

Abstract
Prader–Willi syndrome (PWS) is caused by loss of paternally expressed genes from the 15q11-q13 region and reportedly rearranged as a cause of autism. Additionally, increased inflammatory markers and features of autism are reported in PWS. Cytokines encoded by genes involved with inflammation, cell proliferation, migration, and adhesion play a role in neurodevelopment and could be disturbed in PWS as abnormal plasma cytokine levels are reported in autism. We analyzed 41 plasma cytokines in a cohort of well-characterized children with PWS between 5 and 11 years of age and unaffected unrelated siblings using multiplex sandwich immunoassays with the Luminex magnetic-bead based platform. Data were analyzed using ANOVA testing for effects of diagnosis, gender, body mass index (BMI) and age on the 24 cytokines meeting laboratory criteria for inclusion. No significant effects were observed for age, gender or BMI. The log-transformed levels of the 24 analyzable cytokines were examined simultaneously using MANOVA adjusting for age and gender and a main effect of diagnosis was found (P-value <0.03). Four of 24 plasma cytokine levels (MCP1, MDC, Eotaxin, RANTES) were significantly higher in children with PWS compared with controls and classified as bioinflammatory chemokines supporting a disturbed immune response unrelated to obesity status. BMI was not statistically different in the two subject groups (PWS or unaffected unrelated siblings) and chemokine levels were not correlated with percentage of total body fat. Additional studies are required to identify whether possible early immunological disturbances and chemokine inflammatory processes found in PWS may contribute to neurodevelopment and behavioral features.
Keywords: Prader-Willi syndrome, children, cytokines, bioin-flammatory chemokines, immune system, neurodevelopment
INTRODUCTION
Prader–Willi syndrome (OMIM 176270) is a complex neurodeve-opmental disorder first reported in 1956 [Prader et al., 1956] with cardinal features of failure to thrive, a poor suck and feeding difficulties during infancy, hypogonadism/hypogenitalism, growth hormone deficiency, and typically a paternal 15q11-q13 chromosome deletion [Butler and Palmer, 1983; Butler etal., 1986]. In early childhood, food seeking behaviors and hyperphagia are noted along with a low metabolic rate and decreased physical activity leading to obesity which can be life-threatening, if not controlled. Short stature and small hands are common and most children with PWS are treated with growth hormone [Butler et al., 2006a]. Mild learning disability is common along with behavioral problems including self-injury (skin picking), temper tantrums, stubbornness, repetitive behavior with features of autism and obsessive-compulsive disorder [Butler, 1990; Butler, 2011; Cassidy et al., 2012]. This rare disorder affects about 1 in 15,000 individuals [Butler, 1990].
Prader–Willi syndrome (PWS) is due to errors in genomic imprinting with lack of paternally expressed genes generally due to a 15q11-q13 deletion (in about 75% of cases) while the second most common genetic finding is maternal disomy 15 with both chromosome 15s inherited from the mother [Nicholls et al., 1989]. Maternal disomy 15 occurs in about 25% of cases while a small percentage of subjects have microdeletions or epimutations of the imprinting center controlling the expression of the imprinted genes influenced by the parent of origin. PWS is considered the most common syndromic cause of life threatening obesity in childhood [Butler, 1990].
Other factors relating to obesity in humans include C-reactive protein (CRP) levels which are high during infections or non-infective inflammatory processes in humans [Abernathy and Avery, 1941; Danesh et al., 2004]. There is also a direct correlation between higher body mass index (obesity) and increased CRP levels [Visser et al., 1999] including significantly higher plasma levels in PWS compared with healthy individuals [Butler et al., 2006b]. In addition, several intercellular signaling proteins including cytokines are released from a wide variety of cells that regulate growth and proliferation of neuronal tissue that modulate host responses to infection, injury, inflammation, and chronic diseases [Skogstrand et al., 2005; Parmely, 2006]. Genes that encode immune-related proteins such as cytokines impact neurological development and disorders including autism spectrum disorders (ASD). ASD is reported in a subset of children with PWS [Butler et al., 2006a; Dykens et al., 2011].
Cytokines are small diverse polypeptides released by cells that serve as mediators of innate and adaptive immunity and hematopoiesis. These include interleukins, chemokines, growth factors, interferons, and colony stimulating factors which stimulate cells through specific receptors. T-helper 1 lymphocytes produce interferons while T-helper 2 cells trigger production of interleukins (e.g., IL4, IL5, IL13) [Parmely, 2006].
Increased plasma cytokine IL1β, IL6, IL8, and IL12p40 levels have been reported in young children with ASD [Ashwood et al., 2011] and disturbed chemokines when compared with unaffected, unrelated siblings [Manzardo et al., 2012]. In addition, abnormalities in the innate immune response have been reported in children with ASD, specifically reduced cytotoxic activity and elevated proinflammatory cytokines (e.g., tumor necrosis factor) which could disrupt neurodevelopment [Croonenberghs et al., 2002]. Elevated IgG levels are also associated with ASD with increased autoantibodies to neuronal elements [Connelly et al., 2006]. Identification of genetic or biologic factors underlying the predisposition and causation of ASD is an important area of study with behavioral problems and features in autism frequently seen in PWS.
