

Abstract
Hepatitis E viral (HEV) infection imposes a heavy health burden worldwide and is common in the United States. Previous investigations of risks addressed environmental and host behavioral/lifestyle factors, but host genetic factors have not been examined. We assessed strength of associations between antibody to HEV (anti-HEV) immunoglobulin G seropositivity indicating past or recent HEV infection and human genetic variants among three major racial/ethnic populations in the United States, involving 2434 non-Hispanic whites, 1919 non-Hispanic blacks, and 1919 Mexican Americans from the Third National Health and Nutrition Examination Survey, 1991–1994. We studied 497 single-nucleotide polymorphisms across 190 genes (particularly those associated with lipid metabolism). The genomic control method was used to adjust for potential population stratification. Non-Hispanic blacks had the lowest seroprevalence of anti-HEV immunoglobulin G (15.3%, 95% confidence interval [CI] 12.3%−19.0%) compared with non-Hispanic whites (22.3%, 95% CI 19.1%−25.7%) and Mexican Americans (21.8%, 95% CI 19.0%−25.3%; P< 0.01). Non-Hispanic blacks were the only population that showed association between anti-HEV seropositivity and functional ε3 and ε4 alleles of the apolipoprotein E (APOE) gene, encoding the apolipoprotein E protein that mediates lipoprotein metabolism. Seropositivity was significantly lower in participants carrying APOE ε4 (odds ratio = 0.5, 95% CI 0.4–0.7; P = 0.00004) and ε3 (odds ratio = 0.6,95% CI 0.4–0.8; P = 0.001) compared to those carrying APOE ε2. No significant associations were observed between other single-nucleotide polymorphisms and anti-HEV seropositivity in non-Hispanic blacks or between any single-nucleotide polymorphisms and anti-HEV seropositivity in non-Hispanic whites or Mexican Americans.
Conclusion:
Both APOE ε3 and ε4 are significantly associated with protection against HEV infection in non-Hispanic blacks; additional studies are needed to understand the basis of protection so that preventive services can be targeted to at-risk persons.
Infection with hepatitis E virus (HEV) can lead to hepatitis E, a disease that is usually self-resolving but can result in liver failure.1,2 Worldwide, of 20 million people estimated to be HEV-infected annually, 57,000 die from liver failure.3,4 In developing countries where waterborne HEV transmission is common, mortality is particularly high among pregnant women.5 In industrialized countries, imported and indigenously acquired infections occur, and chronic HEV infection is being increasingly recognized among immunocompromised persons, especially solid-organ transplant recipients in whom progression to cirrhosis has been observed.6
A serological study of participants of the Third National Health and Nutrition Examination Survey (NHANES III) has suggested that one-fifth of the US population may have been exposed to or infected by HEV and that certain host demographic and lifestyle factors predispose a person to HEV infection.7 Unlike for viral hepatitis A, B, and C, human genetic risk factors for hepatitis E are not well understood.8–11 It has been shown, however, that host-cell lipid metabolism influences the initiation of HEV infection, replication, and release from infected cells 12,13 and that the liver is the principal site of lipid, lipoprotein, and apolipoprotein metabolism; as such, patients with acute hepatitis E show increased blood levels of apolipoprotein E (apoE),14,15 and HEV-infected swine accumulate apoE in the liver.16 Because human genetic variants account for variation in lipid metabolism, genetic variations in lipid-related genes might be associated with differential host response to HEV infection.17 We selected candidate genes on the basis of their possible roles in altering lipid and lipoprotein profiles in HEV-infected persons and in modulating the immune response against HEV.
We determined variations in the genetic makeup of humans potentially associated with increased risk for HEV infection in a large candidate gene study using a cross-sectional study of the three major racial/ethnic populations in the United States: non-Hispanic whites, non-Hispanic blacks, and Mexican Americans. A better understanding of the genetic influence on susceptibility to HEV infection can help identify and target high-risk populations appropriate for prevention.
Methods
Ethics Statement.
All data collection and laboratory procedures were approved by the Centers for Disease Control and Prevention’s National Center for Health Statistics Ethics Review Board, and written informed consent was obtained from all participants. Though the original consent did not specifically mention using stored blood specimens for future genetic research, the National Center for Health Statistics Ethics Review Board has approved the use of these samples for this purpose based on the guidelines in the August 1999 National Bioethics Advisory Commission report.18 To ensure privacy of the study participants and confidentiality of their data, analysis was conducted at a secure environment, in the National Center for Health Statistics Research Data Center in Atlanta, Georgia.
Study Population.
