%) of the wildtype group (P = 1.00). Of the patients in the mutation group, 88% had low-grade tumor compared with 80% in the wildtype group (P = 1.00). Local recurrence at 10 years postoperative was 62.5% in the mutation group compared with 63.1% in the wildtype group (Table 3).
Table 3.
Subgroup analysis specific to spinal chordomas
| Variables | TERT Promoter Mutation | P-value | |
|---|---|---|---|
| No (n = 84) | Yes (n = 8) | ||
| Sex (n = 92) | 0.43* | ||
| Male | 58 (69.1) | 4 (50.0) | |
| Female | 26 (30.9) | 4 (50.0) | |
| Age at time of surgery (y) (n = 92) | 60.4 ± 13.9 | 54.6 ± 14.0 | 0.27† |
| Spinal Level (n = 92) | 0.05* | ||
| Mobile | 32 (38.1) | 0 (0.0) | |
| Fixed | 52 (61.9) | 8 (100.0) | |
| Ethnicity (n = 80) | 0.34* | ||
| Non-caucasian | 13 (18.1) | 0 (0.0) | |
| Caucasian | 59 (81.9) | 8 (100.0) | |
| Adjuvant Therapy Given (n = 90) | 0.10* | ||
| No | 56 (68.3) | 8 (100.0) | |
| Yes | 26 (31.7) | 0 (0.0) | |
| Timing of Chemotherapy (n = 90) | 1.00* | ||
| Preop | 2 (2.4) | 0 (0.0) | |
| Postop | 3 (3.7) | 0 (0.0) | |
| Both | 1 (1.2) | 0 (0.0) | |
