

Abstract
Background: A novel prion variant, PRNP p.Tyr225Cys (c.674A>G; p.Y225C), was identified in an atypical Creutzfeldt–Jakob disease (CJD) patient. The patient had a 5-year history of progressive cognitive impairment with speech and gait disturbances. From the basic neurological examination at his first hospital visit, rigidity and myoclonic jerks in all limbs were observed without focal weakness. Electroencephalogram showed the diffuse slow continuous delta activity in the bilateral cerebral hemisphere. Magnetic resonance imaging revealed abnormalities in the brain, such as cortical signal changes and edema in the frontotemporoparietal lobes and the basal ganglia. Cerebrospinal fluid 14–3-3 protein analysis showed a weakly positive signal. Family history remained unclear, but the patient’s mother and sister were diagnosed with cognitive impairment but both refused genetic testing.
Methods: Targeted next generation sequencing (NGS) was performed on 50 genes, involved in different neurodegeneratives diseases, such as Alzheimer's, Parkinson's, frontotemporal dementia or prion diseases. In silico analyses and structure predictions were performed on the potential patohgenic mutations.
Results: NGS and standard sequencing revealed the novel PRNP p.Tyr225Cys mutation in the patient. Structure predictions revealed that this may make the helix more flexible. In addition, the extra cysteine residue in TM-III of prion protein may result in disturbances of natural disulfide bond.
Conclusion: Hence, the pathogenicity of PRNP p.Tyr225Cys was not fully confirmed at present, and its penetrance was suggested to be low. However, its possible pathogenic nature in prion diseases cannot be ignored, since Tyr/Cys exchange could disturb the helix dynamics and contribute to conformational alteration and disease progression.
Keywords: prion, PRNP, Tyr225Cys mutation, atypical Creutzfeldt–Jakob disease, Gerstmann–Sträussler–Scheinker syndrome, sequencing, diagnosis
Introduction
Human prion diseases are fatal neurodegenerative disorders with diverse phenotypes, including, but not limited to Creutzfeldt–Jakob disease (CJD), Gerstmann–Sträussler–Scheinker syndrome (GSS), fatal familial insomnia, and kuru.1,2 Various clinical symptoms may appear inprion diseases, such as cognitive dysfunctions, psychiatric symptoms, motoric impairment (ataxia, dysarthria), myoclonus, visual problems, or weakness.3,4 Genetics plays a role in prion diseases, but only 10–15% of all cases can be explained by genetic mutations.5 Prion protein (PrP), consisting of 253 amino acids, is encoded by the prion gene (PRNP), located on chromosome 20. The exact role of prions remains to be elucidated, but they may be involved in different brain functions, such as neuronal protection, cell adhesion, and signaling, or circadian rhythm regulation.6 More than 30 pathogenic mutations were reported within the PRNP gene (NC_000020.11), and the inheritance of prion mutations may follow a Mendelian autosomal dominant pattern. For example, p.Glu200Lys was a quite common causative mutation for familial CJD,7 but several additional mutations, such as p.Asp178Asn,8 p.Val180Ile,9 p.Val210Ile,10 p.Tyr226Ter,11 or p.Met232Arg,12 were also associated with familial form of the disease. Interestingly, the limited variants in PRNP were also reported without prior family history of the disease (de novo disease cases),5 such as p.Glu196Ala13 and p.Glu200Gly.14 Classic CJD was usually rapidly progressive disease, and patients may die within one year after the disease onset. Clinical features could be the rapid decline of the cognitive functions, ataxia, visual and cerebellar problems, pyramidal and extrapyramidal dysfunctions, and myoclonus. In the cerebrospinal fluid (CSF), the 14-3-3 protein levels may be elevated.15 Several CJD cases were discovered with longer duration of the disease, younger age of onset, atypical findings in electroencephalogram (EEG) signal or or negativity for 14-3-3 protein marker in CSF. Patients with atypical CJD may be easily misdiagnosed as other neurological diseases, such as Alzheimer’s disease (AD) or the frontotemporal dementia (FTD).16,17
In this study, a novel prion variant, PRNP p.Tyr225Cys was reported in a patient with atypical CJD. Diagnosis of atypical CJD was considered due to his slow disease progression with slow cognitive decline. Family history of the patient remained unknown; however, the chance of positive family history cannot be ruled out. An extensive genetic screen on this patient was performed with 50 neurodegenerative disease-associated genes.16 In silico simulations were also performed to uncover possible pathogenic mechanisms of the mutation.
Case presentation
The study was performed in accordance with the latest revision of the Declaration of Helsinki and the Good Clinical Practice: Consolidated Guideline, approved by the International Conference on Harmonization and applicable to national and local laws and regulations. The patient and caregivers provided written informed consent. The patient was examined by the Korean CJD surveillance center. This report was approved by the Institutional Review Board (IRB) of Veteran Health Service Medical Center (IRB No. 2016-05-022).