The NHANES III was conducted between 1988 and 1994, and the survey design, population, DNA bank, genotyping, and quality control have been described elsewhere.18,19 Briefly, the NHANES III used a stratified, multistage probability design to select subjects for household interviews, physical examinations, and laboratory tests to provide nationally representative estimates of the health and nutritional status of the civilian, noninstitutionalized population aged ≥2 months. A DNA bank was created from blood samples collected during the second phase (1991–1994) from 7159 participants aged ≥12 years. Genetic data were combined with behavioral, environmental, and clinical information available in NHANES III. We included in our study 6272 (87.6%) participants who self-reported as being non-Hispanic white (n = 2434), non-Hispanic black (n = 1919), or Mexican American (n = 1919, excluding Hispanic persons of other origins) and who had been tested for antibody to HEV (anti-HEV) immunoglobulin G (IgG). The serostatus of participants was determined by the absence or presence of anti-HEV IgG,7 the latter indicating past or recent HEV infection.
Selection of Polymorphisms and Genotyping.
We selected candidate genes on the basis of their possible roles in altering lipid and lipoprotein profiles in HEV-infected persons and in modulating the immune response against HEV. These genes were selected from a candidate list (Supporting Table S1) generated from the published literature.12–16 A total of 190 genes encompassing 497 single-nucleotide polymorphisms (SNPs), genotyped through the NHANES genetic program as of 2011, were selected and are available in a restricted-access database through the National Center for Health Statistics Research Data Center.18 Detailed genotyping methods and quality control criteria have been described and are available online.18,19
Statistical Analysis.
All data were weighted to the US population in SAS-Callable SUDAAN 9.01 and SAS 9.3.20 Weighted allele frequencies and their 95% confidence intervals (CIs) were calculated using the NHANES III genetic sample weights for the 7159 participants.19 The Taylor series linearization approach was used to correct for the design effect and the possibility of genetic relatedness of persons sampled from the same household. The logistic regression model was used to test for association between each SNP and anti-HEV seropositivity for each race/ethnicity group.21,22 An additive genetic model (AA versus Aa versus aa), which treats the marker as a continuous variable, was assumed for regression analyses to test the null hypothesis that anti-HEV seropositivity does not vary with increasing copy numbers of the minor allele a.
The genomic control (GC) method was used to adjust for potential population stratification.23 Satterthwaite χ2 statistics generated from a supplemental set of 267 SNPs from the NHANES III genetic data set were used to derive empirical estimates of the null distribution that accounted for the effects of population structure (i.e., the presence of a significant difference in allele frequencies within US racial/ethnic subpopulations due to different ancestry). These SNPs were chosen without any prior knowledge of associations with anti-HEV seropositivity and to be at least 1 million base pairs apart from each other in order to reduce the likelihood that they would be in linkage disequilibrium. A list of these SNPs is provided in Supporting Table S2. The variance inflation factor, λ (calculated as the median value from the null distributions divided by the median value of the theoretical χ2 statistics distribution, 0.4549), was used to readjust the Satterthwaite χ2 statistics from the regression models for SNPs in the candidate genes for potential confounding by population structure.
The P values from the GC-corrected χ2 Satterthwaite statistics were then further adjusted for multiple testing using the false discovery rate (FDR) as estimated by the PROC MULTTEST procedure in SAS.24 A significant association was defined as the GC-and FDR-adjusted P value (GC/FDR-P) < 0.05 for all SNPs and anti-HEV seropositivity.
Results
Descriptive Analysis.
Of 7159 possible subjects, 6262 (87.5%) met the inclusion criteria (Table 1). Overall, the seropositivity rate was 21.6% in participants aged ≥12 years, which varied significantly among the three racial/ethnic populations (P< 0.01). It was lower among non-Hispanic blacks (15.3%, 95% CI 12.3%−19.0%) compared with non-Hispanic whites (22.3%, 95% CI and 19.1%−25.7%) or Mexican Americans (21.8%, 95% CI 19.0%−25.3%). Seropositivity was strongly associated with age, rising significantly with increasing age across all three subpopulations and peaking in the >65-year age group. Seropositivity was significantly associated with region of residence, poverty income ratio, education attained, and country of birth (Table 1).
Table 1.