Two related components seen in PWS stimulated our investigation of cytokines including: (i) Many individuals with PWS have features of ASD with reported involvement of the 15q11-q13 region in both disorders; i.e., paternal deletion in PWS and a maternal duplication in ASD. Cytokine disturbances are reported in children with ASD and neurodevelopmental problems linking genes that encode these immune-related proteins [Garbettetal.,2008] and(ii) Obesity, a cardinal feature seen in PWS occurring in early childhood is often associated with inflammatory processes with co-morbidities (diabetes mellitus, hypertension, cardiovascular disease, strokes) and high C-reactive protein levels [Butler et al., 2006b]. Furthermore, Viardot et al. [2010] reported an association in PWS of activation of the innate immune system independent of central obesity and insulin resistance. Herein, we examine plasma cytokine levels in a cohort of children with PWS between 5 and 11 years of age and unaffected unrelated siblings at similar ages to compare cytokine levels and examine patterns between the two subject groups.
MATERIALS AND METHODS
Subjects
Plasma (50 uL) was obtained from 13 males (mean age ± SD = 7.7 ± 1.9 years, age range = 5–11 years) and 10 females (mean age ± SD =8.5 ± 1.9years, age range = 5–11 years) with PWS, diagnosed clinically and confirmed with genetic testing (N = 15 with 15q11-q13 deletion; N = 8 with maternal disomy 15 or imprinting defect). All children with PWS were treated with growth hormone (e.g., initially at 0.5mg/m2/day administered subcutaneously with subsequent adjustments toward 1.0mg/m2/day or 0.24 mg/kg/weekly over 3–6 months and monitored with plasma IGF-1 levels [Deal et al., 2013]). No PWS child was treated with sex steroids. Four PWS children (Patients # 12, 16, 19, 22) had a history of insulin resistance. Three PWS children (Patients # 12, 16, 19) had a history of hypothyroidism requiring treatment. Only one PWS child (Patient #13) was prescribed an atypical anti-psychotic medication. Our patients were enrolled after obtaining valid consent approved by the local Human Subjects Committee from a large ongoing multi-site rare disease consortium on PWS. The control subjects consisted of 10 healthy unaffected unrelated male siblings (mean age ± SD = 8.3 ± 2.5 years, age range = 5–11 years) and eight healthyunaffected unrelated female siblings (meanage ± SD = 8.2 ± 2.1 years, age range = 5–11 years). Height (cm) and weight (kg) were obtained routinely on each patient in the clinic setting and body mass index (BMI) calculated. The mean BMI of the PWS group was 20.1 with a mean Z score of 1.0. The mean BMI of the sibling group was 18.1 with a mean Z score of 0.5. Body composition and total percentage body fat were determined using dual-energy x-ray absorptiometry (DXA) and the Lunar DXA Scanner (General Electric, Atlanta, GA), as a component of the rare disease consortium. Peripheral blood was collected in anti-coagulant vacutainer tubes (e.g., EDTA) in the morning after an overnight fast monitored by parents and plasma immediately separated then stored at —80°C.
Cytokine Assay and Analysis
Cytokine levels were determined from the plasma samples using the multiplex sandwich immunoassays with the Milliplex Human 41 Cytokine/Chemokine Premixed Kit (Millipore; Billerica, MA) and established protocols following manufacturer’s guidelines [Man-zardo et al., 2012]. The 41 cytokines included in the kit belong to four families: hematopoietin (IL1α; IL1β; IL1RA; IL2; IL3; IL4; IL5; IL6; IL7; IL9; IL10; IL12p40; IL12p70; IL13; IL15; IL17; sCD40L; Flt3 ligand; GCSF; and GMCSF); chemokines (EGF; Eotaxin; FGF2; Fractalkine; GRO; IL8; IP10; MCP1; MCP3; MDC; MIP1α; MIP1β; TGFα; VEGF; PDGFAA; PDGFBB; and RANTES); tumor necrosis factors (TNFα and TNFβ) and interferons (IFNα2 and IFNγ).
