

ABSTRACT
T-cell malignancies expressing the γδ T-cell receptor (TCR) are often associated with poor prognosis. Here, we determined the clinical outcome of pediatric patients with T-cell acute lymphoblastic leukemia (T-ALL) expressing the γδ TCR. Of 100 newly diagnosed T-ALL patients, 93 had γδ TCR analysis performed at diagnosis. Repertoire was evaluated by paired sequencing of the rearranged TCR. All patients received intensified chemotherapy and those with minimal residual disease (MRD) ≥ 1% on day 42–46 became candidates for hematopoietic cell transplantation. Of the 93 T-ALL patients, 12 (13%) had γδ T-ALL and 11 (12%) had early T-cell precursor (ETP) ALL. Compared to the remaining 70 T-ALL patients, the γδ T-ALL patients were more likely to have MRD ≥ 1% on day 15–19 (67% vs. 33%, P = 0.03) and day 42–49 (33% vs. 7%; P = 0.007) of remission induction. The 10-year overall survival for γδ T-ALL patients (66.7% ± 22.2%) were lower than that of T-ALL patients (93.3% ± 7.3%, P = 0.001). TCR analysis demonstrated a conserved clonotype. In conclusion, the data suggest that children with γδ T-ALL may have a poor response to remission induction, based on MRD levels and decreased survival than the other T-ALL patients, despite receiving risk-directed therapy.
KEYWORDS: γδ T cells, TCR repertoire, γδ T-ALL, pediatric T-ALL, HCT, risk stratification
Introduction
Two distinct T-cell lineages, which are based on the use of αβ or γδ heterodimers in the T-cell receptor (TCR) complex, diverge early during thymic development and during the course of the β-, γ-, and δ-chain rearrangement. Lineage commitment toward γδ over αβ T-cell and subsequent γδ thymocyte development is favored by strong TCR signaling. Recent studies have shown that the developmental and proliferative programs as well as the expression of genes regulating apoptosis differ between the γδ and αβ lineages.1,2 Certain neoplasms derived from the γδ T cell lineage such as primary cutaneous and hepatosplenic lymphomas are clinically aggressive with bleak prognoses and are recognized as being biologically distinct.3–8 For patients with T-cell acute lymphoblastic leukemia (T-ALL) expressing the γδ TCR, case reports have suggested that they are at higher risk for poor outcomes.7,9–20 Recently, identification of γδ T-cell lineage-specific genetic alterations leading to the fusion transcripts, SET-NUP214 and CALM-AF10, have been associated with chemotherapy resistance and poor prognosis, respectively, suggesting a biological link to the clinical outcome for patients with T-ALL.19,20
For patients with T-ALL, one of the most significant prognostic indicator is the level of minimal residual disease (MRD) during and after remission induction chemotherapy.21 Despite intensification of therapy, approximately 10% of patients with T-ALL continue to have elevated MRD or fail to obtain a morphologic remission after remission induction.21–25 Timely identification of patients with poor response to remission induction allows them to potentially benefit from early intensification treatment.26 Risk stratification of T-ALL has greatly improved by integrating immunophenotypic, cytogenetic, and/or molecular aberration, although the time restraints of genetic profiling remain problematic.27–30 However, flow cytometry (FCM) can rapidly detect TCR expression on lymphoblasts at diagnosis.
The TCR is formed by the recombination of the variable (V) gene to a joining (J) segment and the diversity (D) gene on the delta (TRD) or gamma (TRG) locus. Although similar in TCR structure, γδ T cells are not restricted by major histocompatibility complex (MHC), and their ability to recognize antigens differs remarkably.31–33 The γδ TCR repertoire is limited by six TRGV and eight TRDV genes.34 The two major γδ T cell subsets are determined by the expressed Vδ chain, Vδ1 and Vδ2. Vδ1 T cells are prominent in organs enriched with epithelial or mucosal surfaces and recognize stress-induced self-antigens, heat shock proteins, glycolipids, and members of the “MHC-like” superfamily.35 Vδ2 T cells are found in peripheral blood (PB) and comprise only 1–10% of the lymphocyte population. Majority of the PB TCR repertoire are Vγ9Vδ2 T c ells, which recognizes nonpeptide antigens commonly expressed by microbes.36,37 Here, we evaluated the clinical presentation and outcomes of patients with T-ALL with or without γδ TCR expression.
Results
Patients
Of the 93 T-ALL patients, 12 (13%) had γδ T-ALL and 11 (12%) had ETP ALL. Table 1 shows the presenting clinical and laboratory features of γδ T-ALL and the other T-ALL. There were no significant differences between γδ T-ALL and the other T-ALL in regards to age, gender, presenting leukocyte count, and CNS involvement at diagnosis. The γδ T-ALL group had a significantly higher percentage of African Americans (42% vs. 16%; P = 0.03) and presence of mediastinal mass (50% vs. 20%; P = 0.03) compared to the other T-ALL group. A higher percentage of γδ T-ALL patients were classified to have high-risk disease compared to other T-ALL patients (33% vs. 7%; P = 0.007) (Table 1).
Table 1.
Comparison of clinical and biologic variables between patients with γδ T-ALL, T-ALL and ETP.