Characteristics of Study Participants Who Tested Seropositive for Anti-HEV IgG in the NHANES III, 1991–1994
| Characteristics | All Participants (n = 6272) |
Non-Hispanic Whites (n = 2434) |
Non-Hispanic Blacks (n = 1919) |
Mexican Americans (n =1919) |
||||
|---|---|---|---|---|---|---|---|---|
| % (95% CI)* | P† | % (95% CI) | P | % (95% CI) | P | % (95% CI) | P | |
| Total | 21.6 (18.6–24.4) | <0.001 | 22.3 (19.1–25.7) | 15.3 (12.3–19.0) | 21.8 (19.0–25.3) | |||
| Sex | ||||||||
| Male | 22.1 (18.4–26.3) | 0.43 | 22.8 (18.5–27.8) | 0.62 | 15.1 (11.2–20.0) | 0.83 | 25.5 (21.8–29.7) | 0.004 |
| Female | 20.7 (18.0–23.7) | 21.7 (18.6–25.2) | 15.5 (12.4–19.3) | 18.0 (14.6–22.1) | ||||
| Age (years) | ||||||||
| 12–19 | 3.6 (2.2–6.6) | 3.3 (1.4–7.4) | 5.1 (2.9–8.7) | 5.0 (3.1–7.9) | ||||
| 20–29 | 8.7 (5.6–13.3) | 7.9 (4.3–514.0) | 7.1 (4.3–11.5) | 17.3 (12.7–23.2) | ||||
| 30–44 | 18.5 (15.1–22.4) | <0.001 | 18.8 (14.7–23.7) | <0.001 | 14.0 (10.7–18.2) | <0.001 | 24.3 (21.7–27.5) | <0.001 |
| 45–64 | 33.3 (28.0–39.1) | 33.6 (27.4–40.3) | 28.3 (23.0–34.4) | 39.4 (33.2–45.9) | ||||
| ≥65 | 40.4 (36.3–44.6) | 41.1 (36.5–45.6) | 32.6 (23.4–43.4) | 44.0 (34.9–53.5) | ||||
| Country of birth‡ | ||||||||
| United States | 20.3 (17.4–23.6) | <0.001 | 21.5 (18.2–25.2) | <0.001 | 15.4 (12.3–19.1) | 0.8 | 10.2 (8.6–11.9) | <0.001 |
| Foreign-born | 32.0 (27.3 −37.1) | 35.9 (26.6–46.4) | 14.0 (6.2–28.6) | 32.5 (28.5–36.8) | ||||
| Region of residence§ | ||||||||
| Northeast | 21.3 (14.1–31.0) | 22.1 (14.0–33.1) | 14.4 (11.6–17.7) | 28.8 (11.3–56.2) | ||||
| Midwest | 24.9 (20.1–30.2) | 0.009 | 25.2 (20.6–30.4) | 0.01 | 21.4 (13.5–32.1) | 0.33 | 25.7 (22.7–29.0) | 0.02 |
| West | 24.7 (18.8–31.8) | 14.8 (11.6–18.7) | 13.8 (9.8–19.1) | 15.7 (11.6–20.8) | ||||
| South | 14.6 (11.7–18.2) | 25.8 (18.4–35.0) | 13.2 (9.8–17.6) | 23.7 (20.2–27.5) | ||||
| Poverty income ratio∥ | ||||||||
| >1 | 22.2 (19.3–25.5) | 0.02 | 23.0 (19.8–26.6) | 0.01 | 15.4 (12.5–18.8) | 0.83 | 20.6 (17.5–24.0) | 0.08 |
| ≤1 | 17.9 (14.7–21.7) | 17.3 (13.6–21.6) | 15.9 (10.7–23.0) | 23.7 (19.8–28.2) | ||||
| Education (years) | ||||||||
| <12 | 19.1 (15.9–22.7) | 0.01 | 18.7 (14.7–23.6) | 0.02 | 15.3 (11.6–20.0) | 0.9 | 25.4 (22.3–28.8) | <0.001 |
| ≥12 | 22.3 (19.5–25.3) | 23.4 (20.3–26.7) | 15.3 (12.4–18.8) | 16.0 (13.2–19.4) | ||||
Frequencies were adjusted using NHANES III genetic sample weights.
P value was calculated using the Wald chi-squared test based on the log OR. Having a pet in the home (P = 0.5); consuming liver or other organ meats (P = 0.27); and levels of alanine aminotransferase (P = 0.92), aspartate aminotransferase (P = 0.1), and C-reactive protein (P = 0.07) were not associated with increased odds of anti-HEV seropositivity.
Persons born outside of the United States and its territories were considered foreign-born.
Region was defined using standard Census Bureau groupings.
Poverty income ratio was calculated by dividing the total family income by the poverty threshold adjusted for family size at year of interview. Values <1 were considered below the poverty line.
Genetic Associations With Anti-HEV Seropositivity.
Among the SNPs, 98% were significantly different in allele frequency across race/ethnicity groups (P < 0.01) (data not shown). The extent of this differentiation varied greatly between SNPs and between racial/ethnic populations. Accordingly, we conducted three parallel analyses, one for each race/ethnicity group for each SNP tested. Excluded were SNPs with a minor allele frequency <1% or a major allele frequency >99%. In the final analysis, 459, 491, and 469 SNPs were included for non-Hispanic whites, non-Hispanic blacks, and Mexican Americans, respectively. The effects of using GC on the results are shown in Supporting Fig. S1. The χ2 value overestimation, as indicated by GC inflation factor λ, was 1.3, 1.0, and 1.8 times higher for non-Hispanic whites, non-Hispanic blacks, and Mexican Americans, respectively (Table 2).