| Neither (no chemo) | 76 (92.7) | 8 (100.0) | |
| Timing of Radiation Therapy (n = 90) | 0.61* | ||
| Preop | 5 (6.1) | 0 (0.0) | |
| Postop | 16 (19.5) | 0 (0.0) | |
| Both | 3 (3.7) | 0 (0.0) | |
| Neither (no radiation) | 58 (70.7) | 8 (100.0) | |
| Tumor Grade (n = 92) | 1.00* | ||
| Low (I) | 67 (79.8) | 7 (87.5) | |
| High (II) | 17 (20.2) | 1 (12.5) | |
| Enneking Appropriateness (n = 89) | 1.00* | ||
| EA | 53 (64.6) | 5 (71.4) | |
| EI | 29 (35.4) | 2 (28.6) | |
| Local Recurrence at 10 Years Postoperative (n = 92) | 1.00* | ||
| No | 53 (63.1) | 5 (62.5) | |
| Yes | 31 (36.9) | 3 (37.5) | |
| Survival at 10 Years Postoperative (n = 92) | 0.10* | ||
| Alive | 56 (66.7) | 8 (100.0) | |
| Dead | 28 (33.3) | 0 (0.0) | |
Data are presented as N (%) or mean ± standard deviation; EA: Enneking appropriate, EI: Enneking inappropriate.
*Fisher’s exact test,†Student’s t-test
Patient Outcomes
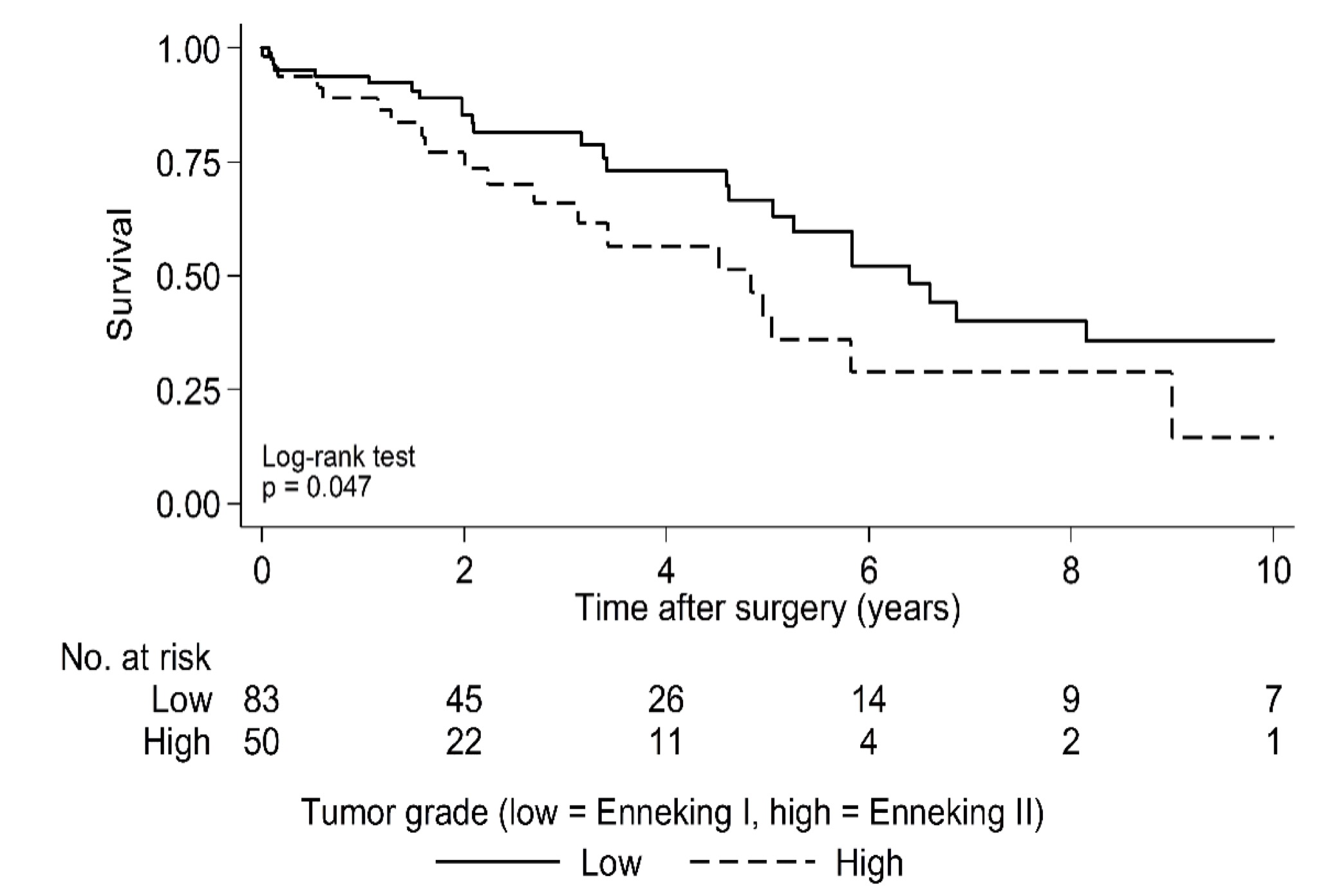
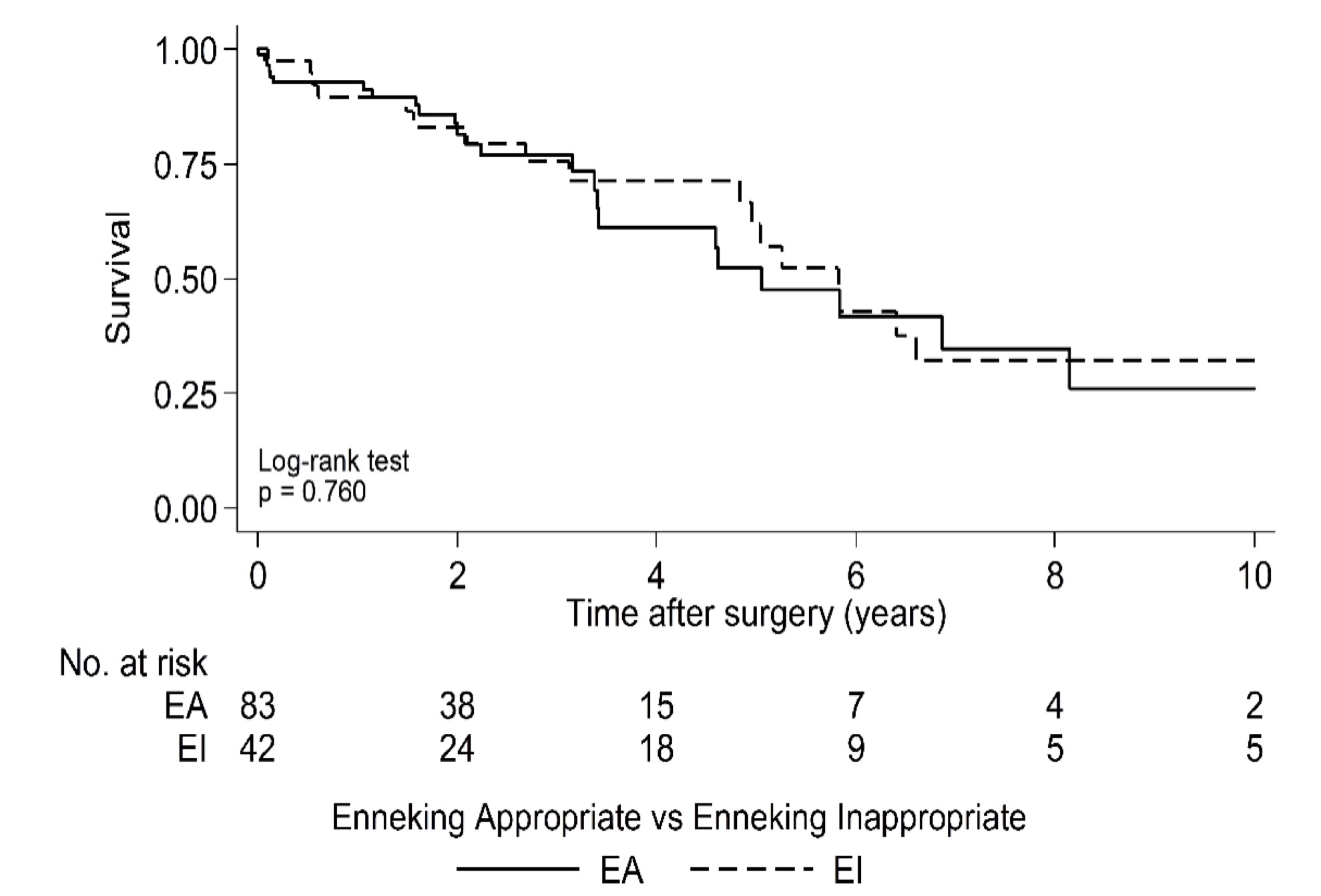
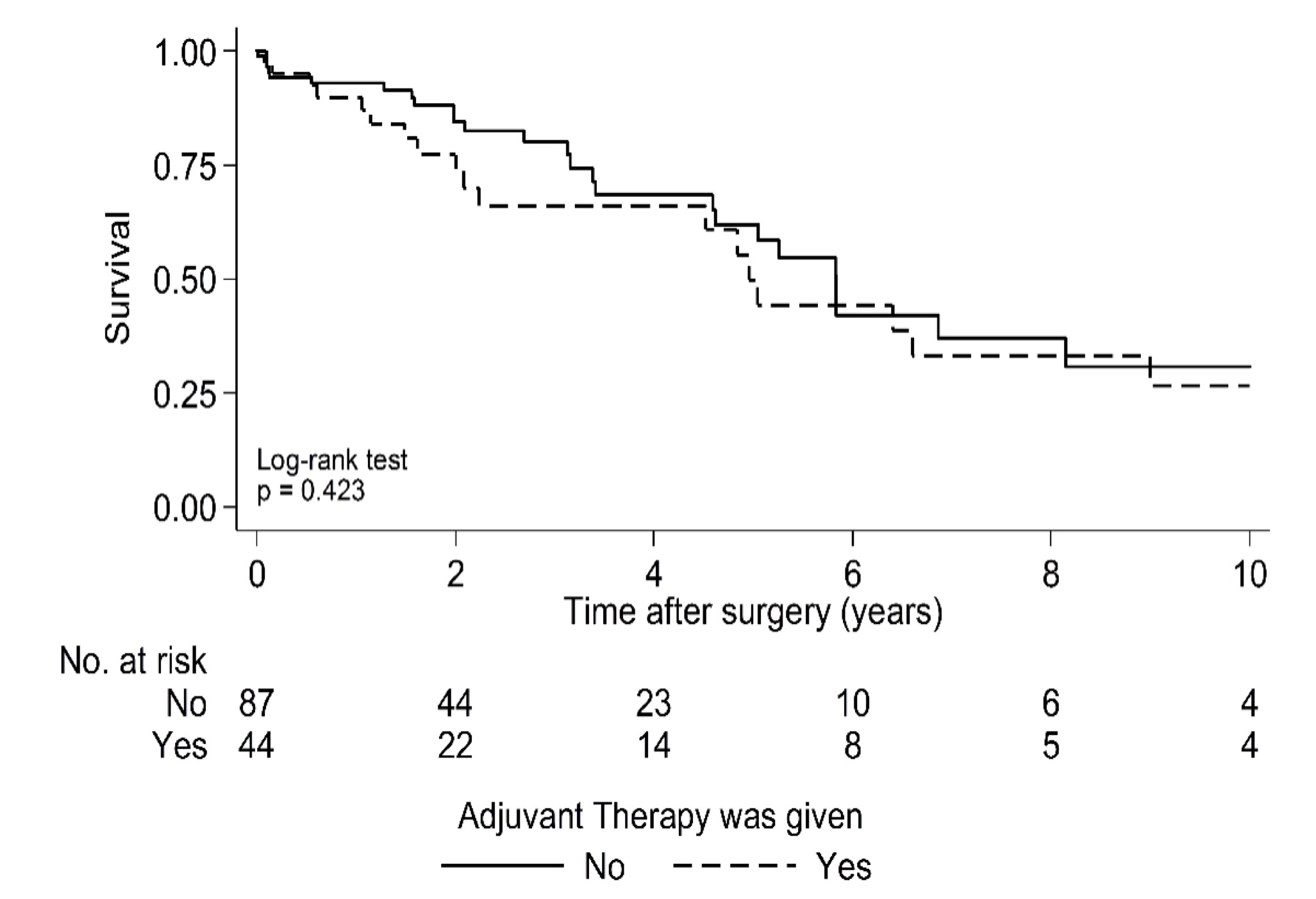