A 59-year-old male was admitted to the Korean Veteran’s Hospital in Seoul, Republic of Korea with a 5-year history of progressive cognitive, speech, and gait impairments. The patient was right-handed person with high school education, and had been living abroad without marriage. He did not have any history of drug addiction/abuse or mental illness. Interestingly, his younger sister noticed and reported his speech impairment during phone conversations. During his first visit to the hospital, he mumbled words that did not fit to the situation or context. Later on, he was unable to communicate properly with other people, often repeating the same words. Two months prior to admission, the patient displayed rapidly progressive speech impairment with occasional mutterings of meaningless words. In the advanced disease stages, the akinetic mutism was observed. He was unable to test for the mini-mental state examination (MMSE).
Patient was initially diagnosed with slowly progressive dementia in a local hospital. Later, the memory decline became rapid and his movement slowed down. Sympathetic dysfunctions and stiffness were observed in both of his upper limbs. The rigidity and myoclonic jerks were observed in all limbs. Tendon reflexes and Babinski signs were normal. He did not use any recreational drugs. Initially, he was able to take care of himself; for example, taking a shower, dressing up, or eating without assistance. Upon examination, he became bedridden with spontaneous eye-opening. He made unintelligible screams and was unable to test for the MMSE. Rigidity and myoclonic jerks affected all limbs and without focal weakness. Tendon reflexes and Babinski signs were normal. Hematological screen, biochemical tests, and chest X-rays were unremarkable. EEG showed diffuse slow continuous delta activity in the bilateral cerebral hemisphere with atypical triphasic waves of CJD (Figure S1, Table S1). In DWI and T2 brain magnetic resonance imaging, high signal intensity findings were observed throughout the cerebral cortex of bilateral basal ganglia, frontal lobe, parietal lobe, and parietal lobe, except cerebellum and occipital lobe (Figure 1A). F-18 FDG positron emission tomography showed severe decreases in the metabolism of both cerebral cortexes, except for the primary sensory motor cortex and occipital lobe. The metabolisms in both basal ganglia, thalamus, and cerebellum were moderately reduced (Figure 1B). There was no leukocyte and red blood cell in CSF upon examination, and glucose and protein levels were normal (Table S1). Results of 14-3-3 protein in CSF by Western blot provided a weakly positive signal. Enzyme-linked immunosorbent assays of Aβ42 and total tau in CSF was performed. The decreased levels of Aβ42 (196.8 pg/mL) with significantly elevated total tau protein concentrations in CSF (1023.7 pg/mL) were observed. Apolipoprotein E (APOE) genotype for the patient was 3/3. The proband (II-1) did not have any previous family history of CJD; however, the exact family history of disease could not be defined. Different disease phenotypes were present in his family (Figure 2). His father died of colon cancer with complications from pneumonia at age 65 (I-1), and did not develop any cognitive dysfunction. His mother presented symptoms of dementia without proper diagnosis and died at the age of 75 (I-2). His sister (II-2, 4 years younger) was diagnosed with mild cognitive decline, but did not present similar phenotypic symptoms as the proband patient. She was registered as an outpatient by the Korean Veteran’s Hospital and refused genetic analyses.
Figure S1.

EEG data of the patient.
Abbreviation: EEG, electroencephalogram.
Table S1.
SF tapping profile of the proband patient
| Marker | Levels | Normal values |
|---|---|---|
| Red blood cell count | 0/mm3 | |
| White blood cell count | 0/mm3 | |
| Glucose | 108 mg/dL | 70–110 mg/dL |
| Albumin | 3.8 g/dL | 3.5–5.0 g/dL |
| Sodium | 139 mEq/L | 137–145 mEq/L |
| Potassium | 3.8 mEq/L | 3.6–5.0 mEq/L |
| Chloride | 103 mEq/L | 98–107 mEq/L |
| CSF microalbumin | 160.00 mg/L (IFCC) | Up to 350 mg/L (IFCC) |
Abbreviations: CSF, cerebrospinal fluid; SF, spinal fluid; IFCC, International Federation of Clinical Chemistry.
Figure 1.
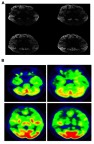
(A) Brain diffusion-weighted-MRI of the proband patient. (B) FDG PET imaging of proband patient.
Abbreviation: MRI, magnetic resonance imaging; FDG, fluorodeoxyglucose; PET, positron emission tomograp.
Figure 2.

Family tree of the patient.
Note: I 1 and I 2, parents of the proband patient; II 1, proband patient; II 2, sister of the proband patient.
Abbreviation: CJD, Creutzfeldt–Jakob disease.