Table 2.
Associations of Genetic Variants in APOA4 and APOE Among Non-Hispanic Blacks With Anti-HEV Seropositivity in the NHANES III, 1991–1994
| Variant | Allele Frequency* (%) | OR (95% CI) | P† | GC/FDR-P‡ |
|---|---|---|---|---|
| Non-Hispanic whites | ||||
| APOA4 rs5110 (A) | 92.7 | 0.8 (0.6–1.1) | 0.17 | 0.99 |
| APOE rs7412 (C) | 8.4 | 0.9 (0.7–1.2) | 0.64 | 0.99 |
| Non-Hispanic blacks | ||||
| APOA4 rs5110 (A) | 98.3 | 0.2 (0.1–0.5) | <0.001 | 0.007 |
| APOE rs7412 (C) | 10.0 | 0.6 (0.5–0.8) | <0.001 | 0.02 |
| Mexican Americans | ||||
| APOA4 rs5110 (A) | 94.9 | 1.2 (0.7–1.9) | 0.53 | 0.99 |
| APOE rs7412 (C) | 3.3 | 0.9 (0.6–1.5) | 0.69 | 0.99 |
Frequencies were adjusted using NHANES III genetic sample weights.
P value was based on logistic regression analysis using an additive genetic model within each race/ethnicity group.
P value was adjusted for population stratification with GC inflation factor λ and multiple comparisons using the FDR in PROC MULTTEST (SAS 9.3). Inflation factor k of the GC was calculated from data of a separate set of 267 SNPs from NHANES III for each race/ethnicity group. λ was 1.31, 1.02, and 1.82 for non-Hispanic whites, non-Hispanic blacks, and Mexican Americans, respectively.
The univariable analysis indicated significant genetic associations with seropositivity and race/ethnicity among non-Hispanic blacks but not among non-Hispanic whites or Mexican Americans (Fig. 1). Two SNPs, rs5110 in apolipoprotein A-IV (APOA4, located in chromosome 11) and rs7412 in APOE (in chromosome 19), were associated with anti-HEV seropositivity among non-Hispanic blacks in the univariable models. Specifically, the alleles APOA4 rs5110-A (odds ratio [OR] = 0.2, 95% CI 0.1–0.5; GC/FDR-P = 0.007) and APOE rs7412-C (OR = 0.6, 95% CI 0.5–0.8; GC/FDR-P = 0.02) were associated with lower odds of sero-positivity (Table 2).
Fig. 1.
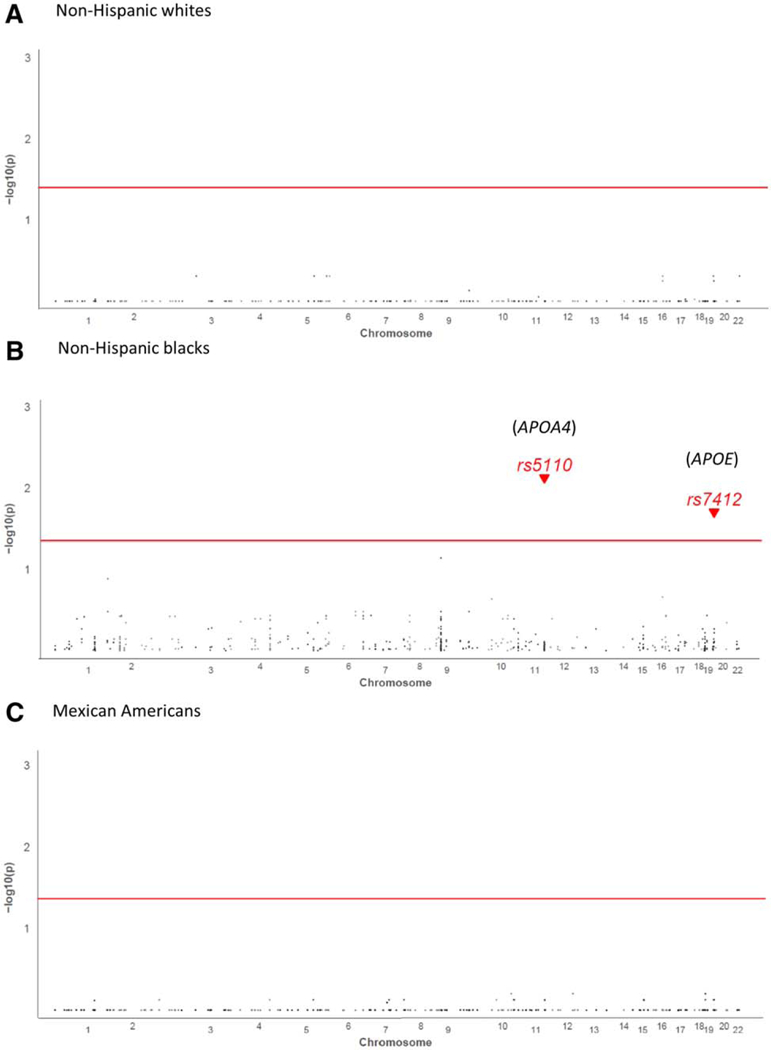
Genetic associations between anti-HEV-seropositivity and race/ethnicity among non-Hispanic blacks, non-Hispanic whites and Mexican Americans by univariable analysis.