Median OS for the entire cohort following surgery was 5.8 years (95% CI: 4.6 to 6.9) (Fig. 1) and median time to first local recurrence was 3.9 years following surgery (95% CI: 2.5 to 6.7) (Fig. 2). OS was worse in the high tumor grade group of Enneking II, with the estimated median OS of 4.8 years following surgery compared with 6.4 years in Enneking I patients (P = 0.05) (Supplementary Fig. 1). There was no statistical difference in OS between EA and EI patients (P = 0.76) (Supplementary Fig. 2). Likewise, whether adjuvant therapy was used had no significant impact on OS following surgery (P = 0.42) (Supplementary Fig. 3).
Fig. 1.

Overall Kaplan–Meier survival curve.
Fig. 2.

Overall time to first local recurrence curve.
Patients who lacked the hTERT mutations in the overall cohort had a significantly worse OS, with an estimated median OS of 5.1 years (95% CI: 4.5 to 6.6) compared with the 12 patients with hTERT mutations, where death was not observed and median OS was not reached by 10 years postoperative (P = 0.03) (Fig. 3). In the chordoma-specific cohort, all 8 patients harboring the mutation were alive at 10 years postoperative compared with 56 (67%) of chordoma patients without the mutation (P = 0.05) (Fig. 4) (Supplementary Fig. 4).
Fig. 3.

Survival curve following surgery by presence of TERT promoter mutation in the overall cohort.
Fig. 4.

Survival curve following surgery by presence of TERT promoter mutation in the chordoma specific cohort.
Discussion
PSCMs are rare lesions, with most studies limited to single-center experiences.37 The current study consists of 133 patients from an international, multicenter database and represents the largest surgical cohort study on the molecular genetics of these rare tumors. Overall, the median OS was 5.8 years and is reflective of the heterogeneous makeup of the cohort, which consists of both indolent tumors like chordomas and more aggressive tumors like Ewing’s sarcoma. In the subgroup analysis, we report the presence of hTERT promoter mutations in 8.7% of spinal chordomas, which is associated with a statistically significant positive OS benefit (100% vs 67% alive at 10 years) without affecting the time to first tumor recurrence.