Genetic screening and in silico analysis
An extensive genetic screening was done using a 50 causative and risk factor genes for different neurodegenerative disorders.16 Next generation sequencing and verification by standard Sanger sequencing were carried out by Theragen Etex Bio Institute (Seoul, Republic of Korea) company and BioNeer Inc. (Dajeon, Republic of Korea), respectively.16 Sequencing data were analyzed using the NCBI Blast (http://blast.ncbi.nlm.nih.gov/Blast.cgi) and the chromatograms were screened using the DNA BASER (http://www.dnabaser.com) tool. Rare variants were screened against reference databases, such as the Korean Reference Genome Database (KRGDB; http://152.99.75.168/KRGDB/menuPages/intro.jsp), Broad Institute’s Genome Association Database (gnomAD, http://gnomad.broadinstitute.org), and 1000 Genomes (http://www.1000genomes.org/) databases. Penetrance of novel prion variant was estimated. The gnomAD database was used as reference, and calculation method was described by Minikel et al (2016), where the upper bound of the 95% confidence interval (CI).18
The possible pathogenic nature of missense variants was predicted using simple online tools, such as PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2), Sorting Intolerant from Tolerant (SIFT; http://sift.jcvi.org/), and PROVEAN (http://provean.jcvi.org) algorithms.19,20
ExPasy analysis was also performed (https://www.expasy.org/) using different parameters, such as Kyte and Doolittle hydrophobicity index, bulkiness, and polarity. Mutant and normal prion protein structures were compared by 3D modeling. Protein structures were built using the Raptor X web server (http://raptorx.uchicago.edu/), while Discovery Studio 3.5 Visualizer (Accelrys, San Diego, USA) was used to display the 3D images.21
Experimental results
A missense variant was found in the PRNP gene, p.Tyr225Cys (c.674A>G; p. Y225C; Figure 3). No literature was available on this variant in regard to any disease phenotype. PRNP Tyr225Cys was not found in KRGDB of asymptomatic Korean individuals. This variant was also absent in the database of 1000 Genomes. In GnomAD database, its frequency was 0.000003978 (“singleton”), and a variant was found in one North-Western European female individual. The patient carried the wild type allele for both codon 129 (Met/Met) and codon 219 (Glu/Glu). No pathogenic or risk mutations in AD, FTD, or Parkinson’s disease) were found. Based on the calculation of Minikel et al (2016)18 and on GnomAD database, the penetrance of the mutation may be low, resulting 0.23% (95% CI 0.0007, 7.3).
Figure 3.

Next generation sequencing data for a patient with PRNP Tyr225Cys, verified by standard sequencing.
PolyPhen-2 predicted the variant to be “possibly damaging” with scores of 0.920 and 0.528 HumDiv and HumVar analyses, respectively. Multiple sequence alignment by PolyPhen-2 revealed Tyr225 as a conservative residue among primates, and the amino acid tyrosine appeared in homologous residue from several species, such as squirrel monkey, marmoset, cynomolgus monkey, and mouse lemur. SIFT also predicted the variant as damaging with a score of 0.001; however, PROVEAN suggested p.Thr225Cys as neutral with a score of −2.42.
ExPASY (Figure 4A) analysis revealed that the Kyte and Doolittle hydrophobicity index showed a significant elevation in hydrophobicity scores due to the variant (from −1.889 to −1.467), which affected the scores of the surrounding amino acids. The bulkiness score of this variant was also decreased from 13.02 to 12.512. Polarity scores were similar between the normal PrP and the PrP with p.Tyr225Cys, but a minor decrease in score could be seen in the case of Cys225 (from 9.167 to 9.089).
Figure 4.

(A) ExPASY analysis of PRNP Tyr225Cys compared with normal PrP protein. (B) Comparison of normal PrP proteins with Tyr225 and mutant Cys225 in terms of distance from Met166. (C) In silico prediction of PRNP Tyr225 and Cys225. Helix-3 in prion proteins may be more flexible in the case of Cys225 due to the smaller size of cysteine.