The APOE gene encodes the apoE protein. This protein combines with fats (lipids) in the body to form lipoproteins. Lipoproteins are responsible for packaging cholesterol and other fats and carrying them through the bloodstream. There are three apoE protein isoforms (apoE2, apoE3, and apoE4), differing from each other by only one or two amino acids at positions 112 and 158. These isoforms are encoded by three allelic variants of APOE: ε2, ε3, and ε4, respectively. Variants ε2, ε3, and ε4 are haplotypes defined by rs7412 and rs42 9 3 5 8.25 In non-Hispanic blacks APOE rs7412 was associated with lower odds of seropositivity, and rs429358 was not statistically significant in the overall scan after adjustments by both the GC for potential population stratification and the FDR for multiple testing. As we were interested in these well-studied functional APOE allelic variants, we further conducted analysis on ε2, ε3, and ε4 to determine any association (P < 0.05) with anti-HEV seropositivity for each race/ethnicity group. The seropositivity rate among non-Hispanic blacks carrying ε4 was significantly lower than in those carrying ε2 (OR = 0.5, 95% CI 0.4–0.7; P = 0.00004), as was the rate in those carrying e3 compared to those carrying ε2 (OR = 0.6, 95% CI 0.4–0.8; P = 0.001) (Table 3).
Table 3.
Protective Effect of APOE Variants Against HEV Infection in Non-Hispanic Blacks* in the NHANES III, 1991–1994
| Variant | Allele Frequency† (%) | OR (95% CI) | P |
|---|---|---|---|
| Non-Hispanic whites | |||
| APOE ε2 | 8.3 | 1 (reference) | |
| APOE ε3 | 76.0 | 0.9 (0.7–1.2) | 0.99 |
| APOE ε4 | 15.7 | 0.9 (0.6–1.6) | 0.99 |
| Non-Hispanic blacks | |||
| APOE ε2 | 10.1 | 1 (reference) | |
| APOE ε3 | 67.9 | 0.6 (0.4–0.8) | 0.001 |
| APOE ε4 | 22.0 | 0.5 (0.4–0.7) | 0.00004 |
| Mexican Americans | |||
| APOE ε2 | 3.3 | 1 (reference) | |
| APOE ε3 | 85.7 | 0.9 (0.6–1.5) | 0.99 |
| APOE ε4 | 11.0 | 0.8 (0.5–1.6) | 0.99 |
Logistic regression analysis within each race/ethnicity group.
Frequencies adjusted using NHANES III genetic sample weight.
Discussion
We have examined associations between human genetic variants and HEV infection in a nationally representative sample of the three major race/ethnicity groups in the United States. Among non-Hispanic blacks, whose anti-HEV seroprevalence rate was lowest compared to non-Hispanic whites and Mexican Americans, APOE ε3 and ε4 were found to be significantly associated with protection against HEV infection.