The interplay between hTERT and OS in chordoma or other primary spinal neoplasms is not well understood; several studies on the topic have focused on hTERT expression and its effects on tumor growth and invasion. In one study, Zou et al reported hTERT expression in 54 spinal chordoma tissue samples but not in 20 nucleus pulposus control samples; hTERT expression was significantly associated with local tumor invasion and cellular proliferation based on Ki-67 staining index.30 Additionally, high hTERT expression was an independent predictor of poor local recurrence-free survival, but no differences were seen in OS. In a series of 26 patients with clival chordomas, Pallini et al found that hTERT mRNA expression was frequently associated with increased doubling time for residual tumor and probability of local tumor recurrence.38 Similarly, Hu et al reported higher expression of telomerase in 20 patients with sacral chordoma recurrence.31 In another study, on chondrosarcomas, Chi et al reported a positive correlation between hTERT overexpression and telomere attrition and concluded that hTERT overexpression is associated with malignant sarcoma potential.39 For both the Hu et al and Chi et al studies, overall survival was not evaluated. These reports suggest an association between hTERT overexpression and tumor aggressiveness, akin to previous studies in melanoma14,40 and papillary thyroid carcinoma.24 Despite these findings, the relationship between hTERT overexpression and OS in chordoma patients remains unclear. To our knowledge, there are no reports that specifically evaluate the role of hTERT promoter mutations in prognosticating survival of PSCM.
In many human cancers, hTERT promoter mutations are associated with higher hTERT expression. The C228T and C250T promoter mutations generate a binding motif for E-twenty-six transcription factors and are found to upregulate the transcriptional activity of hTERT by 2- to 6-fold in human melanomas.23,24 Another study, in 48 GBMs, also reported significantly higher hTERT expression in C228T or C250T mutated tumors.41 Although hTERT promoter mutations are associated with poor prognosis in many human cancers, there are reports in CNS tumors suggesting a survival benefit in patients harboring these mutations. hTERT mutations are seen in as many as 75% of glioma and GBM patients and do not appear to be a prognostic biomarker when evaluated in isolation;27 despite this, patients with both MGMT methylation and hTERT promoter mutation are found to have improved survival (OS 28.3 vs 15.9 mo).27 Conversely, hTERT mutations were found to be negatively prognostic in MGMT unmethylated GBM patients. In a study by You et al, patients with hTERT mutations exhibited improved prognosis when paired with IDH 1 or 2 (IDH1/2) mutations and 1p/19q loss of heterozygosity.28 Poor prognosis was seen when hTERT mutations were paired with mesenchymal subtype or tumor protein p53 and epidermal growth factor receptor alteration.28 In gliomas, the prognostic influence of hTERT promoter mutation appears to be dependent on the coexistence of other molecular biomarkers and drivers.42
This dual prognostic nature of the hTERT promoter mutation can help explain incongruent results in the literature, which mainly suggest that hTERT mutation is a negative prognostic biomarker. The pro-survival benefit of hTERT mutations observed in our study is also likely related to yet to be determined molecular pathways. A recent study by Tarpey et al defined the somatic molecular drivers of 104 cases of sporadic chordoma and found duplication of the notochordal transcription factor brachyury in 27% of cases. In addition, phosphatidylinositol-3 kinase signaling mutations and LYST inactivation mutations were implicated as potential novel oncologic markers.43 The complex interactions between hTERT and other molecular drivers in PSCMs are still undefined and warrant further investigation.
Despite the use of large-scale, population-based, multicenter data, the current study remains limited by the rarity of PSCM and chordomas, variability between institutions with respect to treatment and follow-up time, and number of viable tissue samples. Intrinsic to the rarity of these tumors, the small sample sizes between the wildtype and hTERT mutation cohorts could potentially mask important differences between the groups that may only be detectable with larger sampling. As such, the 2 small groups being evenly matched is not definitively a negative lack of association. The compound analysis of all PSCM in the overall cohort can be confounded by biological differences and tumor heterogeneity, which prompted a subgroup analysis specific to spinal chordoma. We also did not specifically evaluate hTERT expression in this study. Further studies are needed to fully elucidate the interplay between hTERT promoter mutations, telomerase expression, and other molecular markers. Nevertheless, this study remains the largest contemporary series of primary spinal column tumors and is the first time the hTERT promoter mutations C228T and C250T have been implicated as a positive prognostic factor for survival in chordoma patients. We report for the first time that hTERT promoter mutations C228T and C250T are present in approximately 8.7% of spinal chordomas. The presence of hTERT mutations conferred a statistically significant survival benefit and could potentially be a valuable positive prognostic molecular marker in spinal chordomas.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgments
AOSpine is a clinical division of the AO Foundation—an independent medically guided nonprofit organization. The AOSpine Knowledge Forums are pathology-focused working groups acting on behalf of AOSpine in their domain of scientific expertise. Each forum consists of a steering committee of up to 10 international spine experts who meet on a regular basis to discuss research, assess the best evidence for current practices, and formulate clinical trials to advance spine care worldwide. Study support is provided directly through AOSpine’s Research department and AO’s Clinical Investigation and Documentation unit.