Blood plasma (25uL), concentration standard, or Milliplex quality control standard was combined with pre-mixed antibody-coupled magnetic beads and assay buffer, followed by overnight incubation at 4°C. All incubation steps were carried out on a micro-titer plate shaker at 300 rpm. The following day, the samples were washed, followed by incubation with secondary detection antibodies for 1 hr at room temperature. The samples were then put through another series of washes followed by addition of the fluorescent Streptavidin-Phycoerythrin detection solution. This mixture was incubated for 30 min at room temperature. Each sample was run in duplicate. Following incubation, sheath fluid was added to each sample well and the plate was read on the Luminex 200™ instrument (Luminex Molecular Diagnostic; Toronto, ON) based on magnetic-bead technology and level of magnetic field to separate the beads. The following cytokines/chemokines were analyzed using the Luminex 200™ v2.3 software with the indicated minimum detectable concentration levels given in pg/ml values: IL1α (9.4), IL1β (0.8), IL1RA (8.3), IL2 (1.0), IL3 (0.7), IL4 (4.5), IL5 (0.5), IL6 (0.9), IL7 (1.4), IL9 (1.2), IL10 (1.1), IL12p40 (7.4), IL12p70 (0.6), IL13 (1.3), IL15 (1.2), IL17 (0.7), sCD40L (5.1), Flt3 ligand (5.4), GCSF (1.8), GMCSF (7.5), EGF (2.8), Eotaxin (4.0), FGF2 (7.6), Fractalkine (22.7), GRO (9.9), IL8 (0.4), IP10 (8.6), MCP1 (1.9), MCP3 (3.8), MDC (3.6), MIP1α (2.9), MIP1β (3.0), TGFα (0.7), VEGF (26.3), TNFα (0.7), TNFβ (1.5), IFNα2 (2.9), IFNγ (0.8), PDGFAA (0.4), PDGFBB (2.2), and RANTES (1.2). Plasma cytokine concentrations were calculated using a standard curve derived from the reference cytokine concentration standards supplied by the manufacturer. The inter-assay coefficient of variation for the cytokines ranged from 3.5 to 18.9% the intraassay coefficient of variation ranged from 1.5 to 4.3%. Plasma samples were analyzed using numbers and blinded as to gender and control versus PWS status during each assay run.
Statistical Analysis
Data were presented as median ± standard deviation of raw and/or log-transformed cytokine levels by diagnosis (PWS or unaffected unrelated siblings). Correlations were examined between cytokine levels, diagnosis, age, sex, BMI, and percentage of total body fat using Spearman rank correlations. One-way analysis of variance (ANOVA) with Bonferroni correction was used to test for differences in age, gender, BMI, and diagnosis using log-transformed cytokine levels. Final analyses considered data that fell within the detection limits of the Luminex assay. Data falling below the detection limits were replaced with values one fourth of the minimum detection level for that cytokine as reported in previous studies [Ashwood et al., 2011; Manzardo et al., 2012; Zerbo et al., 2014] and as recommended by the manufacturer with samples below level of detection. Cytokines containing fewer than two SD or 68% of values within the detectable concentration range for the Luminex assay were not analyzed. Log-transformed data met the necessary statistical criteria for the assumption of normality showing equal variance and near linear residual plots. The main effect of diagnosis on log-transformed values considering all 24 cytokines meeting criteria for inclusion were also examined using multivariate analysis of variance (MANOVA) adjusting for age and gender. Findings with P-values of <0.05 were considered significant. Statistical analyses including descriptive statistics and Spearman rank correlations were generated using SAS statistical analysis software version 9.4 (SAS Inc., Cary, NC). In addition, a prior analysis indicated that 12 subjects would be required to provide 95% power to detect an effect size of 0.25 at a 0.05 alpha level of significance using ANOVA with repeated measures, within and between, group interactions.
RESULTS
Table I shows the baseline subject characteristics with height, weight, and body composition measures and chemokine levels found to be disturbed in PWS compared with unaffected unrelated sibling controls. The average age ± SD for the PWS children and unrelated siblings was 8.0 ± 2.0 years and 8.2 ± 2.3 years. The average height ± SD for the PWS children and unrelated siblings was 130.1 ± 12.7 cm and 133.5 ± 16.9 cm, respectively. The average weight ± SD for the PWS children and unrelated siblings was 35.1 ± 12.7kg and 33.7± 13.6kg, respectively. The average BMI ± SD for the PWS children and unrelated siblings was 20.1 ± 5.1 and 18.1 ± 3.2, respectively. The average percent body fat ± SD for the PWS children and unrelated siblings was 33.1 ± 13.8 and 24.0 ± 10.1, respectively. No significant differences or correlations were seen for age, weight, height, or BMI for PWS children or sibling controls, but PWS children had significantly higher percent body fat (F = 5.46; P < 0.02). The raw data and statistical results for each of the 24 analyzable cytokines meeting laboratory requirements for inclusion in our 41 children (N = 23, PWS; N = 18, unaffected unrelated siblings of children with PWS) between 5 and 11 years of age categorized by diagnosis (PWS and unrelated siblings) are presented in Figure 1. Seventeen (TGFα, Flt3 ligand, IL10, IL13, IL15, IL1RA, IL1α, IL1β, IL9, IL2, IL3, IL4, IL5, IL6, IL8, MIP1α, and TNFβ) of the 41 cytokines contained fewer than two SD or 68% of values within the detectable concentration range for the Luminex assay. As often the case in pediatric investigations of rare diseases, the plasma sample quantity is limited. The use of other assay methods for replication or to reassess cytokine levels below detection, such as ELISA, would require additional plasma.
TABLE I.