Compared to normal PrP, 3D modeling revealed structural changes in the PrP with p.Tyr225Cys (Figure 4B–C). It was predicted that Tyr225 may have long-range hydrophobic interactions with Met166, which could play a crucial role in prion protein structure.22 Superimposing normal and mutant prion proteins revealed longer distances between Met166 and Cys225 in comparison with Met166 and Tyr225. The smaller and less hydrophobic cysteine residue results in abnormal movement of the α3 helix (Figure 4B). Interactions inside the protein were also altered due to the variant. In the normal PrP, Tyr225 can form a hydrogen bond with Gly229 only. In the case of Cys225, the contact with Gly229 may disappear. In addition, Tyr/Cys exchange could generate new hydrogen bonds with Gln227 and Arg228, which changed the shape of α-3 helix. In the mutant protein, the last fold of α-3 helix may tend closer to the C-terminal loop region, resulted by the stronger contact with the loop (Figure 4C)
Discussion
In this study, a novel PRNP variant p.Tyr225Cys was described in a patient with atypical CJD. This variant was previously unrecognized, and it did not appear among Asian populations confirmed by KRGDB, 1000 Genomes, and GnomAD databases. In GnomAD, one European individual possessed the p.Tyr225Cys variant. Thus far, no report was available on p.Tyr225Cys in regard to any disease phenotype, and it was defined as “singleton” or a very low-frequency variant.18,23 Penetrance of this variant was suggested to be small. Several rare missense variants in PRNP appeared in control group in the GnomAD database. Rare variants found in few patients and/or unaffected individuals could be difficult to classify especially without family history. It would be complicated to estimate the pathogenicity of prion variants, neutral or possibly progressions into disease phenotype.18,23
The patient had a quite long duration of disease (more than 5 years), suggesting an atypical phenotype of CJD with slowly progressing dementia or atypical triphasic waves in EEG. Several PRNP variants could be associated with atypical form of CJD with longer duration of the disease and diverse unusual phenotypes (Table 1).24 Several cases of PRNP Val180Ile were also reported as atypical CJD cases with slow progressions, negative for 14-3-3 protein marker test, and without periodic sharp wave complexes in EEG signal.25–27 Interestingly, a PRNP Glu196Lys mutation was observed from an Italian CJD patient who developed rapidly progressive cognitive decline and personality changes without motor dysfunction. EEG was negative for periodic activities, and the family history was unclear. This patient was initially diagnosed with FTD.28 Secondly, PRNP Gly114Val was detected in a young onset CJD patient (24 years of age) with long duration of the disease (5 years). Penetrance of this variant could not be confirmed, and the patient was negative for the 14-3-3 marker.29 Thirdly, a heterozygous octapeptide insertion with long disease progression (4–13 years) was reported from American families with early onset CJD (25–35 years). Studies from brain tissues revealed a low degree of PrP amyloid accumulation.29
Table 1.
Examples of mutations, observed in atypical CJD cases
| Mutation | Family history | AOO (y) | Disease duration | Symptoms | Imaging data/EEG | Biomarker data |
|---|---|---|---|---|---|---|
| Tyr225Cys | Unknown | 59 | 5 years | Speech impairment, slow progressive dementia, rigidity myoclonic jerks | MRI: cortical signal changes in frontotemporoparietal region EEG: atypical triphasic waves |
CSF: Weak positive for 14-3-3 protein CSF: reduced Aβ42 and elevated total Tau levels |
| Val180Ile25–27 | Negative or positive | 70 | More than 1 year | Neurological signs with dementia | EEG: Periodic sharp waves may be absent | Can be negative for 14-3-3 protein, but positive cases were also found |
| Glu196Lys28 | Probably negative | 75 | 18 months | Dementia, but no motor signs, misdiagnosed as FTD | MRI: larger cortical sulci in the frontal and temporal lobes EEG: normal |
CSF: Positive for 14-3-3 protein, normal for Tau |
| Gly114Val29 | Probable positive | 24 | 5 years | Cognitive decline, movement impairment, aphasia, ataxia | EEG: sharp wave-like morphology MRI: restricted diffusion in the cortical ribbon |
CSF: 14-3-3 protein, Total Tau and Aβ42 were normal |
| Octapeptide insertion30 | Positive | 23–35 | 4–13 years | CJD | NA | NA |
Abbreviations: CJD, Creutzfeldt–Jakob disease; EEG, electroencephalogram; MRI, magnetic resonance imaging; CSF: cerebrospinal fluid; AOO, age of onset; NA, not available.