Conflict of interest statement
• Chetan Bettegowda reports consulting for Depuy-Synthes, which is outside of the submitted work.
• Stephen Yip reports reports consulting and royalties from Bayer, which is outside of the submitted work.
• Bowen Jiang reports consulting for Longeviti Neuro Solutions, which is outside the submitted work.
• C. Rory Goodwin reports research grants from the NIH/NINDS K12 NRCDP Physician Scientist Award, North Carolina Spine Society, and the Robert Wood Johnson’s Harold Amos Medical Faculty Development Program, which are all outside the submitted work.
• Daniel M. Sciubba reports consulting and royalties from Medtronic, Depuy-Synthes, Stryker, Nuvasive, K2M, which are all outside the submitted work.
• Niccole M. Germscheid reports employment with AO Foundation.
• Arjun Sahgal reports past educational seminars with Elekta AB, Accuray Inc, and Varian medical systems; research grant with Elekta AB; travel accommodations and expenses from Elekta and Varian; and belongs to the Elekta MR Linac Research Consortium, which are all outside the submitted work.
• Ziya L. Gokaslan reports research support from AOSpine North America and stock ownership of Spinal Kinetics, which are all outside the submitted work.
• Stefano Boriani reports educational commitments with K2M, which are outside the submitted work.
• Charles Fisher reports consulting and royalties from Medtronic; research grants from OREF; and fellowship support paid to institution from AOSpine and Medtronic, which are all outside the submitted work.
• Laurence Rhines reports educational commitments with Stryker, which are outside the submitted work.
No other authors report a conflict of interest.
Authorship statement
• All authors contributed to writing the manuscript
• Analysis and interpretation: CB, SY, BJ, MZ, NMG, LR
• Data acquisition: CB, SY, WW, MJC, AL MG, MZ, CRG, DMS, JW, EM, ZLG, SB, PPV, CF, LR
• Experimental design: CB, SY, WW, AL, MG, NMG, AS, ZLG, SB, PPV, CF, LR
References
- 1. Arutyunyan GG, Clarke MJ. Management of primary and metastatic spinal tumors. J Neurosurg Sci. 2015;59(2):181–193. [PubMed] [Google Scholar]
- 2. Ozaki T, Flege S, Liljenqvist U, et al. . Osteosarcoma of the spine: experience of the Cooperative Osteosarcoma Study Group. Cancer. 2002;94(4):1069–1077. [PubMed] [Google Scholar]
- 3. Schoenfeld AJ, Hornicek FJ, Pedlow FX, et al. . Osteosarcoma of the spine: experience in 26 patients treated at the Massachusetts General Hospital. Spine J. 2010;10(8):708–714. [DOI] [PubMed] [Google Scholar]
- 4. Ailon T, Torabi R, Fisher CG, et al. . Management of locally recurrent chordoma of the mobile spine and sacrum: a systematic review. Spine. 2016; 41(Suppl 20):S193–S198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Pham M, Awad M. Outcomes following surgical management of cervical chordoma: a review of published case reports and case series. Asian J Neurosurg. 2017;12(3):389–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Potluri S, Jefferies SJ, Jena R, et al. . Residual postoperative tumour volume predicts outcome after high-dose radiotherapy for chordoma and chondrosarcoma of the skull base and spine. Clin Onco. 2011;23(3):199–208. [DOI] [PubMed] [Google Scholar]
- 7. Stuer C, Schramm J, Schaller C. Skull base chordomas: management and results. Neurol Med Chir. 2006;46(3):118–124; discussion 124–125. [DOI] [PubMed] [Google Scholar]
- 8. Jiang B, Veeravagu A, Feroze AH, et al. . CyberKnife radiosurgery for the management of skull base and spinal chondrosarcomas. J Neurooncol. 2013;114(2):209–218. [DOI] [PubMed] [Google Scholar]
- 9. Indelicato DJ, Rotondo RL, Begosh-Mayne D, et al. . A prospective outcomes study of proton therapy for chordomas and chondrosarcomas of the spine. Int J Radiat Oncol Biol Phys. 2016;95(1):297–303. [DOI] [PubMed] [Google Scholar]