| γδ T-ALL |
T-ALL |
vs. γδ T-ALL |
ETP ALL |
vs. γδ T-ALL |
||
|---|---|---|---|---|---|---|
| Category | Group | N = 12 (13%) | N = 70 (75%) | P | N = 11 (12%) | P |
| Age | 1–10 years | 8 (67%) | 44 (63%) | 0.8 | 6 (55%) | 0.6 |
| >10 years | 4 (33%) | 26 (37%) | 5 (45%) | |||
| Mean ± sem Median (Range) |
8.7 ± 1.3 8.5 (1.4–15.1) |
9.3 ± 0.6 8.2 (1.1–18.6) |
11.7 ± 1.1 10.6 (6.1–18.7) |
|||
| Gender | Male | 9 (75%) | 49 (70%) | 0.7 | 7 (67%) | 0.6 |
| Female | 3 (25%) | 21 (30%) | 4 (33%) | |||
| Race | White | 7 (58%) | 59 (80%) | 0.03 | 7 (64%) | 0.8 |
| Black | 5 (42%) | 11 (16%) | 4 (36%) | |||
| Leukocyte count 103/mm3 |
<10 | 2 (17%) | 12 (17%) | 0.8 | 2 (18%) | 0.8 |
| 10–50 | 4 (33%) | 15 (21%) | 4 (36%) | |||
| ≥50–100 | 2 (17%) | 10 (14%) | 3 (27%) | |||
| ≥100 | 4 (33%) | 33 (47%) | 2 (18%) | |||
| Mean ± sem Median (Range) |
107 ± 39 46 (4.2–401) |
171 ± 22 90 (1.9–657) |
0.1 | 50 ± 17 27 (3.9–182) |
0.6 | |
| CNS | CNS1 | 5 (42%) | 35 (50%) | 0.7 | 7 (64%) | 0.6 |
| CNS 2 | 5 (42%) | 22 (31%) | 3 (27%) | |||
| CNS 3 | 0 | 5 (7%) | 0 | |||
| Traumatic | 2 (17%) | 8 (11%) | 1 (9%) | |||
| Mass | Present | 6 (50%) | 14(20%) | 0.03 | 2 (18%) | 0.1 |
| Absent | 6 (50%) | 56 (80%) | 9 (82%) | |||
| Risk | Standard | 7 (58%) | 65 (93%) | 0.0007 | 4 (36%) | 0.3 |
| High | 5 (42%) | 5 (7%) | 7 (64%) | |||
| MRD Day 15–19 |
<1% vs .≥ 1% | 33% vs. 67% | 67% vs. 33% | 0.03 | 18% vs. 82% | 0.4 |
| <.01% | 1 (8%) | 18 (26%) | 0 | |||
| .01–0.1% | 3 (25%) | 10 (14%) | 0 | |||
| 0.1–1% | 0 | 18 (26%) | 2 (18%) | |||
| ≥ 1% | 8 (67%) | 23 (33%) | 9 (82%) | |||
| Mean ± sem Median (Range) |
27 ± 8.5 23 (0.05–79) |
6 ± 1.8 0 (≤.001–69) |
28 ± 7.6 28 (0.83–87) |
|||
| <1% vs. ≥ 1% | 67% vs. 33% | 93% vs 7% | 0.007 | 73% vs 27% | 0.7 | |
| MRD Day 42–46 |
<.01% | 7(58%) | 55 (79%) | 3 (27%) | ||
| .01–0.1% | 1 (8%) | 5 (7%) | 3 (27%) | |||
| 0.1–1% | 0 | 4 (6%) | 2 (18%) | |||
| ≥ 1% | 4 (33%) | 5 (7%) | 3 (27%) | |||
| Mean ± sem Median (Range) |
1.9 ± 0.9 0 (≤.001–7.1) |
0.13 ± 0.05 0 (≤.001–2.1) |
0.63 ± 0.4 0.03 (≤.001–3.8) |
|||
| HSCT | Yes | 5 (42%) | 7 (10%) | 0.004 | 8 (73%) | 0.1 |
| No | 7 (58%) | 63 (90%) | 3 (27%) | |||
| STATUS | Alive | 8 (67%) | 66 (94%) | 0.003 | 7 (64%) | 0.9 |
| Expired | 4 (33%) | 4 (6%) | 4 (36%) |
Abbreviations: T-ALL: T-cell acute lymphoblastic leukemia; γδ: γδ T-All; ETP: early T-cell progenitor; WBC: white blood count; CNS: central nervous system; MRD: minimal residual disease; sem: standard error of mean; Med.: media
Phenotypic alterations of γδ T-ALL
All of the lymphoblasts expressed γδ TCR, CD3, cyCD3, CD45, and CD7 and lacked αβ TCR, cyMPO, CD19, CD20, CD61, CD64, CD65, CD117, and CD45RA expression. The diverse maturation stages were reflected by the heterogeneous profile of CD2, CD1a, and CD34. The expression of CD4 and CD8 included double positive CD4+CD8+ (18%), double negative CD4−CD8− (27%), single positive CD4+ (18%), and CD8+ (36%) lymphoblasts. Patients with CD56 expression (5) had variable coexpression of CD4 (1), CD8 (3), or CD4/CD8 (1). One patient (No. 9) fulfilled criteria for both γδ T-ALL and ETP ALL, with full immunophenotype details provided in Table 2.
Table 2.
Immunophenotype of γδ T-ALL.
Abbreviations: S: song; B: bright; M: moderate; C: complex; D: dim; W: weak; *ETP: early T-cell progenitor, cy: cytoplasmic
TCR repertoire in γδ T-ALL lymphoblast
The V-(D)-J gene rearrangement for the TRG and TRD loci expressed by the lymphoblast was identified using single-cell PCR as previously described.38,39 Appropriate consents and samples were available for 9 of the 12 patients with γδ T-ALL (Table 3). A dominant clonal population of lymphoblast was identified for all patients. A biclonal population was observed in one patient where one TRDV region paired with two unique TRGV regions. The combinatory diversity of the TRG genes showed a bias toward TRGJ segments from the distal region 2 (JP2 and J2) (89%) joining to the terminal constant region (TRGC2). The use of the proximal region 1 (TRGJ1 and TRGC1) was rare. The TRGV regions detected were TRGV9 (33%), TRGV5 (33%), TRGV4 (20%), TRGV2 (10%), and TRGV8 (10%). Despite variability in the TRGV regions the CDR3γ regions were similar. The CDR3γ region contained an average of 14.2 ± 2.2 amino acids and each patient had an average of 3.3 ± 2.6 unique amino acids and shared an average of 11 ± 0.5 amino acids (88%). In contrast, the TRDV regions expressed were predominantly TRDV1 (67%). Non-TRDV1 included TRDV3 (11%), TRDV5 (11%), and TRDV8 (11%). All in frame sequences used the TRDJ1 segment.
Table 3.