Originally recognized for its importance in lipoprotein metabolism and cardiovascular disease, apoE, whose isoforms are apoE4, apoE3 and apoE2, are increasingly known to play key roles in biological processes in addition to lipid transport, such as cognitive function and immunoregulation.26,27 Although the isoforms differ by single–amino acid substitutions, these changes can lead to profound functional consequences, including variability in domain interactions, protein stability, and protein folding, which in turn can influence susceptibility to and the natural history of disease. Thus, individuals carrying the APOE ε4 allele are reported to be 10 times more likely to develop late-onset Alzheimer’s disease than those carrying other variants.28
Among the non-Hispanic blacks in our study population, the association between lower APOE ε4 carriage and seropositivity rate for anti-HEV likely reflects the protective effect of apoE4 against primary HEV infection in the liver. Apolipoprotein E is synthesized mainly by the liver, and in vitro studies have shown that apoE4 exhibits greater binding affinity to cultured hepatoma cells than apoE3.30 Possible explanations for how apoE influences the protection against HEV infection include (1) HEV using the same hepatocyte entry mediators as those of apoE, (2) the incorporation of apoE in the HEV envelope, and (3) apoE modulating the immune response against HEV.29 First, apoE is able to bind to several cell surface receptors, including heparan sulfate proteoglycans (HSPGs) and low-density lipoprotein receptors. Several pathogens, such as hepatitis C virus,31 herpes simplex virus,32 human immunodeficiency virus,33 and the malaria protozoon,34 have been reported to bind to one or another of these receptor sites to gain entry to target cells. Indeed, apoE can inhibit such binding by competing with HSPGs.27,35 Also, HSPGs have been shown to be requisite for the HEV capsid protein to bind to facilitate cell entry,36 so any difference in isoform affinity for HSPGs conferred by the APOE genotype may affect the extent to which HEV enters hepatocytes. Second, HEV virions circulating in the blood are covered with an envelope that possibly incorporates apoE in the lipid membrane as it exits from the host cell,16 similar to other enveloped viruses (e.g., human immunodeficiency virus, hepatitis C virus, and herpes simplex virus).37 The envelope protects the virus against neutralizing antibodies. Moreover, being essential for the transport of cholesterol into and out of the liver and a critical component of the viral envelope, apoE may be essential for viral entry and assembly.38 Third, as HEV-specific T-cell responses play critical roles in controlling the course of HEV infection,39 apoE may modulate the immune response against HEV by regulating T-lymphocyte activation and proliferation.29
This study is among the first to apply the GC method to correct for potential confounding from population substructures in the NHANES, which may be particularly important as race/ethnicity was based on self-report. Confounding can result from stratification or recent admixture of distinct subpopulations.40 Genetic admixture is commonly encountered among Mexican American and non-Hispanic black populations (substantially less among non-Hispanic whites).41,42 Given the GC inflation factors in the current study, Mexican Americans were most likely to be affected by population structure, followed by non-Hispanic whites and non-Hispanic blacks. Applying a GC, as was done in this study, lends confidence to validating the detected associations.
This study has several limitations. First, the outcome of the study, seropositivity to anti-HEV IgG, is cumulative without knowledge of when the antibody first appeared, its persistence, or to which HEV genotype it was elicited against. Second, overly conservative P values may be generated by FDR, which may decrease our ability to identify true associations. Unlike unadjusted P values expressing the probability of a false-positive result for a single test, the FDR gives a conservative estimate of the proportion of false positives among variants with significant association. Finally, the statistical power to detect significant associations with uncommon genetic variants was limited. Although this cross-sectional study was adequately powered to detect in each racial/ethnic group the two crude associations between anti-HEV seropositivity and rs5110-A or rs74l2-C that were found for non-Hispanic blacks (Supporting Table S3), the possibility that these SNPs are proxies for true causal variants cannot be excluded. Differences in linkage disequilibrium between races/ethnicities thus may hamper our ability to detect associations across all three groups, if they exist.
In conclusion, this NHANES-based cross-sectional study found that APOE ε3 and ε4 may be associated with protection against HEV infection in non-Hispanic blacks but not non-Hispanic whites or Mexican Americans. No other data have yet been generated to suggest that certain individuals or populations are biologically more resistant or susceptible to HEV infection. Therefore, replication of our study’s findings is encouraged, as is investigation into the functional relevance of the genes uncovered. Discovery of the basis of protection against HEV can inform the development of targeted preventative measures for at-risk populations.
Supplementary Material
Acknowledgment:
We sincerely thank all the study subjects who participated in NHANES III. Special thanks to staff at the Research Data Center in the National Center for Health Statistics for their data support and assistance in disclosure review.
Supported by the Centers for Disease Control and Prevention (Atlanta, GA). The findings and conclusions in this report are those of the authors and do not necessarily represent the views of the Centers for Disease Control and Prevention.
Abbreviations:s
- anti-HEV
antibody to HEV
- APOA4
apolipoprotein A-IV
- apoE
apolipoprotein E
- CI
confidence interval
- FDR
false discovery rate
- GC
genomic control
- GC/FDR-P
P value adjusted for genomic control and FDR
- HEV
hepatitis E virus
- HSPG
heparan sulfate proteoglycan
- IgG
immunoglobulin G
- NHANES III
Third National Health and Nutrition Examination Survey
- OR
odds ratio
- SNP
single-nucleotide polymorphism
Footnotes
Potential conflict of interest: Nothing to report.
Supporting Information
Additional Supporting Information may be found at onlinelibrary.wiley.com/doi/10.1002/hep.27938/suppinfo.
References
- 1.Teo CG. Much meat, much malady: changing perceptions of the epide-miology of hepatitis E. Clin Microbiol Infect 2010;16:24–32. [DOI] [PubMed] [Google Scholar]
- 2.Kamar N, Bendall R, Legrand-Abravanel F, Xia NS, Ijaz S, Izopet J, et al. Hepatitis E. Lancet 2012;379:2477–2488. [DOI] [PubMed] [Google Scholar]
- 3.Rein DB, Stevens GA, Theaker J, Wittenborn JS, Wiersma ST. The global burden of hepatitis E virus genotypes 1 and 2 in 2005. Hepatology 2012;55:988–997. [DOI] [PubMed] [Google Scholar]
- 4.World Health Organization. Hepatitis E, fact sheet no. 280. http://www.who.int/mediacentre/factsheets/fs280/en/. Updated June 2014. Accessed September 23, 2014.