Baseline Subject Characteristics and Disturbed Chemokine Levels in Prader–Willi Syndrome (N = 23) and Unaffected Unrelated Siblings (N = 18)
| Genetic | Age | Weight | Height | BMI | BMI Z | Total | Eotaxin | MCP1 | MDC | RANTES | |||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Subject | Group | subtype | Sex | (yr) | (kg) | (cm) | (kg/m2) | Score | Body Fat (%) | (pg/mL) | (pg/mL) | (pg/mL) | (pg/mL) |
| 1 | PWS | Deletion | Male | 5 | 18.2 | 112.0 | 14.5 | −1.4 | 20.4 | 78.5 | 111 | 650 | 1920 |
| 2 | PWS | Maternal disomy | Male | 5 | 24.7 | 107.6 | 21.3 | 2.8 | 39.7 | 73.1 | 172 | 1408 | 3970 |
| 3 | PWS | Deletion | Female | 5 | 21.0 | 113.0 | 16.4 | 0.8 | 12.8 | 77.9 | 164 | 810 | 2206 |
| 4 | PWS | Deletion | Male | 6 | 19.7 | 116.3 | 14.6 | −0.8 | 13.6 | 69.5 | 114 | 1338 | 345 |
| 5 | PWS | Maternal disomy | Female | 6 | 18.9 | 112.5 | 14.9 | −0.2 | 19.2 | 68.4 | 102 | 494 | 1183 |
| 6 | PWS | Deletion | Male | 7 | 18.1 | 116.1 | 13.4 | −2.0 | 12.9 | 71.3 | 103 | 470 | 141 |
| 7 | PWS | Deletion | Female | 7 | 51.3 | 125.7 | 32.5 | 2.9 | 54.0 | 70.4 | 173 | 967 | 11597 |
| 8 | PWS | Maternal disomy | Male | 7 | 42.0 | 135.5 | 22.9 | 2.3 | 46.0 | 69.4 | 108 | 830 | 5382 |
| 9 | PWS | Deletion | Male | 7 | 40.4 | 132.8 | 22.9 | 2.3 | 45.5 | 67.8 | 141 | 1646 | 429 |
| 10 | PWS | Imprinting defect | Male | 8 | 27.3 | 126.4 | 17.1 | 0.8 | 21.2 | 73.4 | 133 | 1530 | 1863 |
| 11 | PWS | Deletion | Female | 8 | 25.1 | 124.5 | 16.2 | 0.3 | 19.3 | 68.6 | 79.3 | 596 | 1997 |
| 12 | PWS | Deletion | Male | 8 | 34.7 | 127.6 | 21.3 | 1.9 | 40.5 | 66.5 | 92.9 | 628 | 551 |
| 13 | PWS | Deletion | Male | 8 | 34.6 | 138.0 | 18.2 | 1.1 | 37.5 | 71.1 | 167 | 430 | 1528 |
| 14 | PWS | Maternal disomy | Male | 8 | 22.0 | 123.8 | 14.4 | −1.2 | 17.0 | 68.3 | 78.2 | 420 | 923 |
| 15 | PWS | Deletion | Female | 9 | 35.5 | 142.9 | 17.4 | 0.4 | 36.4 | 64.4 | 75.0 | 573 | 595 |
| 16 | PWS | Deletion | Female | 10 | 58.3 | 142.7 | 28.6 | 2.4 | 54.0 | 61.7 | 94.2 | 458 | 908 |
| 17 | PWS | Deletion | Female | 10 | 31.3 | 138.5 | 16.3 | −0.1 | 27.4 | 70.0 | 159 | 869 | 4503 |
| 18 | PWS | Maternal disomy | Male | 10 | 46.8 | 151.0 | 20.5 | 1.4 | 31.7 | 64.5 | 68.7 | 449 | 680 |
| 19 | PWS | Deletion | Female | 10 | 52.2 | 140.8 | 26.3 | 2.2 | 48.1 | 65.7 | 57.4 | 448 | 842 |
| 20 | PWS | Deletion | Female | 10 | 49.1 | 134.7 | 27.1 | 2.2 | 45.8 | 67.8 | 88.8 | 586 | 807 |
| 21 | PWS | Deletion | Male | 10 | 47.9 | 141.0 | 24.1 | 1.9 | 48.0 | 65.2 | 57.0 | 361 | 1143 |