V-(D)-J rearrangement and CDR3 region.
| PT |
T-cell receptor gamma (TRG) rearrangement |
T-cell receptor delta (TRD) rearrangement |
||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| # | V | J | C | CDR3γ | AA | V | J | D | CDR3δ | AA | ||||
| 2 | 5*0 | 1*0 | 2*0 | CAT | WDRH | YKKLF | 12 | 8*0 | 1*0 | 1*0 | CA | YRSAR LPDDT | DKLIF | 17 |
| 4 | 5*0 | 2*0 | 2*0 | CAT | WDRR | YKKLF | 12 | 1*0 | 1*0 | 1*0 | CA | LGELNTLRGGEVT | DKLIF | 20 |
| 3*0 | 2*0 | 2*0 | CAT | WDRRDY | YKKLF | 14 | ||||||||
| 5 | 9*0 | 2*0 | 2*0 | CAL | WEVHY | YKKLF | 13 | 5*0 | 1*0 | 3*0 | CA | ASE LYWGNSTAQL | DKLIF | 15 |
| 6 | 9*0 | 1*0 | 2*0 | CAL | WEVHVGAQLD | KKLF | 17 | 5*0 | 2*0 | 3*0 | CA | A LPTGLGGVGDSAA | QLFF | 20 |
| 7 | 4*0 | P2*0 | 2*0 | CAT | WDDG | SDWIKTF | 14 | 1*0 | 1*0 | 3*0 | CA | LGDSTGGYT | DKLIF | 14 |
| 9 | 8*0 | P2*0 | 2*0 | CAT | WDMG | SDWIKTF | 14 | 1*0 | 1*0 | 2*0 | CA | LGELNPSKLGDMG | LIF | 18 |
| 10 | 2*0 | 2*0 | 2*0 | CAT | WDGHLKTKNY | YKKLF | 18 | 1*0 | 1*0 | 3*0 | CA | LGKGGFS | DKLIF | 14 |
| 11 | 5*0 | P2*0 | 2*0 | CAT | WAYSL | SDWIKTF | 15 | 3*0 | 1*0 | 2*0 | CA | FP LSYNSGGRKC | DKLIF | 19 |
| 12 | 9*0 | 2*0 | 2*0 | CAL | WEG | YKKLF | 11 | 1*0 | 1*0 | 2*0 | CA | LGEQPSPWGIRN | KLIF | 18 |
Abbreviations: TRGV: T-cell receptor gamma variable; TRGJ: T-cell receptor gamma joining; TRGC: T-cell receptor gamma constant; CDR3: complementarity determining region 3; TRDV: T-cell receptor delta variable, TRDJ: T-cell receptor delta joining; TRDD: T-cell receptor delta diversity; AA: amino acid
Cytogenetic alterations
Majority of patients with γδ T-ALL had complex cytogenetic abnormalities (91%) and one patient had normal karyotype (Table 4). Hyperdiploidy was common (50%), but only one patient had high hyperdiploidy with a DNA index of 1.17. Chromosomal abnormalities involving the TRD locus on chromosome 14 (14q11.2) or TRG locus on chromosome 7 (7p14) were not detected by conventional cytogenetics. Cytogenetic aberrations were detected on chromosome 1 (1q23 and 1p36.1), 6q (6q13q21, 6q13q23, 6q21q23, and 6q21), 11 (11p11.2, 11p13, and 11q22), 12p (12p11.2 and 12p13,), and 14q (14q13, 14q32, and 14q32.1). Deletions involving 6q (q13-23) and/or 12p11-13 were observed in over half the patients (60%). Translocation included t(11;14)(p13;q32), t(10;11)(p12;q22), and t(11;14)(p11.2;q32.1), with the breakpoint on chromosome 14 centromeric to the IGH locus.
Table 4.
Clinical and laboratory characteristics of the 12 patients with γδ T-ALL.
| Patient | Gender | Age (Yrs) | Race | WBC 103/mm3 |
Mass | CNS | °Risk | MRD 15 | MRD D42 | HSCT | Status | Years | Disease | Ploidy | Chrom. # | DNA Index | Cytogenetics |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | M | 9.1 | W | 5.1 | N | T | H | 63 | 5.82 | MSD | A | 10 | CR | QNS | QNS | 1 | 2 Metaphases Appear Normal |
| 2 | M | 3.5 | AA | 102 | Y | 2 | S | 1.6 | <0.01* | URD | E | 0.2 | R | PD | 46 | 1 | 46,XY,del(6)(q21q23),del(11)(p13) 46,add(1)(q23),add(14)(q13) |
| 3 | F | 13.9 | W | 10 | N | 1 | S | 0.01 | <0.01 | A | 8.8 | CR | HD | 47–50 | 1 | 48,XX,+17,add(18)(p11.3),+19;48,idem,-17,+i(17)(q10) | |
| 4 | F | 6.1 | W | 64 | Y | T | H | 31 | >1%** | # | E | 1.3 | IF | HD | 47–48 | 1 | 47,XX,del(6)(q13q23),t(10;11)(p12;q22),del (12)(p13),der(17); +del(22)(q11.2); 48,idem,+del(22)(q11.2) |
| 5 | M | 5.5 | AA | 266 | Y | 1 | S | 0.02 | <0.01 | A | 4.6 | CR | HD | 47–50 | 1 | 47,XY,del(6)(q13q21),t(11;14)(p13;q32),+20 | |
| 6 | M | 9.7 | AA | 8.5 | N | 1 | S | 19 | 0.014 | E | 0.9 | CR | PD | 46 | 46,XY,del(7)(q22),del(12)(p11.2) | ||
| 7 | M | 1.4 | AA | 401 | Y | 2 | H | 28 | 7.1 | UCB | E | 0.3 | R | HD | 47 | 47,XY,add(7)(p22),+10 | |
| 8 | M | 13.6 | W | 9 | Y | 2 | S | <0.01 | <0.01 | A | 2.7 | CR | HD | 47 | 47,XY,del(10)(q26),del(12)(p13),+mar | ||
| 9 | M | 8.0 | W | 15 | N | 2 | H | 70.8 | <0.01 | URD | A | 3.0 | CR | HD | 53 | 1.17 | 53,XY,+7,+8,+10,+11,+13,+17,+21 |
| 10 | M | 12.9 | W | 30 | Y | 1 | S | 24 | <0.01 | A | 1.8 | CR | PD | 46 | 1 | 46,XY,del(6)(q21) | |
| 11 | F | 15.2 | AA | 284 | N | 1 | H | 80 | 3.8 | URD | A | 2.5 | CR | PD | 46 | 1 | 46,XX,t(11;14)(p11.2;q32.1) |
| 12 | M | 6.1 | W | 87 | N | 1 | S | 0.02 | <0.01 | A | 9.5 | CR | PD | 46 | 1 | 46,XY,add(1)(p36.1),del(12)(p11.2) |
Abbreviations: M: male; F: female; W: white; AA: African American; N: no; Y: yes; 1: CNS1; 2: CNS2; 3:CNS3; T: traumatic tap with blast; R: relapse; MSD: matched sibling donor; URD: unrelated donor; UCB: umbilical cord blood; HSCT: hematopoietic stem cell; PD: pseudodiploidy; HD: hyperdiploidy; DP: double positive CD4+CD8+; DN: double negative CD4−CD8−; A: alive; E: expired; CR: clinical remission; IF: induction failure. Risk group based on MRD levels measured on day 15 and 42; *MRD < 0.01 but relapsed after induction; ** MRD by PCR; # Expired prior to HSCT.
