
Fig. 4.
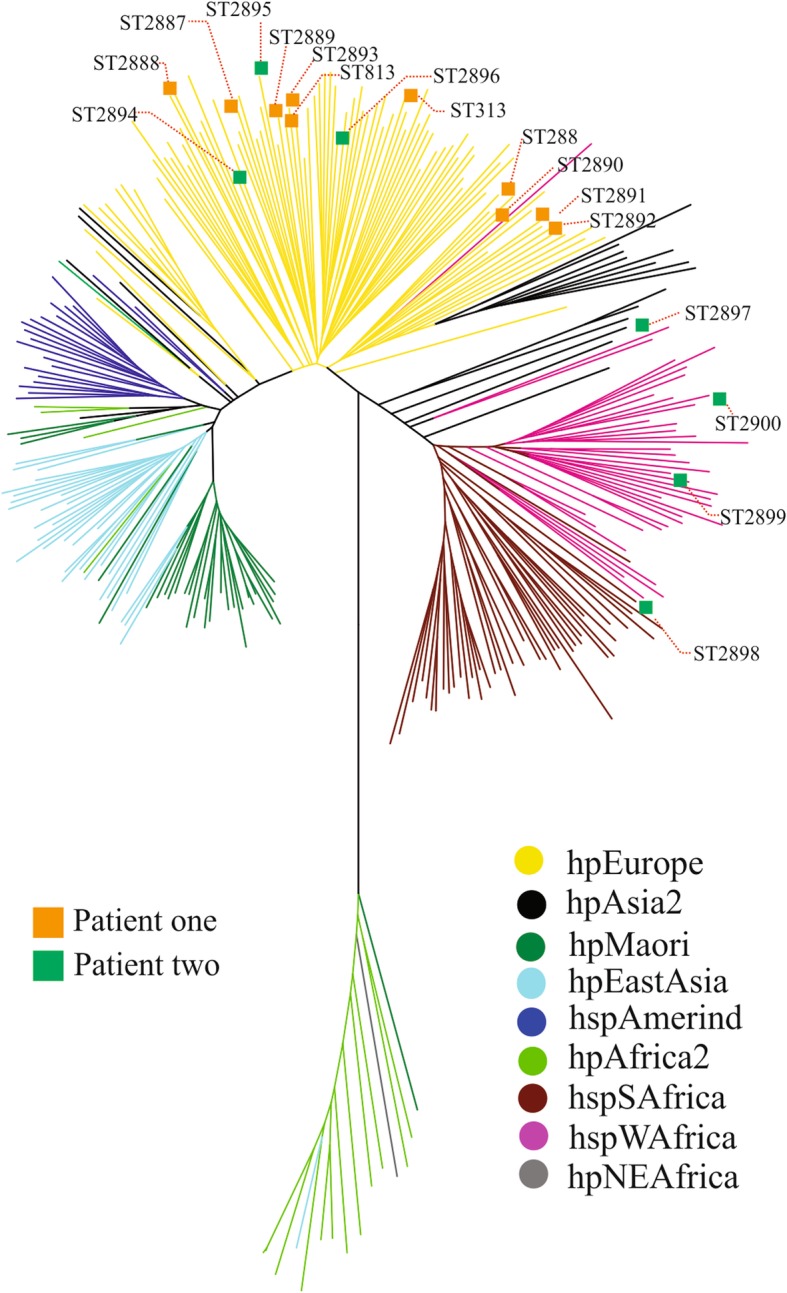
Phylogeography of the analysed STs. The phylogeography was inferred using the neighbour-joining method [27]. The optimal tree with a summed branch length of 3.97653233 is shown. The tree is drawn to scale; branch lengths with the same units as the evolutionary distances are used to infer the phylogenetic tree. The evolutionary distances were computed using the Kimura 2-parameter method [28], and the units are the number of base substitutions per site. The rate variations among the sites were modeled with a gamma distribution (shape parameter = 1). The analysis included 316 reference STs from the H. pylori MLST database [http://pubmlst.org/helicobacter/]. All ambiguous positions were removed for each sequence pair. The major H. pylori populations were identified according to the assigned population available at PubMLST and the identification and positions of the 17 STs identified in this study are shown in the tree. There were 3402 positions in the final dataset. Phylogeography analyses were conducted using MEGA6 [29]