| 22 | PWS | Maternal disomy | Female | 11 | 41.0 | 140.3 | 20.8 | 1.0 | 29.6 | 70.7 | 143 | 926 | 4688 |
| 23 | PWS | Maternal disomy | Male | 11 | 46.8 | 147.6 | 21.5 | 1.2 | 39.8 | 66.4 | 102 | 357 | 865 |
| 24 | Sibling | Normal | Male | 5 | 27.7 | 111.2 | 22.4 | 2.9 | 32.0 | 68.8 | 121 | 433 | 289 |
| 25 | Sibling | Normal | Female | 5 | 15.5 | 112.5 | 12.2 | −3.6 | 10.3 | 66.2 | 79.3 | 579 | 1194 |
| 26 | Sibling | Normal | Male | 5 | 19.2 | 112.5 | 15.2 | −0.2 | 9.7 | 69.6 | 89.6 | 588 | 404 |
| 27 | Sibling | Normal | Male | 5 | 22.6 | 118.5 | 16.4 | 0.5 | 20.5 | 68.8 | 86.2 | 572 | 1053 |
| 28 | Sibling | Normal | Female | 6 | 24.8 | 115.4 | 18.6 | 1.6 | 29.9 | 65.7 | 87.4 | 408 | 1281 |
| 29 | Sibling | Normal | Male | 6 | 19.6 | 113.9 | 15.1 | −0.3 | 10.7 | 68.6 | 117 | 478 | 76.0 |
| 30 | Sibling | Normal | Female | 8 | 33.2 | 135.1 | 18.2 | 1.1 | 24.4 | 69.3 | 74.8 | 459 | 415 |
| 31 | Sibling | Normal | Female | 8 | 30.0 | 131.5 | 17.3 | 0.8 | 28.8 | 66.0 | 134 | 512 | 190 |
| 32 | Sibling | Normal | Female | 8 | 29.7 | 134.5 | 16.4 | 0.2 | 22.5 | 68.3 | 66.6 | 381 | 598 |
| 33 | Sibling | Normal | Male | 9 | 32.4 | 134.5 | 17.9 | 0.8 | 25.1 | 64.3 | 63.2 | 366 | 1087 |
| 34 | Sibling | Normal | Female | 9 | 28.5 | 133.6 | 16.0 | −0.3 | 18.4 | 65.0 | 78.9 | 545 | 733 |
| 35 | Sibling | Normal | Male | 10 | 35.0 | 142.6 | 17.2 | 0.4 | 18.6 | 67.8 | 96.1 | 447 | 1538 |
| 36 | Sibling | Normal | Male | 10 | 30.2 | 133.9 | 16.8 | 0.1 | 11.9 | 67.3 | 70.9 | 255 | 764 |
| 37 | Sibling | Normal | Female | 10 | 57.0 | 163.1 | 21.4 | 1.3 | 37.9 | 66.3 | 100 | 432 | 823 |
| 38 | Sibling | Normal | Male | 11 | 45.6 | 143.7 | 22.1 | 1.5 | 29.2 | 67.0 | 86.8 | 436 | 795 |
| 39 | Sibling | Normal | Male | 11 | 67.7 | 156.2 | 25.8 | 2.0 | 46.7 | 69.5 | 74.0 | 504 | 1186 |
| 40 | Sibling | Normal | Female | 11 | 39.5 | 152.2 | 17.0 | −0.2 | 22.6 | 65.0 | 84.5 | 458 | 1427 |
| 41 | Sibling | Normal | Male | 11 | 48.0 | 158.2 | 19.8 | 0.7 | 33.1 | 67.3 | 89.2 | 434 | 765 |
Fig. 1.
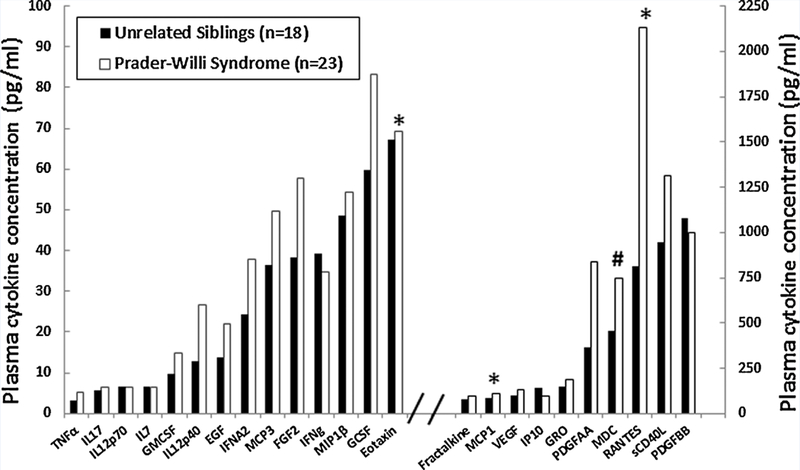
Histograms representing raw data and statistical analysis for each of the 24 detectable cytokines meeting the laboratory requirements for inclusion and grouped by diagnosis. The cytokines belong to four families: tumor necrosis factor (TNFα), hematopoietin (IL17, IL12p70, IL7, GMCSF, IL12p40, GCSF, and sCD40L), chemokine (EGF, MCP3, FGF2, MIP1β Eotaxin, Fractalkine, MCP1, VEGF, IP10, GRO, PDGFAA, MDC, RANTES, and PDGFBB), and interferon (IFNα2 and IFNγ). Significant values are indicated: *P < 0.05; #P < 0.01.