Decreased levels of Aβ42 (196.8 pg/mL) with significantly elevated total tau protein concentrations in CSF (1023.7 pg/mL) were observed, which could suggest both AD or CJD. Even though elevated levels of total tau in CSF could generally be markers for neurofibrillary lesions and tangles in both AD and CJD, higher tau levels were observed from CJD patients than AD. Oftentimes, the neurofibrillary pathology may not be prominent in CJD patients, and the increased total tau concentrations in CSF would suggest and correlate with significant neuronal loss.31 Similar to patients with AD, patients with CJD may also present with reduced Aβ42 concentrations in CSF.31 However, the reduced Aβ42 levels in patients with CJD may not correlate with amyloid deposits. Reasons of reduced Aβ42 in patients with CJD remained unclear: a possible carrier may mask the Aβ42 epitopes. The other putative explanation could be a putative alternative cleavage of Aβ42 peptides. An additional hypothesis suggested the influences in the metabolism of amyloid beta productions by normal PrP.32
Family history of the proband patient remained unclear, since all living family members refused genetic testing, and no additional relative of patient was reported to have similar symptoms. Even though his mother and sister were diagnosed with dementia and mild cognitive decline, respectively, the diagnosis could not be confirmed, prohibiting the segregation analyses. The possible familial case of mutation should not be ruled out since diverse disease phenotypes may be possible in genetic prion disorders. For example, Pro102Leu was verified as the most common GSS-causing mutation, but age of onset and phenotypes could vary. Besides the GSS patients, additional phenotypes such as CJD or AD-like symptoms could also possibly appear in the same family.33,34 Codon 129 may be a genetic modifier in terms of human prion disorders, and may be different in familial, sporadic or variant CJD patients. A genotype of codon 129 carried the homozygous Met/Met allele in the proband patient. Due to the long duration of the disease, the age of onset, cognitive decline, and the clinical symptoms were observed to be similar to the cases in the sporadic CJD with MM2 haplotype. Interestingly, the homozygous Met/Met phenotype may slightly favor the familial form of the disease.35
Cys179 and Cys214 were two cysteine residues in the α2-α3 helices in stabilizing the structure. Since disturbing the disulfide bridge may play a critical role in PrPC to PrPSc conversion, our Cys225 may thus disturb or destabilize the conformation of PrP.36–38 It may be also possible that an additional cysteine in α2 or α3 helices (such as Cys225) residue could increase the risk for shuffling of disulfide bridge.39,40 Disulfide bridge could affect the conversion of PrPC to PrPSc, and its disulfide bridge exchange may increase the prion propagation or its cleavage may enhance the formation of PrPSc. In addition, cleavage of native disulfide bond could result in intermolecular disulfide bridge, forming dimer.41 Exchange of cysteine to or from another residue in NOTCH3 protein was responsible for impaired structure and functions of the protein, leading to the cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy (CADASIL). An unpaired cysteine could result in possible abnormal intra-or intermolecular interactions of NOTCH3 protein.42,43
PRNP Tyr225Cys mutation is located in the C-terminal region of 3rd helix in prion protein. Four pathogenic or probable pathogenic mutations were described in the nearby region, such as Gln217Arg, Tyr218Asn, Tyr226Ter, and Gln227Ter (Table 2.).11,44–46 None of them were associated with typical GSS or CJD, but FTD-like or AD-like symptoms were common in them. Instead, patients with these presented diverse disease phenotypes, such as AD-like pathology, FTD or GSS. Patient with Tyr225Cys was diagnosed with atypical CJD, but FTD-like symptoms (language impairment, behavioral changes) and brain atrophy also affected the frontotemporoparietal region. Patients developed the disease phenotypes in their late 40s or early 60s. All of these mutations were associated with relatively long disease duration, when age of death occurred 2–13 years after the age of onset. Expect of Tyr226Ter, all mutations were associated with athropy or dysfunctions of frontal, temporal, or parietal regions.11,25,44–46 It may be possible that mutations, located in the C-terminal region of 3rd transmembrane domain of PrP could result in structure disturbances, resulting in atypical prion diseases. These mutations may prove the possible pathological overlap between prion diseases, AD and FTD. Genetic screening for PRNP should also be performed in AD and FTD patients.25,44
Table 2.
Mutations, located in the C-terminal region of 3rd helix of prion protein
| Mutation | Family history | AOO (y) | Disease duration | Symptoms | Imaging/neurological |
|---|---|---|---|---|---|
| Glu217Arg44 | Probable positive | 45 | Over 10 years | GSS, with FTD-like symptoms | MRI: mild cortical atrophy affecting the frontal, temporal, and parietal lobes, depigmentation in substania nigra Microscopy: large amyloid plaques with fibrillary cores |
| Tyr218Asn45 | Probable positive | 61 | 6 years | GSS, AD and FTD-like symptoms | EEG: focal frontotemporal slowing during hyperventilation, but no periodic short-wave complexes. PET: asymmetric bifrontal and parieto-occipital hypoperfusion Neuropathology: Spongiform changes, PrPres and Tau pathology |
| Tyr225Cys | Unknown | 59 | 5 years | Atypical CJD | MRI: cortical signal changes in frontotemporoparietal region |
| Tyr226Ter11,46 | Probable positive | 55 | 27 months | AD-like dementia | EEG: generalized slowing and typical pattern of periodic synchronous wave complexes Neuropathology: amyloid deposition, CAA, Tau deposits |
| Gln227Ter11 | Probable positive | 42 | 72 months | FTD, extrapyramidal signs | SPECT: hypoperfusion in the left frontal and temporal cortex Neuropathology: PrP positive plaques, amyloid plaques, neurofibrillary tangles |
Abbreviations: GSS: Gerstmann–Sträussler–Scheinker syndrome; FTD, frontotemporal dementia; MRI, magnetic resonance imaging; AD: Alzheimer’s disease; EEG, electroencephalogram; PET; positron emission tomography; CJD, Creutzfeldt–Jakob disease; AOO, age of onset; CAA, cerebral amyloid angiopathy.