- 10. Bielack S, Carrle D, Casali PG; ESMO Guidelines Working Group Osteosarcoma: ESMO clinical recommendations for diagnosis, treatment and follow-up. Ann Oncol. 2009;20(Suppl 4):137–139. [DOI] [PubMed] [Google Scholar]
- 11. Paulussen M, Bielack S, Jürgens H, Casali PG; ESMO Guidelines Working Group Ewing’s sarcoma of the bone: ESMO clinical recommendations for diagnosis, treatment and follow-up. Ann Oncol. 2009;20(Suppl 4):140–142. [DOI] [PubMed] [Google Scholar]
- 12. Kim NW, Piatyszek MA, Prowse KR, et al. . Specific association of human telomerase activity with immortal cells and cancer. Science. 1994;266(5193):2011–2015. [DOI] [PubMed] [Google Scholar]
- 13. Casuscelli J, Becerra MF, Manley BJ, et al. . Characterization and impact of TERT promoter region mutations on clinical outcome in renal cell carcinoma. Eur Urol Focus. 2017. pii:S2405-4569(17)30212-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Bai X, Kong Y, Chi Z, et al. . MAPK Pathway and TERT promoter gene mutation pattern and its prognostic value in melanoma patients: a retrospective study of 2,793 cases. Clin Cancer Res. 2017;23(20):6120–6127. [DOI] [PubMed] [Google Scholar]
- 15. Wang X, Lopez-Beltran A, Osunkoya AO, et al. . TERT promoter mutation status in sarcomatoid urothelial carcinomas of the upper urinary tract. Future Oncol. 2017;13(8):705–714. [DOI] [PubMed] [Google Scholar]
- 16. Liu X, Wu G, Shan Y, Hartmann C, von Deimling A, Xing M. Highly prevalent TERT promoter mutations in bladder cancer and glioblastoma. Cell Cycle. 2013;12(10):1637–1638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Chang KP, Wang CI, Pickering CR, et al. . Prevalence of promoter mutations in the TERT gene in oral cavity squamous cell carcinoma. Head Neck. 2017;39(6):1131–1137. [DOI] [PubMed] [Google Scholar]
- 18. Liu X, Bishop J, Shan Y, et al. . Highly prevalent TERT promoter mutations in aggressive thyroid cancers. Endocr Relat Cancer. 2013;20(4):603–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Johanns TM, Fu Y, Kobayashi DK, et al. . High incidence of TERT mutation in brain tumor cell lines. Brain Tumor Pathol. 2016;33(3):222–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Koelsche C, Sahm F, Capper D, et al. . Distribution of TERT promoter mutations in pediatric and adult tumors of the nervous system. Acta Neuropathol. 2013;126(6):907–915. [DOI] [PubMed] [Google Scholar]
- 21. Horn S, Figl A, Rachakonda PS, et al. . TERT promoter mutations in familial and sporadic melanoma. Science. 2013;339(6122):959–961. [DOI] [PubMed] [Google Scholar]
- 22. Killela PJ, Reitman ZJ, Jiao Y, et al. . TERT promoter mutations occur frequently in gliomas and a subset of tumors derived from cells with low rates of self-renewal. Proc Natl Acad Sci U S A. 2013;110(15):6021–6026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Arita H, Narita Y, Fukushima S, et al. . Upregulating mutations in the TERT promoter commonly occur in adult malignant gliomas and are strongly associated with total 1p19q loss. Acta Neuropathol. 2013;126(2):267–276. [DOI] [PubMed] [Google Scholar]
- 24. Huang FW, Hodis E, Xu MJ, Kryukov GV, Chin L, Garraway LA. Highly recurrent TERT promoter mutations in human melanoma. Science. 2013;339(6122):957–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Nasirden A, Saito T, Fukumura Y, et al. . In Japanese patients with papillary thyroid carcinoma, TERT promoter mutation is associated with poor prognosis, in contrast to BRAF V600E mutation. Virchows Arch. 2016;469(6):687–696. [DOI] [PubMed] [Google Scholar]