No significant differences or correlations were found in cytokine levels based upon age, PWS genetic subtype, sex or BMI. However, logIL7 level was significantly positively correlated with percentage of total body fat in the combined PWS and sibling subject groups (rho = 0.33; P = 0.033) and log Eotaxin was significantly negatively correlated with percentage total body fat in PWS children only (rho = —0.51; P = 0.014). The remaining log-transformed data met criteria for analysis using ANOVA. Four cytokines (i.e., MCP1, MCD, Eotaxin, and RANTES) were significantly higher (P < 0.05) in the children with PWS compared with unaffected unrelated siblings without PWS [PWS (N = 23); unaffected unrelated sibling controls (N = 18)] (Fig 2). The four disturbed cytokines were classified as chemokines. A statistically significant main effect of diagnosis was identified (F = 2.6, df num = 23, df den = 16, P< 0.03) using MANOVA adjusting for the effects of age and gender on cytokine level. Scatter plots of data with linear regression lines were generated to illustrate the relationship between the four disturbed chemokine levels and percentage total body fat for both subject groups (PWS vs. unrelated unaffected siblings, see Fig 3).
Fig. 2.
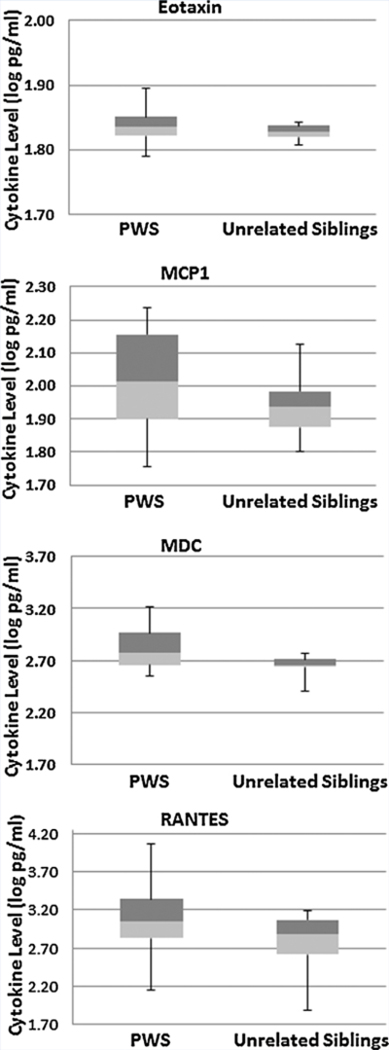
Representative box plots from selected cytokines in subjects grouped by diagnosis. Box plots represent median (junction between light and dark shading) and interquartile ranges (25%ile range, light shading; 75%ile range, dark shading). Bars represent maximum and minimum values from log transformed data for each subject group.
Fig. 3.
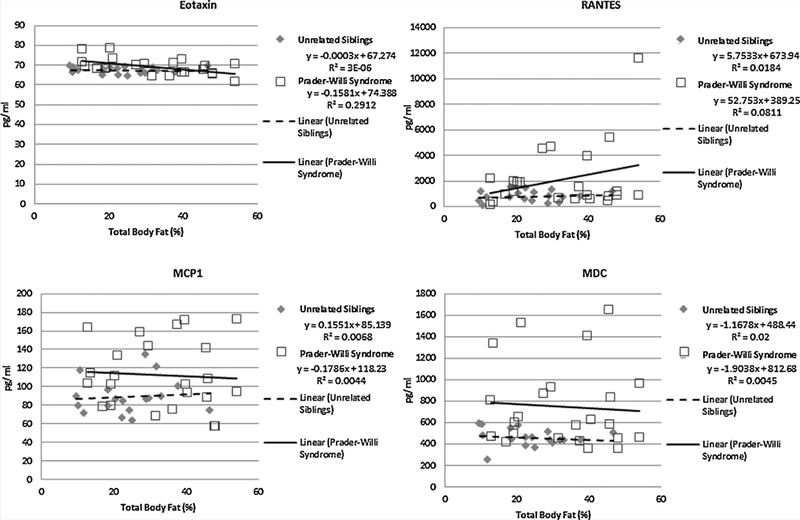
Scatter plot representations of data illustrating the relationship between the disturbed chemokine levels (Eotaxin, MCP1, MDC, RANTES) as the dependent variable and percentage total body fat as the explanatory variable for both subject groups (Prader–Willi syndrome vs. unrelated unaffected siblings). Linear equations and their derived best fit regression lines and are presented with R-squared values for both subject groups.