- 5.Teo CG. Fatal outbreaks of jaundice in pregnancy and the epidemic history of hepatitis E. Epidemiol Infect 2012;140:767–787. [DOI] [PubMed] [Google Scholar]
- 6.Kamar N, Izopet J, Rostaing L. Hepatitis E virus infection. Curr opin Gastroenterol 2013;29:271–278. [DOI] [PubMed] [Google Scholar]
- 7.Kuniholm MH, Purcell RH, McQuillan GM, Engle RE, Wasley A, Nelson KE. Epidemiology of hepatitis E virus in the United States: results from the Third National Health and Nutrition Examination Survey, 1988–1994. J Infect Dis 2009;200:48–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhang L, Yesupriya A, Hu DJ, Chang MH, Dowling NF, Ned RM, et al. Variants in ABCB1, TGFB1, and XRCC1 genes and susceptibility to viral hepatitis A infection in Mexican Americans. Hepatology 2012;55:1008–1018. [DOI] [PubMed] [Google Scholar]
- 9.Kamatani Y, Wattanapokayakit S, ochi H, Kawaguchi T, Takahashi A, Hosono N, et al. A genome-wide association study identifies variants in the HLA-Dp locus associated with chronic hepatitis B in Asians. Nat Genet 2009;41:591–595. [DOI] [PubMed] [Google Scholar]
- 10.Thomas DL, Thio CL, Martin MP, Qi Y, Ge D, O’Huigin C, et al. Genetic variation in IL28B and spontaneous clearance of hepatitis C virus. Nature 2009;461:798–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Thompson AJ. Genetic factors and hepatitis C virus infection. Gastroenterology 2012;142:1335–1339. [DOI] [PubMed] [Google Scholar]
- 12.Feng Z, Lemon SM. Peek-a-boo: membrane hijacking and the pathogenesis of viral hepatitis. Trends Microbiol 2014;22:59–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yamada K, Takahashi M, Hoshino Y, Takahashi H, Ichiyama K, Nagashima S, et al. ORF3 protein of hepatitis E virus is essential for virion release from infected cells. J Gen Virol 2009;90:1880–1891. [DOI] [PubMed] [Google Scholar]
- 14.Luo L, Pu X, Wang Y, Xu N. Impaired plasma lipid profiles in acute hepatitis. Lipids Health Dis 2010;9:5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Taneja S, Ahmad I, Sen S, Kumar S, Arora R, Gupta VK, et al. Plasma peptidome profiling of acute hepatitis E patients by MALDI-TOF/TOF. Proteome Sci 2011;9:5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lee G, Han D, Song JY, Kim JH, Yoon S. Proteomic analysis of swine hepatitis E virus (sHEV)–infected livers reveals upregulation of apolipoprotein and downregulation of ferritin heavy chain. FEMS Immunol Med Microbiol 2011;61:359–363. [DOI] [PubMed] [Google Scholar]
- 17.Pirruccello J, Kathiresan S. Genetics of lipid disorders. Curr Opin Cardiol 2010;25:238–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Centers for Disease Control and Prevention. NHANES genetic data. http://www.cdc.gov/nchs/nhanes/genetics/genetic.htm. Updated January 12, 2011. Accessed January 30, 2014.