- 26. Jung SJ, Kim DS, Park WJ, et al. . Mutation of the TERT promoter leads to poor prognosis of patients with non-small cell lung cancer. Oncol Lett. 2017;14(2):1609–1614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Griewank KG, Murali R, Puig-Butille JA, et al. . TERT promoter mutation status as an independent prognostic factor in cutaneous melanoma. J Natl Cancer Inst. 2014; 106(9). pii:dju246. doi:10.1093/jnci/dju246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Nguyen HN, Lie A, Li T, et al. . Human TERT promoter mutation enables survival advantage from MGMT promoter methylation in IDH1 wild-type primary glioblastoma treated by standard chemoradiotherapy. Neuro Oncol. 2017;19(3):394–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. You H, Wu Y, Chang K, et al. . Paradoxical prognostic impact of TERT promoter mutations in gliomas depends on different histological and genetic backgrounds. CNS Neurosci Ther. 2017;23(10):790–797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Zou MX, Lv GH, Li J, She XL, Jiang Y. Upregulated human telomerase reverse transcriptase (hTERT) expression is associated with spinal chordoma growth, invasion and poor prognosis. Am J Transl Res. 2016;8(2):516–529. [PMC free article] [PubMed] [Google Scholar]
- 31. Hu H, Yang HL, Lu J, et al. . Association of telomerase expression with recurrence of sacral chordoma. Ann Oncol. 2012;23(10):2772. [DOI] [PubMed] [Google Scholar]
- 32. Butler MG, Dahir GA, Hedges LK, Juliao SF, Sciadini MF, Schwartz HS. Cytogenetic, telomere, and telomerase studies in five surgically managed lumbosacral chordomas. Cancer Genet Cytogenet. 1995;85(1):51–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Bettegowda C, Yip S, Lo SL, et al. ; AOSpine Knowledge Forum Tumor Spinal column chordoma: prognostic significance of clinical variables and T (brachyury) gene SNP rs2305089 for local recurrence and overall survival. Neuro Oncol. 2017;19(3):405–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Varga PP, Szövérfi Z, Fisher CG, et al. . Surgical treatment of sacral chordoma: prognostic variables for local recurrence and overall survival. Eur Spine J. 2015;24(5):1092–1101. [DOI] [PubMed] [Google Scholar]
- 35. Fisher CG, Goldschlager T, Boriani S, et al. . An evidence-based medicine model for rare and often neglected neoplastic conditions. J Neurosurg Spine. 2014;21(5):704–710. [DOI] [PubMed] [Google Scholar]
- 36. Jawad MU, Scully SP. In brief: classifications in brief: enneking classification: benign and malignant tumors of the musculoskeletal system. Clin Orthop Relat Res. 2010;468(7):2000–2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Stacchiotti S, Sommer J; Chordoma Global Consensus Group Building a global consensus approach to chordoma: a position paper from the medical and patient community. Lancet Oncol. 2015;16(2):e71–e83. [DOI] [PubMed] [Google Scholar]
- 38. Pallini R, Maira G, Pierconti F, et al. . Chordoma of the skull base: predictors of tumor recurrence. J Neurosurg. 2003;98(4):812–822. [DOI] [PubMed] [Google Scholar]
- 39. Chi YK, Shen Q, Wang JC, Zheng XZ, Hou L, Zhang B. [Correlation of telomere length and the expression of its regulating proteins in mesenchymal sarcomas]. Beijing Da Xue Xue Bao Yi Xue Ban. 2008;40(4):363–368. [PubMed] [Google Scholar]
- 40. Bagheri S, Nosrati M, Li S, et al. . Genes and pathways downstream of telomerase in melanoma metastasis. Proc Natl Acad Sci U S A. 2006;103(30):11306–11311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Park CK, Lee SH, Kim JY, et al. . Expression level of hTERT is regulated by somatic mutation and common single nucleotide polymorphism at promoter region in glioblastoma. Oncotarget. 2014;5(10):3399–3407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Mancini A, Xavier-Magalhaes A, Woods WS, et al. . Disruption of the beta1L isoform of GABP reverses glioblastoma replicative immortality in a TERT promoter mutation-dependent manner. Cancer Cell. 2018; 34(3):513–528.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Tarpey PS, Behjati S, Young MD, et al. . The driver landscape of sporadic chordoma. Nat Commun. 2017;8(1):890. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.