DISCUSSION
Data from our study in children with PWS indicate a disturbed subset of cytokines with a potential resultant compromised immune system, particularly with chemokine involvement as chemo-attractants for eosinophils, monocytes, natural killer cells, and dendritic cells. Specifically, four chemokines (MCP1, MDC, Eotaxin, RANTES) were found to be higher in the PWS subjects compared with an unaffected healthy and unrelated group of sibling controls when age, sex, height, weight, and BMI were controlled. Chemokines are coded by genes and perform as cell signaling proteins secreted by cells to induce chemotaxis and direct movement of immune cells to the site of infection, presence of microbes or related to tissue growth. Chemokines are small peptides with a molecular weight of 8–10 kDa and involved with neutrophils that leave the vascular system and migrate with later recruitment of monocytes and immature dendritic cells. Specific chemokines have roles in development, promoting angiogenesis or in guiding cells to tissues to provide signals critical for cellular maturation. Chemokines can be proinflammatory when activated during the immune response or while controlling cell migration as part of normal tissue growth and maintenance.
Proinflammatory chemokines are released from a wide variety of cells in response to infections and agents that cause physical damage to promote wound healing. The chemokines found disturbed with higher plasma levels in our study were bioinflammatory and included RANTES, MDC, MCP1, and Eotaxin.
RANTES is an acronym for Regulated on Activation, Normal T Expressed and Secreted also known as CCL5 while MCP1 (Monocyte Chemotactic Protein 1) is known as CCL2 [Cocchi et al., 1995; Ignatov et al., 2006]. RANTES (CCL5) and MCP1 (CCL2) represent distinct members of the 27 chemokine ligands (CC1-CC28) that induce the migration of monocytes and other cell types including natural killer (NK) cells and dendritic cells. RANTES (CCL5) attracts cells such as T cells, eosinophils and basophils that express the receptor, CCR5, a co-receptor for human immunodeficiency virus. RANTES is also a member of the interleukin-8 family of cytokines [Maghazachi et al., 1996]. MCP1 (CCL2) induces monocytes to leave the bloodstream and enter the surrounding tissue to be converted to tissue macrophages. MDC (Macrophage Derived Chemokine) is a chemoattractant for monocytes and dendritic cells, NK cells and activated T lymphocytes. Eotaxin (also known as Small Inducible Cytokine A1I) is a chemokine that attracts eosinophils [Daugherty et al., 1996].
The current standard of care treatment of PWS may also impact upon cytokine and chemokine function through the effects of growth hormone. Nearly all infants and young children with PWS are treated with growth hormone which is effective in increasing growth and promotes the development of lean muscle mass to reduce and prevent obesity. Our PWS children were treated with growth hormone which effectively normalized height, weight and BMI in relationship to unaffected unrelated siblings at comparable ages, but percent body fat remained significantly higher in the PWS group.
Growth hormone (GH) can act as a cytokine in controlling survival and proliferation of immune cells particularly in times of stress [Jeay et al., 2002]. GH is classified as a peptide hormone synthesized and secreted in the anterior pituitary but with the ability to stimulate growth of the skeleton and soft tissues including proliferation of lymphoid cells [Kooijman et al., 1996; Kiess and Gallaher, 1998]. This process potentially modulates cytokine production and enhances B cell development leading to antibody production, neutrophil adhesion and monocyte migration [Hatton et al., 2009].
Bozzola et al. [2003] investigated the effect of exogenously administered growth hormone serum interleukins levels (i.e., IL1β, IL2, IL12, TNFα, and IFNγ) and their relationship with IGF-1 levels in 23 non-syndromic short stature children with an average age of 12 years. Significant increases were found in five cytokines indicating that short-term growth hormone administration in normal children can influence the immune system. Other studies have shown that growth hormone enhances prion-flammatory cytokine production by monocytes in whole blood [Uronen-Hansson et al., 2003] and stimulates production of INFγ in peripheral blood mononuclear cells [Mustafa et al., 1997]. IFNγ, IL12, and TNFα are primarily involved in cell mediated immunity [Morel and Oriss, 1988]. GH induces the production of MIP1α and RANTES and proinflammatory responses in macrophages mediated by the JAK2/PI3K/PKC/ ERK1/2/JAK2/JNK and JAK1/STAT signaling cascades [reviewed by Sodhi and Tripathi, 2008a, b] while the GH-induced ERK1/2 cascade regulates production of TNFα and IL1β in macrophages [Tripathi and Sodhi, 2009].
Studies of biomarkers for inflammation and infection in ASD have examined cytokine and chemokine levels. For example, Zerbo et al. [2014] studied a collection of archived neonatal blood specimens from a cohort of infants diagnosed at a later age with ASD (N = 84), with developmental delay but without ASD (N = 49) or controls from the general population (N = 159). The specimens were initially collected for routine newborn screening from the state of California and then analyzed commercially with available cytokine and chemokine assay kits developed for a multiplex bead-based platform, similar to our study. Cytokine levels were generally not detected in the vast majority of newborn samples studied regardless of their diagnosis but the chemokine MCP1 was elevated and the chemokine RANTES was detected but decreased in ASD subjects compared to unaffected controls from the general population. MIP1a and RANTES levels were also detected but decreased in children with developmental delay compared with unaffected controls.