Several limitations were observed in this study. Firstly, since the patient’s family members did not agree to undergo genetic testing, segregation analysis could not be performed. Although no family member was affected with CJD, it could not be verified whether the variant was present only in the proband patient. Moreover, a brain biopsy could not be performed to confirm CJD diagnosis. Another limitation was the lack of cell or animal model studies on PRNP p.Tyr225Cys; thus, putative pathogenic mechanisms of the mutation could not be defined. Structural predictions could be helpful in making hypotheses on the putative disturbing mechanisms of novel mutations; however, these models may not be consistent with in vivo disturbances. In silico modeling on Tyr225Cys could not fully demonstrate the damaging impact of mutation on prion protein.23
Conclusion
In this study, a novel PRNP p.Tyr225Cys variant was discovered from an atypical CJD patient with detailed clinical studies. Even though few limitations with family agreements in further genetic testing and functional studies, this report focused on reporting a unique mutation of Cys amino acid. The familial form of CJD cannot be concluded, even the mother and sister of the patient were both diagnosed with cognitive dysfunction, due to the strong refusal of the genetic testing for the segregation analysis. The putative protein structural changes of hydrophobic interactions between Tyr/Cys225 and Met166 were predicted, which may affect the protein conformations of two helices. An extra cysteine residue beside the natural disulfide bond could enhance the risk of disulfide bridge shuffling and could also affect the protein misfolding and aggregation. In the future, a follow-up cellular functional studies will be performed. PRNP Tyr225Cys will be introduced to the cell by the CRISPR/Cas system for its role in the pathogenicity and cellular functions.
Acknowledgments
Written informed consent was obtained from the patient and family members for publication of this genetic report. The authors would like to express their gratitude to the proband patient and his family members for their time and support. This research was supported by a National Research Foundation of Korea (NRF) Grants awarded by the Korean government (MEST, Nos. 2017R1A2B4012636 and 2017R1C1B5017807).
Disclosure
The authors report no conflicts of interest in this work.
Supplementary materials
References
- 1.Mastrianni JA, Iannicola C, Myers RM, DeArmond S, Prusiner SB. Mutation of the prion protein gene at codon 208 in familial Creutzfeldt-Jakob disease. Neurology. 1996;47:1305–1312. doi: 10.1212/wnl.47.5.1305 [DOI] [PubMed] [Google Scholar]
- 2.Escandón-Vargas K, Zorrilla-Vaca A, Corral-Prado RH. Positive 14-3-3 and tau proteins in a sporadic Creutzfeldt-Jakob disease case and a brief perspective of prion diseases in Colombia. Biomedica. 2016;36:29–36. doi: 10.7705/biomedica.v36i3.2729 [DOI] [PubMed] [Google Scholar]
- 3.Brown K, Mastrianni JA. The prion diseases. J Geriatr Psychiatry Neurol. 2010;23:277–298. doi: 10.1177/0891988710383576 [DOI] [PubMed] [Google Scholar]
- 4.Kübler E, Oesch B, Raeber AJ. Diagnosis of prion diseases. Br Med Bull. 2003;66:267–279. doi: 10.1093/bmb/66.1.267 [DOI] [PubMed] [Google Scholar]
- 5.Mastrianni JA. The genetics of prion diseases. Genet Med. 2010;12:187–195. doi: 10.1097/GIM.0b013e3181cd7374 [DOI] [PubMed] [Google Scholar]
- 6.Shen L, Ji HF. Mutation directional selection sheds light on prion pathogenesis. Biochem Biophys Res Commun. 2011;410:159–163. doi: 10.1016/j.bbrc.2011.06.007 [DOI] [PubMed] [Google Scholar]
- 7.Jeong BH, Ju WK, Huh K, et al. Molecular analysis of prion protein gene (PRNP) in Korean patients with Creutzfeldt-Jakob disease. J Korean Med Sci. 1998;13:234–240. doi: 10.3346/jkms.1998.13.3.275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Marcon G, Indaco A, Di Fede G, et al. Panencephalopathic Creutzfeldt-Jakob disease with distinct pattern of prion protein deposition in a patient with D178N mutation and homozygosity for valine at codon 129 of the prion protein gene. Brain Pathol. 2014;24:148–151. doi: 10.1111/bpa.12095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Deguchi K, Takamiya M, Deguchi S, et al. Spreading brain lesions in a familial Creutzfeldt-Jakob disease with V180I mutation over 4 years. BMC Neurol. 2012;12:144. doi: 10.1186/1471-2377-12-144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mastrianni JA, Capellari S, Telling GC, et al. Inherited prion disease caused by the V210I mutation: transmission to transgenic mice. Neurology. 2001;57:2198–2205. doi: 10.1212/wnl.57.12.2198 [DOI] [PubMed] [Google Scholar]