- 19.Chang MH, Lindegren ML, Butler MA, Chanock SJ, Dowling NF. Gallagher M, et al. Prevalence in the United States of selected candidate gene variants: Third National Health and Nutrition Examination Survey, 1991–1994. Am J Epidemiol 2009;169:54–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Plan and operation of the Third National Health and Nutrition Examination Survey, 1988–94. Series 1: programs and collection procedures. Vital Health Stat 1 1994:1–407. [PubMed] [Google Scholar]
- 21.Binder DA. On the variances of asymptotically normal estimators from complex surveys. International Statistical Review 1983;51:279–292. [Google Scholar]
- 22.Woodruff RS. A simple method for approximating the variance of a complicated estimate. J Am Stat Assoc 1971;66:411–414. [Google Scholar]
- 23.Devlin B, Bacanu SA, Roeder K. Genomic control to the extreme. Nat Genet 2004;36:1129–1130; author reply 1131. [DOI] [PubMed] [Google Scholar]
- 24.Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc Series B Stat Methodol 1995;57:289–300. [Google Scholar]
- 25.Zannis VI, Breslow JL, Utermann G, Mahley RW, Weisgraber KH, Havel RJ, et al. Proposed nomenclature of apoE isoproteins, apoE genotypes, and phenotypes. J Lipid Res 1982;23:911–914. [PubMed] [Google Scholar]
- 26.Mahley RW, Rall SC Jr. Apolipoprotein E: far more than a lipid transport protein. Annu Rev Genomics Hum Genet 2000;1:507–537. [DOI] [PubMed] [Google Scholar]
- 27.Kuhlmann I, Minihane AM, Huebbe P, Nebel A, Rimbach G. Apolipo-protein E genotype and hepatitis C, HIV and herpes simplex disease risk: a literature review. Lipids Health Dis 2010;9:8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rhinn H, Fujita R, Qiang L, Cheng R, Lee JH, Abeliovich A. Integrative genomics identifies APOE epsilon4 effectors in Alzheimer’s disease. Nature 2013;500:45–50. [DOI] [PubMed] [Google Scholar]
- 29.Mahley RW. Apolipoprotein E: cholesterol transport protein with expanding role in cell biology. Science 1988;240:622–630. [DOI] [PubMed] [Google Scholar]
- 30.Mamotte CD, Sturm M, Foo JI, van Bockxmeer FM, Taylor RR. Comparison of the LDL-receptor binding of VLDL and LDL from apoE4 and apoE3 homozygotes. Am J Physiol Endocrinol Metab 1999; 276:E553–E557. [DOI] [PubMed] [Google Scholar]
- 31.Barth H, Schafer C, Adah MI, Zhang F, Linhardt RJ, Toyoda H, et al. Cellular binding of hepatitis C virus envelope glycoprotein E2 requires cell surface heparan sulfate. J Biol Chem 2003;278:41003–41012. [DOI] [PubMed] [Google Scholar]
- 32.Itzhaki RF, Wozniak MA. Herpes simplex virus type 1, apolipoprotein E, and cholesterol: a dangerous liaison in Alzheimer’s disease and other disorders. Prog Lipid Res 2006;45:73–90. [DOI] [PubMed] [Google Scholar]
- 33.Burt TD, Agan BK, Marconi VC, He W, Kulkarni H, Mold JE, et al. Apolipoprotein (apo) E4 enhances HIV-1 cell entry in vitro, and the APOE epsilon4/epsilon4 genotype accelerates HIV disease progression. Proc Natl Acad Sci USA 2008;105:8718–8723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shakibaei M, Frevert U. Dual interaction of the malaria circumsporozoite protein with the low density lipoprotein receptor–related protein (LRP) and heparan sulfate proteoglycans. J Exp Med 1996;184:1699–1711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mahley RW, Ji ZS. Remnant lipoprotein metabolism: key pathways involving cell-surface heparan sulfate proteoglycans and apolipoprotein E. J Lipid Res 1999;40:1–16. [PubMed] [Google Scholar]
- 36.Kalia M, Chandra V, Rahman SA, Sehgal D, Jameel S. Heparan sulfate proteoglycans are required for cellular binding of the hepatitis E virus ORF2 capsid protein and for viral infection. J Virol 2009;83:12714–12724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nagashima S, Takahashi M, Jirintai S, Tanggis, Kobayashi T, Nishizawa T, et al. The membrane on the surface of hepatitis E virus particles is derived from the intracellular membrane and contains trans-Golgi network protein 2. Arch Virol 2014;159:979–991. [DOI] [PubMed] [Google Scholar]
- 38.Liao Z, Graham DR, Hildreth JE. Lipid rafts and HIV pathogenesis: virion-associated cholesterol is required for fusion and infection of susceptible cells. AIDS Res Hum Retroviruses 2003;19:675–687. [DOI] [PubMed] [Google Scholar]
- 39.Suneetha PV, Pischke S, Schlaphoff V, Grabowski J, Fytili P, Gronert A, et al. Hepatitis E virus (HEV)–specific T-cell responses are associated with control of HEV infection. Hepatology 2012;55:695–708. [DOI] [PubMed] [Google Scholar]
- 40.Dadd T, Weale ME, Lewis CM. A critical evaluation of genomic control methods for genetic association studies. Genet Epidemiol 2009;33: 290–298. [DOI] [PubMed] [Google Scholar]
- 41.McKeigue PM, Carpenter JR, Parra EJ, Shriver MD. Estimation of admixture and detection of linkage in admixed populations by a Bayesian approach: application to African-American populations. Ann Hum Genet 2000;64:171–186. [DOI] [PubMed] [Google Scholar]
- 42.Salari K, Choudhry S, Tang H, Naqvi M, Lind D, Avila PC, et al. Genetic admixture and asthma-related phenotypes in Mexican American and Puerto Rican asthmatics. Genet Epidemiol 2005;29:76–86. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.