Response to remission induction and overall survival
MRD was monitored during induction treatment. A higher portion of γδ T-ALL patients had MRD ≥ 1% compared to other T-ALL patients on day 15–19 (67% vs. 33%, P = 0.03) and on day 42–49 (33% vs. 7%; P = 0.007) of remission induction. There was no difference in the percent of patients with γδ T-ALL and ETP ALL with MRD ≥ 1% on day 15–19 (67% vs. 82%; P = 0.4) and on day 42–46 (33% vs. 27%; P = 0.7) of remission induction.
T-ALL patients with relapse/refractory disease or MRD ≥ 1% at the end of induction were referred to HSCT. The proportion γδ T-ALL patients who received a HSCT (42%) was significantly higher than other T-ALL patients (10%; P = 0.004). The γδ T-ALL patients referred for an allogeneic HSCT were due to elevated MRD ≥ 1% at the end of induction (n; No. 1, 7, and 11), relapse after induction (No. 2), refractory disease (No. 4), or γδ/ETP ALL diagnosis (No. 9). Patient No. 9 was stratified as high-risk, underwent a HSCT, and remains in CR. The one patient with refractory disease (No. 4) died from disease progression prior to undergoing an HSCT. Three patients (No. 1, 9, and 11) are in complete remission 10, 3, and 2.5 years post-HSCT, respectively and two patients relapsed (No. 2 and 7) on day 70 and day 90 after HSCT.
With a median follow-up of 9.3 ± 1.4 years (range; 7.1–15.5 years), the 10-year OS for patients with γδ T-ALL was less compared to the other T-ALL patients (66.7% ± 22.2% vs. 93.3% ± 7.3%) (P = 0.001). The 10-year EFS for patients with γδ T-ALL was lower as compared to the other T-ALL patients (66.7% ± 22.2% vs. 81.2% ± 12.4%), albeit not significant (P = 0.11) (Figure 1). The cumulative incidence of relapse was not significantly different for patients with γδ T-ALL (18.2% ± 12.3%) compared to the other T-ALL patients (13.3% ± 4.2%) (P = 0.58). In multivariable analyses, adjusting for age, race, presenting leukocyte count, and MRD level on day 15–19 or day 42–46, γδ T-ALL was independently associated with poor survival (hazard ratio, 6.95; 95% CI, 1.2–40.2; P = 0.03; Table 5).
Figure 1.
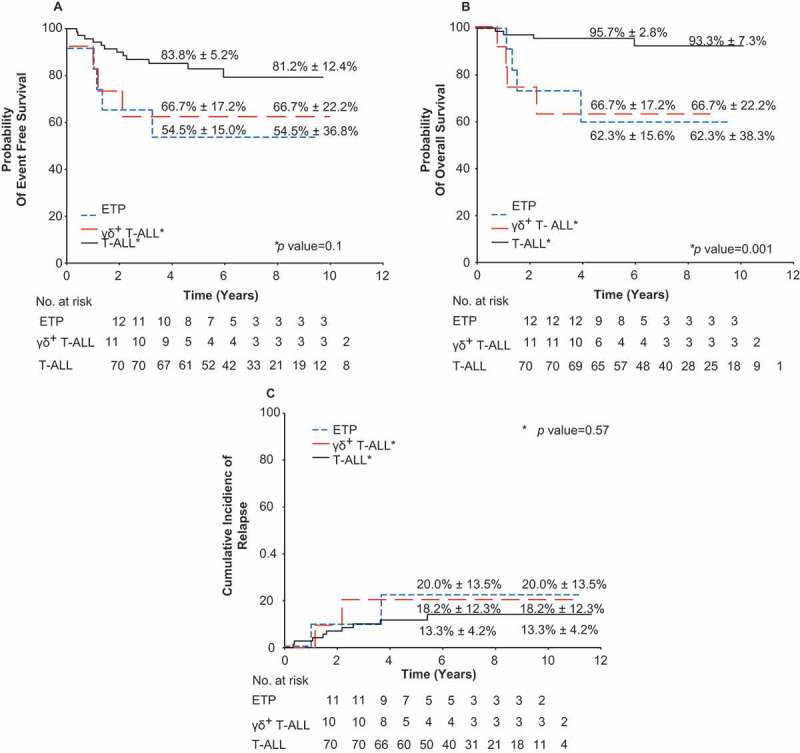
Probability of event-free survival, overall survival, and cumulative incidence of relapse for patients with γδ T-ALL. Kaplan-Meier estimates of (a) event-free survival, (b) overall survival, and (c) cumulative incidence of relapse for patients with γδ T-ALL, early T cell progenitor (ETP) ALL and T-ALL are shown. *P values are for γδ T-ALL vs. T-ALL. Five- and 10-year rates reported as means ± standard error.
Table 5.
EFS, OS, and risk of relapse for patients with γδ T-ALL compared to T-ALL and ETP ALL.