- 11.Jansen C, Parchi P, Capellari S, et al. Prion protein amyloidosis with divergent phenotype associated with two novel nonsense mutations in PRNP. Acta Neuropathol. 2010;119:189–197. doi: 10.1007/s00401-009-0609-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lim JG, Oh E, Park S, Kim YS, Lee A. Familial Creutzfeldt-Jakob disease with M232R mutation presented with corticobasal syndrome. Neurol Sci. 2015;36:1291–1293. doi: 10.1007/s10072-014-2038-4 [DOI] [PubMed] [Google Scholar]
- 13.Shi Q, Zhou W, Chen C, et al. Rare E196A mutation in PRNP gene of 3 Chinese patients with Creutzfeldt-Jacob disease. Prion. 2016;10:331–337. doi: 10.1080/19336896.2016.1190897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kim MO, Cali I, Oehler A, et al. Genetic CJD with a novel E200G mutation in the prion protein gene and comparison with E200K mutation cases. Acta Neuropathol Commun. 2013;1:80. doi: 10.1186/2051-5960-1-80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sanchez-Juan P, Green A, Ladogana A, et al. CSF tests in the differential diagnosis of Creutzfeldt-Jakob disease. Neurology. 2006;67:637–643. doi: 10.1212/01.wnl.0000230159.67128.00 [DOI] [PubMed] [Google Scholar]
- 16.Giau VV, An SS, Bagyinszky E, Kim SY. Gene panels and primers for next generation sequencing studies on neurodegenerative disorders. Mol Cell Toxicol. 2015;11:89–143. doi: 10.1007/s13273-015-0011-9 [DOI] [Google Scholar]
- 17.Mackenzie G, Will R. Creutzfeldt-Jakob disease: recent developments. F1000Res. 2017;6:2053. doi: 10.12688/f1000research.12681.1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Minikel EV, Vallabh SM, Lek M. Quantifying prion disease penetrance using large population control cohorts. Sci Transl Med. 2016;8:322ra9. doi: 10.1126/scitranslmed.aaf0746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Adzhubei IA, Schmidt S, Peshkin L, et al. A method and server for predicting damaging missense mutations. Nat Method. 2010;7:248–249. doi: 10.1038/nmeth0410-248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ng PC, Henikoff S. SIFT: predicting amino acid changes that affect protein function. Nucleic Acids Res. 2003;31:3812–3814. doi: 10.1093/nar/gkg509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Källberg M, Wang H, Wang S, et al. Template-based protein structure modeling using the RaptorX web server. Nat Protoc. 2012;7:1511–1522. doi: 10.1038/nprot.2012.085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ilc G, Giachin G, Jaremko M, et al. NMR structure of the human prion protein with the pathological Q212P mutation reveals unique structural features. PLoS One. 2010;5:e11715. doi: 10.1371/journal.pone.0011715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mok TH, Koriath C, Jaunmuktane Z, et al. Evaluating the causality of novel sequence variants in the prion protein gene by example. Neurobiol Aging. 2018;71:265.e1–265.e7. doi: 10.1016/j.neurobiolaging.2018.05.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Amano Y, Kimura N, Hanaoka T, et al. Creutzfeldt-Jakob disease with a prion protein gene codon 180 mutation presenting asymmetric cortical high-intensity on magnetic resonance imaging. Prion. 2015;9:29–33. doi: 10.1080/19336896.2015.1017703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bagyinszky E, Giau VV, Youn YC, An SSA, Kim SY. Characterization of mutations in PRNP (prion) gene and their possible roles in neurodegenerative diseases. Neuropsychiatr Dis Treat. 2018;14:2067–2085. doi: 10.2147/NDT.S165445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yeo MJ, Lee SH, Lee SY, et al. Familial Creutzfeldt-Jakob disease with a mutation at codon 180 presenting with an atypical phenotype. J Clin Neurosci. 2013;20:180–182. doi: 10.1016/j.jocn.2012.01.044 [DOI] [PubMed] [Google Scholar]
- 27.Jin K, Shiga Y, Shibuya S, et al. Clinical features of Creutzfeldt-Jakob disease with V180I mutation. Neurology. 2004;62:502–505. doi: 10.1212/01.wnl.0000106954.54011.80 [DOI] [PubMed] [Google Scholar]