| Factor | Year | γδ T-ALL N = 12 Mean ± SE (%) |
T-ALL N = 70 Mean ± SE (%) |
γδ T-ALL vs T-ALL P |
ETP ALL N = 11 Mean ± SE (%) |
γδ T-ALL vs ETP ALL P |
|---|---|---|---|---|---|---|
| EFS | 5 | 66.7 ± 17.2 | 83.8 ± 5.2 | 0.11 | 54.5 ± 15.0 | 0.63 |
| 10 | 66.7 ± 22.2 | 81.2 ± 12.4 | 54.5 ± 36.8 | |||
| OS | 5 | 66.7 ± 17.2 | 95.7 ± 2.8 | 0.001 | 62.3 ± 15.6 | 0.98 |
| 10 | 66.7 ± 22.2 | 93.3 ± 7.3 | 62.3 ± 38.3 | |||
| Relapse | 5 | 18.2 ± 12.3 | 13.3 ± 4.2 | 0.58 | 20.0 ± 13.5 | 0.98 |
| 10 | 18.2 ± 1 2.3 | 13.3 ± 4.2 | 20.0 ± 13.5 |
Abbreviations: EFS: event-free survival; SE: standard error; OS: overall survival; ETP: early T-cell progenitor; SE: standard error
Discussion
We found the incidence of γδ T-ALL to be ~15% in childhood T-ALL, which is within the range of 10–26% reported in other studies.9,40 The proportion of African Americans and patients with mediastinal mass was higher in the γδ T-ALL group compared to the other T-ALL group. Ethnic differences in the frequency of γδ T cells, with a higher proportion of Vδ1 T cells in healthy African Americans compared to Caucasians have been reported.41–43 Furthermore, MICA genetic polymorphisms in African Americans have significant linkage disequilibria with HLA-B and has been implicated in various disease susceptibilities, but its role in T-ALL remains elusive.44
Patients with γδ T-ALL are more likely to have a poor response to treatment as reflected by the high MRD levels during remission induction therapy. A large proportion (67%) of patients with γδ T-ALL had MRD levels ≥ 1% after 15–19 days of induction therapy and received additional chemotherapy. Despite intensification of therapy, a third of the γδ T-ALL patients still had elevated MRD levels ≥ 1% at the end of induction therapy and became candidates for HSCT. Patients with T-ALL expressing the γδ TCR demonstrated a poor response to remission induction treatment, which has been highly predictive for extremely poor outcomes.23,24 The 10-year OS for patient with γδ T-ALL was lower compared to other T-ALL patients (66.7% vs. 93.3%; P = 0.001). The lack of significant difference in the cumulative risk of relapse between the two groups may be due to a higher proportion of γδ T-ALL patients undergoing HSCT.
Deletion of 6q is observed in approximately 10–20% of T-ALL patients and was frequently observed in patients with γδ T-ALL. However, the t(11;14)(p11;q32) translocation observed are more often reported in patients with relatively aggressive splenic marginal zone B cell lymphoma but is uncommon in T-ALL. The specific breakpoint on chromosome 14 centromeric to the IGH locus occurs at high frequency in patients with ataxia telangiectasia and mature T cell diseases but less common in T-ALL. Guiterrez et al.30 showed that absence of biallelic deletion (ABD) of the TCRγ locus was a robust predictor of induction failure and associated with poor overall survival. Given that TCRγ rearrangements occur early in development, the authors found an overlap between patient with ABD and ETP. Patients with γδTCR have VJ recombination on at least one TCRγ allele occurring later in development and would not fulfill the definition of ABD. In summary, majority of patients with γδ T-ALL had complex cytogenetic abnormalities and additional studies are needed to detect the cryptic lesions associated with γδ T-ALL.
Here, we describe the outcomes for the largest cohort of γδ T-ALL patients reported to date. The incidence of γδ T-ALL ~10–15% was comparable to other reports. Similarly, we observed that a higher proportion of γδ T-ALL patients had aggressive disease. Patients who relapsed were refractory to chemotherapy and often died from disease progression. Due to the rare occurrence and limited cases of γδ T-ALL, the adverse prognostic significance should be confirmed in an independent cohort of children with T-ALL.
For certain lymphomas, the biologic behavior of neoplasms derived from γδ T cells have been recognized as being distinct according to the World Health Organization (WHO) classification of lymphoid neoplasms. Comparably for patients with T-ALL, recent studies have identified γδ T cell specific genetic alterations that is predictive of poorer response to therapy or outcomes. Here, we present corroborating data suggesting that patients with γδ T-ALL are also at risk for poorer response to therapy or outcomes. Understanding the mechanism driving leukemic transformation and identifying factors contributing to the poor treatment response may help to develop treatment strategies to improve clinical outcome of these patients.
Patient and methods
Patient selection
Between 2000 and 2014, 100 consecutive newly diagnosed T-ALL patients were enrolled onto the Total Therapy XV (NCT00137111) or XVI (NCT00549848) study at St. Jude Children’s Research Hospital (SJCRH). Of the 100 patients, 93 had diagnostic samples with γδ TCR data and were evaluable for follow up. The studies were approved by the SJCRH Institutional Review Board and are in accordance with the Helsinki Declaration of 1975; written informed consents were obtained from the parents, guardians, or the patients, and assent from the patients, as appropriate.
Diagnosis and risk classification
ALL diagnosis was based on morphologic, immunophenotypic, and genetic features of leukemic blast cells while early T-cell precursor (ETP) ALL, a subtype of T-cell ALL generally associated with poor prognosis, was diagnosed by immunophenotype as previously described.45 MRD was determined by FCM and/or PCR, as previously described.21 All T-ALL patients were provisionally classified to have standard-risk ALL to receive intensive chemotherapy. Those with MRD levels ≥1% at the end of remission induction or relapsed during therapy were candidates for hematopoietic stem-cell transplantation (HSCT). Patients with ETP ALL treated in Total Therapy XVI were all considered to have high-risk disease and were candidates for HSCT.
Risk-adapted treatment
Details of the treatment regimen for the protocols have been described previously.45,46 Treatment consisted of remission induction, consolidation, and continuation. In brief, patients with MRD ≥ 1% in the bone marrow (BM) on day 15–19 of induction were given three additional doses of native E. coli asparaginase in Total Therapy XV or one dose of peg-asparaginase in Total Therapy XVI. At the end of induction (days 42–46), BM aspiration was performed to assess MRD level, and consolidation therapy with high-dose methotrexate (5 g/m2 per dose) and daily mercaptopurine was given for four courses. Standard-risk patients then received intensive continuation chemotherapy, and high-risk patients were offered the option of HSCT. All patients received triple intrathecal therapy administered early during remission induction and throughout the first 2 years of continuation treatment.
TCR repertoire by single-cell-nested PCR
Nested PCR was carried out as previously described.38,39 Briefly, γδ T cells (CD3+TCRγδ+CD14−CD19−) were single cell sorted using a BD FACSAria III or Sony iCytSy3200 (Sony Biotechnology) into 96-well PCR plates (Eppendorf) preloaded with 2.5 μL of reverse-transcription master mix (iScript cDNA Synthesis Kit, Bio-Rad Laboratories/SuperScript VILO cDNA Synthesis Kit, Life Technologies) containing 0.5 μL 5X iScript reaction mix, 0.5 μL iScript reverse transcriptase, and 0.1% Triton X-100 (Sigma-Aldrich). Columns 11 and 12 of each PCR plate were left empty to serve as controls. Plates were sealed, spun down, and reverse transcription was performed on a Bio-Rad C1000 Thermo Cycler using the following protocol: 5 min at 25°C, 60 min at 42°C, 5 min at 85°C, hold 4°C. First round PCR was then carried out with 5 μM of all forward TCRγ variable (TRGV) and δ variable (TRDV) external (ext) primers and 20 μM all reverse external primers (EuroFins Genomics) using the Taq DNA Polymerase Kit (QIAGEN) according to manufacturer’s instructions. Second-round reactions were then electrophoresed on a 2% agarose gel (Bio-Rad) to confirm presence of amplicons. Prior to sequencing, all PCR reactions were purified with exonuclease I-shrimp alkaline phosphatase according to the manufacturer’s instructions (Affymetrix USB). A total of 20 μM of the γ or δ internal reverse primers were added to the appropriate purified PCR products, and sequenced using a ABI Big Dye sequencer (Applied Biosystems). Sequences were then blasted using the IMGT database.
Statistical analysis
Event-free survival (EFS) and overall survival (OS) were estimated by the method of Kaplan-Meier, with associated standard errors calculated by the method of Peto and Pike.47 The cumulative incidence functions of relapse were estimated according to Kalbfleisch and Prentice, and compared with Gray’s test.48,49 Deaths in remission were considered competing events in the estimation of cumulative incidence of relapse. Statistical analyses were performed with SAS software (v9.3).
Supplementary Material
Funding Statement
This work was supported in part by NIH P30 CA021765, R56 AI091938, R37 CA36401, U01 GM92666, P50 GM115279, F32 CA141762, and R37 A36401. This work was also supported by the NCI Cancer Center Support Grant CA 21765, the American Association for Cancer Research, St. Baldrick’s Foundation,Individual Biomedical Research Award from The Hartwell Foundation, American Lebanese Syrian Associated Charities (ALSAC), and Angie Fowler Adolescent & Young Adult Cancer Institute (The Char and Chuck Fowler Family Foundation).
AUTHORSHIP
Conception and design
Mari Hashitate Dallas and Ching-Hon Pui.
Funding
Mari Hashitate Dallas, Paul Thomas, and Ching-Hon Pui.
Provision of study material or patients
Sima Jeha, Hiroto Inaba, Ching-Hon Pui, John Choi, and Paul Thomas.
Collection and assembly of data
Mari Hashitate Dallas, Suzanne Tomchuck, Scarlett Evans, Susana Raimondi, John Choi, Deqing Pei, and Ching-Hon Pui.
Data analysis and interpretation
Mari Hashitate Dallas, Cheng Cheng, Deqing Pei, Suzanne Tomchuck, Scarlett Evans, Susana Raimondi, John Choi, Paul Thomas, and Ching-Hon Pui
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- 1.Zarin P, Wong GW, Mohtashami M, Wiest DL, Zuniga-Pflucker JC.. Enforcement of γδ -lineage commitment by the pre-T-cell receptor in precursors with weak γδ -TCR signals. Proc Natl Acad Sci USA. 2014;111:5658–5663. doi: 10.1073/pnas.1312872111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ribeiro ST, Tesio M, Ribot JC, Macintyre E, Barata JT, Silva-Santos B.. Casein kinase 2 controls the survival of normal thymic and leukemic γδ T cells via promotion of AKT signaling. Leukemia. 2017;31:1603–1610. doi: 10.1038/leu.2016.363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Vega F, Medeiros LJ, Gaulard P. Hepatosplenic and other γδ T-cell lymphomas. Am J Clin Pathol. 2007;869–880. doi: 10.1309/LRKX8CE7GVPCR1FT. [DOI] [PubMed] [Google Scholar]
- 4.Toro JR, Liewehr DJ, Pabby N, Sorbara L, Raffeld M, Steinberg SM, Jaffe ES. Gamma-delta T-cell phenotype is associated with significantly decreased survival in cutaneous T-cell lymphoma. Blood. 2003;101:3407–3412. doi: 10.1182/blood-2002-05-1597. [DOI] [PubMed] [Google Scholar]
- 5.Sugimoto T, Imoto S, Matsuo Y, Kojima K, Yasukawa M, Murayama T, Kohfuku J, Mizuno I, Yakushijin K, Sada A, et al. T-cell receptor γδ T-cell leukemia with the morphology of T-cell prolymphocytic leukemia and a postthymic immunophenotype. Ann Hematol. 2001;80:749–751. doi: 10.1007/s00277-001-0381-z. [DOI] [PubMed] [Google Scholar]
- 6.Smith JL, Haegert DG, Hodges E, Stacey GN, Howell WM, Wright DH, Jones DB. Phenotypic and genotypic heterogeneity of peripheral T-cell lymphoma. Br J Cancer. 1988;58:723–729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Saito T, Matsuno Y, Tanosaki R, Watanabe T, Kobayashi Y, Tobinai K. γδ T-cell neoplasms: a clinicopathological study of 11 cases. Ann oncol. 2002;13:1792–1798. doi: 10.1093/annonc/mdf293. [DOI] [PubMed] [Google Scholar]
- 8.Belhadj K, Reyes F, Farcet JP, Tilly H, Bastard C, Angonin R, Deconinck E, Charlotte F, Leblond V, Labouyrie E, et al. Hepatosplenic γδ T-cell lymphoma is a rare clinicopathologic entity with poor outcome: report on a series of 21 patients. Blood. Vol 102 2003. p. 4261–4269. doi: 10.1182/blood-2003-05-1675 [DOI] [PubMed] [Google Scholar]