- 28.Clerici F, Elia A, Girotti F, et al. Atypical presentation of Creutzfeldt-Jakob disease: the first Italian case associated with E196K mutation in the PRNP gene. J Neurol Sci. 2008;275:145–147. doi: 10.1016/j.jns.2008.06.036 [DOI] [PubMed] [Google Scholar]
- 29.Cali I, Mikhail F, Qin K, et al. Impaired transmissibility of atypical prions from genetic CJD G114V. Neurol Genet. 2018;4:e253. doi: 10.1212/NXG.0000000000000253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Brown P, Goldfarb LG, McCombie WR, et al. Atypical Creutzfeldt-Jakob disease in an American family with an insert mutation in the PRNP amyloid precursor gene. Neurology. 1992;42:422–427. doi: 10.1212/wnl.42.2.422 [DOI] [PubMed] [Google Scholar]
- 31.Kapaki E, Kilidireas K, Paraskevas GP, Michalopoulou M, Patsouris E. Highly increased CSF tau protein and decreased beta-amyloid (1–42) in sporadic CJD: a discrimination from alzheimer’s disease? J Neurol Neurosurg Psychiatry. 2001;71:401–403. doi: 10.1136/jnnp.71.3.401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mollenhauer B, Esselmann H, Roeber S, et al. Different CSF β-amyloid processing in alzheimer’s and Creutzfeldt-Jakob disease. J Neural Transm (Vienna). 2011;118:691–697. doi: 10.1007/s00702-010-0543-z [DOI] [PubMed] [Google Scholar]
- 33.Chi NF, Lee YC, Lu YC, Wu HM, Soong BW. Transmissible spongiform encephalopathies with P102L mutation of PRNP manifesting different phenotypes: clinical, neuroimaging, and electrophysiological studies in Chinese kindred in Taiwan. J Neurol. 2010;257:191–197. doi: 10.1007/s00415-009-5290-4 [DOI] [PubMed] [Google Scholar]
- 34.Rosenthal NP, Keesey J, Crandall B, Brown WJ. Familial neurological disease associated with spongiform encephalopathy. Arch Neurol. 1976;33:252–259. [DOI] [PubMed] [Google Scholar]
- 35.Gambetti P, Kong Q, Zou W, Parchi P, Chen SG. Sporadic and familial CJD: classification and characterisation. Br Med Bull. 2003;66:213–239. doi: 10.1093/bmb/66.1.213 [DOI] [PubMed] [Google Scholar]
- 36.Gendoo DM, Harrison PM. The landscape of the prion protein’s structural response to mutation revealed by principal component analysis of multiple NMR ensembles. PLoS Comput Biol. 2012;8:e1002646. doi: 10.1371/journal.pcbi.1002646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hermann LM, Caughey B. The importance of the disulfide bond in prion protein conversion. Neuroreport. 1998;9:2457–2461. doi: 10.1097/00001756-199808030-00006 [DOI] [PubMed] [Google Scholar]
- 38.Maiti NR, Surewicz WK. The role of disulfide bridge in the folding and stability of the recombinant human prion protein. J Biol Chem. 2001;276(4):2427–2431. doi: 10.1074/jbc.M007862200 [DOI] [PubMed] [Google Scholar]
- 39.Mossuto MF. Disulfide bonding in neurodegenerative misfolding diseases. Int J Cell Biol. 2013;2013:318319. doi: 10.1155/2013/318319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Welker E, Wedemeyer WJ, Scheraga HA. A role for intermolecular disulfide bonds in prion diseases? Proc Natl Acad Sci U S A. 2001;98:4334–4336. doi: 10.1073/pnas.071066598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Li Y, Yan J, Zhang X, Huang K. Disulfide bonds in amyloidogenesis diseases related proteins. Proteins. 2013;81:1862–1873. doi: 10.1002/prot.24338 [DOI] [PubMed] [Google Scholar]
- 42.Dichgans M, Herzog J, Gasser T. NOTCH3 mutation involving three cysteine residues in a family with typical CADASIL. Neurology. 2001;57:1714–1717. doi: 10.1212/wnl.57.9.1714 [DOI] [PubMed] [Google Scholar]
- 43.Arboleda-Velasquez JF, Rampal R, Fung E, et al. CADASIL mutations impair notch3 glycosylation by fringe. Hum Mol Genet. 2005;14:1631–1639. doi: 10.1093/hmg/ddi171 [DOI] [PubMed] [Google Scholar]
- 44.Woulfe J, Kertesz A, Frohn I, Bauer S, George-Hyslop PS, Bergeron C. Gerstmann-Sträussler-Scheinker disease with the Q217R mutation mimicking frontotemporal dementia. Acta Neuropathol. 2005;110:317–319. doi: 10.1007/s00401-005-1054-0 [DOI] [PubMed] [Google Scholar]
- 45.Alzualde A, Indakoetxea B, Ferrer I, et al. A novel PRNP Y218N mutation in Gerstmann-Sträussler-Scheinker disease with neurofibrillary degeneration. J Neuropathol Exp Neurol. 2010;69:789–800. doi: 10.1097/NEN.0b013e3181e85737 [DOI] [PubMed] [Google Scholar]
- 46.Kovač V, Zupančič B, Ilc G, Plavec J, Čurin Šerbec V. Truncated prion protein PrP226* – a structural view on its role in amyloid disease. Biochem Biophys Res Commun. 2017;484:45–50. doi: 10.1016/j.bbrc.2017.01.078 [DOI] [PubMed] [Google Scholar]