- 9.Alfsen GC, Beiske K, Holte H, Hovig E, Deggerdal A, Sandlie I, Widing E, Slordahl S, Klepper LK, Sizoo W, et al. T-cell receptor tau delta +/CD3+4-8-T- cell acute lymphoblastic leukemias: a distinct subgroup of leukemias in children. A report of five cases. Blood. Vol 77 1991. p. 2023–2030. [PubMed] [Google Scholar]
- 10.Gonzalez-Sarmiento R, LeBien TW, Bradley JG, Greenberg JM, Seidman JG, Ang S, Kersey JH. Acute leukemia expressing the gamma gene product of the putative second T cell receptor. J Clin Invest. 1987;79:1281–1284. doi: 10.1172/JCI112949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Langerak AW, Wolvers-Tettero IL, van Den Beemd MW, van Wering ER, Ludwig WD, Hahlen K, Necker A, van Dongen JJ. Immunophenotypic and immunogenotypic characteristics of TCRgammadelta+ T cell acute lymphoblastic leukemia. Leukemia. 1999;13:206–214. [DOI] [PubMed] [Google Scholar]
- 12.de Villartay JP, Pullman AB, Andrade R, Tschachler E, Colamenici O, Neckers L, Cohen DI, Cossman J. γδ lineage relationship within a consecutive series of human precursor T-cell neoplasms. Blood. 1989;74:2508–2518. [PubMed] [Google Scholar]
- 13.Schott G, Sperling C, Schrappe M, Ratei R, Martin M, Meyer U, Riehm H, Ludwig WD. Immunophenotypic and clinical features of T-cell receptor γδ + T-lineage acute lymphoblastic leukaemia. Br J Haematol. 1998;101:753–755. [DOI] [PubMed] [Google Scholar]
- 14.Zheng H, Wang X, Ma Y, Xu B, Chen S, Yang L, Wu X, Przybylski GK, Huang S, Ye T, et al. The TCR γδ repertoire and relative gene expression characteristics of T-ALL cases with biclonal malignant Vδ1 and Vδ2 T cells. DNA Cell Biol. 2014;33:49–56. doi: 10.1089/dna.2013.2199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wilhelm M, Tony HP. Characterization of an acute T lymphoblastic leukemia expressing the γδ T-cell receptor. Blut. 1990;61:213–218. [DOI] [PubMed] [Google Scholar]
- 16.Morales Madrid La A, Ouyang K, Raca G, Jamali M, Hyjek E, McNeer JL, Anastasi J. A case of pediatric γδ T-cell malignancy with t(8;14)(q24;q11)/MYC-TCR successfully treated with pulse type chemotherapy followed by stem cell transplant. Leuk Lymphoma. 2013;54:403–405. doi: 10.3109/10428194.2012.708930. [DOI] [PubMed] [Google Scholar]
- 17.Breit TM, Wolvers-Tettero IL, Hahlen K, van Wering ER, van Dongen JJ. Limited combinatorial repertoire of γδ T-cell receptors expressed by T-cell acute lymphoblastic leukemias. Leukemia. 1991;5:116–124. [PubMed] [Google Scholar]
- 18.Clemente MJ, Przychodzen B, Jerez A, Dienes BE, Afable MG, Husseinzadeh H, Rajala HL, Wlodarski MW, Mustjoki S, Maciejewski JP. Deep sequencing of the T-cell receptor repertoire in CD8+ T-large granular lymphocyte leukemia identifies signature landscapes. Blood. 2013;122:4077–4085. doi: 10.1182/blood-2013-05-506386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Asnafi V, Radford-Weiss I, Dastugue N, Bayle C, Leboeuf D, Charrin C, Garand R, Lafage-Pochitaloff M, Delabesse E, Buzyn A, et al. CALM-AF10 is a common fusion transcript in T-ALL and is specific to the TCRγδ lineage. Blood. 2003;102:1000–1006. doi: 10.1182/blood-2002-09-2913. [DOI] [PubMed] [Google Scholar]
- 20.Ben Abdelali R, Roggy A, Leguay T, Cieslak A, Renneville A, Touzart A, Banos A, Randriamalala E, Caillot D, Lioure B, et al. SET-NUP214 is a recurrent γδ lineage-specific fusion transcript associated with corticosteroid/chemotherapy resistance in adult T-ALL. Blood. 2014;123:1860–1863. doi: 10.1182/blood-2013-08-521518. [DOI] [PubMed] [Google Scholar]
- 21.Pui CH, Pei D, Coustan-Smith E, Jeha S, Cheng C, Bowman WP, Sandlund JT, Ribeiro RC, Rubnitz JE, Inaba H, et al. Clinical utility of sequential minimal residual disease measurements in the context of risk-based therapy in childhood acute lymphoblastic leukaemia: a prospective study. Lancet Oncol. 2015;16:465–474. doi: 10.1016/S1470-2045(15)70082-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Duval M, Klein JP, He W, Cahn JY, Cairo M, Camitta BM, Kamble R, Copelan E, de Lima M, Gupta V, et al. Hematopoietic stem-cell transplantation for acute leukemia in relapse or primary induction failure. J Clin Oncol. 2010;28:3730–3738. doi: 10.1200/JCO.2010.28.8852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Oudot C, Auclerc MF, Levy V, Porcher R, Piguet C, Perel Y, Gandemer V, Debre M, Vermylen C, Pautard B, et al. Prognostic factors for leukemic induction failure in children with acute lymphoblastic leukemia and outcome after salvage therapy: the FRALLE 93 study. J Clin Oncol. 2008;26:1496–1503. doi: 10.1200/JCO.2007.12.2820. [DOI] [PubMed] [Google Scholar]
- 24.Schrappe M, Hunger SP, Pui CH, Saha V, Gaynon PS, Baruchel A, Conter V, Otten J, Ohara A, Versluys AB, et al. Outcomes after induction failure in childhood acute lymphoblastic leukemia. N Engl J Med. 2012;366:1371–1381. doi: 10.1056/NEJMoa1110169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pui CH, Evans WE. Treatment of acute lymphoblastic leukemia. N Engl J Med. 2006;354:166–178. doi: 10.1056/NEJMra052603. [DOI] [PubMed] [Google Scholar]
- 26.Herold R, von Stackelberg A, Hartmann R, Eisenreich B, Henze G. Acute lymphoblastic leukemia-relapse study of the Berlin-Frankfurt-Munster Group (ALL-REZ BFM) experience: early treatment intensity makes the difference. J Clin Oncol. 2004;22:569–570. author reply 70-1. doi: 10.1200/JCO.2004.99.153. [DOI] [PubMed] [Google Scholar]
- 27.Aifantis I, Raetz E, Buonamici S. Molecular pathogenesis of T-cell leukaemia and lymphoma. Nat Rev Immunol. 2008;8:380–390. doi: 10.1038/nri2304. [DOI] [PubMed] [Google Scholar]
- 28.Ferrando A. NOTCH mutations as prognostic markers in T-ALL. Leukemia. 2010;24:2003–2004. doi: 10.1038/leu.2010.237. [DOI] [PubMed] [Google Scholar]
- 29.Ferrando AA, Look AT. Clinical implications of recurring chromosomal and associated molecular abnormalities in acute lymphoblastic leukemia. Semin Hematol. 2000;37:381–395. [DOI] [PubMed] [Google Scholar]
- 30.Gutierrez A, Dahlberg SE, Neuberg DS, Zhang J, Grebliunaite R, Sanda T, Protopopov A, Tosello V, Kutok J, Larson RS, et al. Absence of biallelic TCRγ deletion predicts early treatment failure in pediatric T-cell acute lymphoblastic leukemia. J Clin Oncol. 2010;28:3816–3823. doi: 10.1200/JCO.2010.28.3390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ciofani M, Zuniga-Pflucker JC. Determining γδ versus αβ T cell development. Nat Rev Immunol. 2010;10:657–663. doi: 10.1038/nri2820. [DOI] [PubMed] [Google Scholar]
- 32.Melichar HJ, Narayan K, Der SD, Hiraoka Y, Gardiol N, Jeannet G, Held W, Chambers CA, Kang J. Regulation of γδ versus αβ T lymphocyte differentiation by the transcription factor SOX13. Science. 2007;315:230–233. doi: 10.1126/science.1135344. [DOI] [PubMed] [Google Scholar]
- 33.Adams EJ, Chien YH, Garcia KC. Structure of a γδ T cell receptor in complex with the nonclassical MHC T22. Science. 2005;308:227–231. doi: 10.1126/science.1106885. [DOI] [PubMed] [Google Scholar]
- 34.Shin S, El-Diwany R, Schaffert S, Adams EJ, Garcia KC, Pereira P, Chien YH. Antigen recognition determinants of γδ T cell receptors. Science. 2005;308:252–255. doi: 10.1126/science.1106480. [DOI] [PubMed] [Google Scholar]
- 35.Uldrich AP, Le Nours J, Pellicci DG, Gherardin NA, McPherson KG, Lim RT, Patel O, Beddoe T, Gras S, Rossjohn J, et al. CD1d-lipid antigen recognition by the γδ TCR. Nat Immunol. 2013;14:1137–1145. doi: 10.1038/ni.2713. [DOI] [PubMed] [Google Scholar]
- 36.Ferrero I, Wilson A, Beermann F, Held W, MacDonald HR. T cell receptor specificity is critical for the development of epidermal γδ T cells. J Exp Med. 2001;194:1473–1483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Allison TJ, Winter CC, Fournie JJ, Bonneville M, Garboczi DN. Structure of a human γδ T-cell antigen receptor. Nature. 2001;411:820–824. doi: 10.1038/35081115. [DOI] [PubMed] [Google Scholar]
- 38.Wang GC, Dash P, McCullers JA, Doherty PC, Thomas PG. T cell receptor αβ diversity inversely correlates with pathogen-specific antibody levels in human cytomegalovirus infection. Sci Transl Med. 2012;4:128ra142. doi: 10.1094/PDIS-11-11-0999-PDN. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dash P, McClaren JL, Oguin TH 3rd, Rothwell W, Todd B, Morris MY, Becksfort J, Reynolds C, Brown SA, Doherty PC, et al. Paired analysis of TCRalpha and TCRbeta chains at the single-cell level in mice. J Clin Invest. 2011;121:288–295. doi: 10.1172/JCI44752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Matos DM, Rizzatti EG, Fernandes M, Buccheri V, Falcao RP. γδ and αβ T-cell acute lymphoblastic leukemia: comparison of their clinical and immunophenotypic features. Haematologica. 2005;90:264–266. [PubMed] [Google Scholar]
- 41.Cairo C, Armstrong CL, Cummings JS, Deetz CO, Tan M, Lu C, Davis CE, Pauza CD. Impact of age, gender, and race on circulating γδ T cells. Hum Immunol. 2010;71:968–975. doi: 10.1016/j.humimm.2010.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hviid L, Akanmori BD, Loizon S, Kurtzhals JA, Ricke CH, Lim A, Koram KA, Nkrumah FK, Mercereau-Puijalon O, Behr C. High frequency of circulating γδ T cells with dominance of the Vδ1 subset in a healthy population. Int Immunol. 2000;12:797–805. [DOI] [PubMed] [Google Scholar]
- 43.Hviid L, Kurtzhals JA, Adabayeri V, Loizon S, Kemp K, Goka BQ, Lim A, Mercereau-Puijalon O, Akanmori BD, Behr C. Perturbation and proinflammatory type activation of Vδ1(+) γδ T cells in African children with Plasmodium falciparum malaria. Infect Immun. 2001;69:3190–3196. doi: 10.1128/IAI.69.5.3190-3196.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tian W, Boggs DA, Uko G, Essiet A, Inyama M, Banjoko B, Adewole T, Wz D, Mohseni M, Fritz R, et al. MICA, HLA-B haplotypic variation in five population groups of sub-Saharan African ancestry. Genes Immun. 2003;4:500–505. doi: 10.1038/sj.gene.6364017. [DOI] [PubMed] [Google Scholar]
- 45.Pui CH, Campana D, Pei DQ, Bowman WP, Sandlund JT, Kaste SC, Ribeiro RC, Rubnitz JE, Raimondi SC, Onciu M, et al. Treating childhood acute lymphoblastic leukemia without cranial irradiation. New Engl J Med. 2009;360:2730–2741. doi: 10.1056/NEJMoa0900386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pui CHCD, Sandlund JT, Bhojwani D, Evans WE, Relling MV, Jeha S. Treatment of childhood acute lymphoblastic leukemia without cranial irradiation. Ann Hematol. 2011;90:S61–S63. [Google Scholar]
- 47.Peto R, Pike MC, Armitage P, Breslow NE, Cox DR, Howard SV, Mantel N, McPherson K, Peto J, Smith PG. Design and analysis of randomized clinical trials requiring prolonged observation of each patient. II. analysis and examples. Br J Cancer. 1977;35:1–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Prentice RL. Methods for the analysis of time to failure data. Prog Clin Biol Res. 1983;132E:69–79. [PubMed] [Google Scholar]
- 49.Gray RJ. A class of K-sample tests for comparing the cumulative incidence of a competing risk. Ann Stat. 1988;16:1141–1154. doi: 10.1214/aos/1176350951. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.