Similarly, Abdallah et al. [2013] studied neonatal chemokine levels and the risk of ASD in a Danish follow-up study using a birth cohort. They examined three inflammatory chemokines (MCP1, MIP1a and RANTES) in neonatal archived dried blood spot samples from 359 subjects (4:1 male to female ratio) diagnosed later with ASD at 6 years of age or older and 741 controls. Measurements of the selected chemokines were performed with an in-house assay panel using the multiplex flow cytometric assay system Luminex MultiAnalyte Profiling Technology. They found evidence for decreased levels of RANTES and cautiously suggested an altered cell-mediated immunity during the early neonatal period in the development of ASD. The increased chemokine levels including RANTES found in plasma samples from our children with PWS compared with unaffected unrelated siblings was in contrast to the decreased levels reported in the archived dried blood spots from the Danish study reported by Abdulla etal. [2013] or in neonatal blood specimens reported by Zerbo etal. [2014] with ASD diagnosed at an older age. It is unclear how the collection process (i.e., archived dried blood spots) would impact chemokine levels. However, our results could support macrophage-activating properties attributable to growth hormone treatment affecting chemokine levels [Warwick-Davies et al., 1995]. The JAK2 signaling cascade pathway plays a central role in mediating proinflammatory responses by macrophages during GH treatment [Tripathi and Sodhi, 2009]. Interestingly, RANTES has been postulated to play a role in psychiatric disorders such as mood disorders, schizophrenia and cognitive impairment [Stuart and Baune, 2014].
All PWS children in our study were treated with growth hormone; thus, comparison with PWS children not taking growth hormone was not possible. One could propose testing PWS children from other countries where growth hormone is not easily accessible to further evaluate our observation of elevated chemokines and the role of growth hormone treatment. The growth hormone doses in our children were not supra-physiologic and growth hormone levels were regularly monitored.
No significant differences were found in plasma cytokine levels in individuals with different PWS genetic subtypes. More psychotic events are reported among those PWS individuals with maternal disomy 15, particularly during young adulthood [Butler et al., 2006a] which could be related to higher cytokine levels or neuro-inflammatory mechanisms [Najjar and Pearlman, 2014].
Our PWS cohort ranged in age from 5 to 11 years, generally too young to experience psychotic events in the PWS individuals with maternal disomy 15. Similarly, the increased fat mass observed in PWS children did not appear to significantly enhance or otherwise stimulate cytokine production as evidenced by the general lack of correlation with the percentage of total body fat and chemokine levels shown in Figure 3. The disturbed cytokines observed in our study were mostly different from those seen in children with autism and without PWS reported by others [Ashwood et al., 2011; Manzardo et al., 2012]. However, cytokine observations reviewed by Masi et al. [2014] in individuals with autism found that 6 of 19 analyzable cytokines were elevated. These included IL1β, IL6, IL8, IFNγ, Eotaxin, and MCP1. Of these, Eotaxin and MCP1 levels were also increased in our individuals with PWS compared with unaffected unrelated siblings. IL1β, IL6, and IL8 were below the level of detection in our study and would require a separate assay for comparison such as ELISA requiring new sample collection and analysis.
Cytokines disturbed in our study were different than those reported by Bozzola et al. [2003] following children with short stature and treated with growth hormone. Five cytokines were significantly increased by growth hormone treatment in children with short stature and two of these cytokines (TNFα and IFNγ) were detectable in our study, as well, but not disturbed compared with the control siblings. The effects of GH treatment and/or impact of disturbed cytokines on neurodevelopment and function will require more research including with a larger number of subjects and possibly including different assay methods (e.g., ELISA).
A recent report with the multiplex method and Luminex technology showed that the detection and measurement of serum antibodies against tumor antigens were comparable to the peptide ELISA method and the inter-assay reproducibility of the multiplex approach was excellent (R2 > 0.98) for 20 sera tested against all tumor antigens. Therefore, multiplex immunoassays represent valuable and readily accessible techniques for detection and quantification of proteins with high sensitivity and gaining popularity in measuring growth factors, hormones, neuropeptides and cytokines [Reuschenbach et al., 2014] as undertaken in our study. In contrast to ELISA, multiplex immunoassays offer simultaneous quantification of many proteins (peptides) across multiple samples using small sample volumes which can save reagents, costs and time [Valekova et al., 2014].
In summary, our study showed higher plasma levels of cytokines in PWS representing the chemokine family acting as chemoattractants for eosinophils (Eotaxin, RANTES), monocytes (MDC, MCP1), T lymphocytes (RANTES, MDC), basophils (RANTES), and natural killer T lymphocytes and dendritic cells (MDC) maintaining a diverse range of physiological and pathologic functions. These attributes are proinflammatory in nature and may contribute to the control, regulation and function of the immune system influencing neurodevelopment and behavior which are abnormal in PWS. There is also an established link between inflammatory/ immune activation and cardiovascular risk which should be further considered given that individuals with PWS have higher cardiovascular risks [Butler et al., 2006a].
The impact of growth hormone therapy in PWS on chemokine levels and immune function is unknown requiring further investigation. Additional research is needed to identify the role of the disturbed chemokines in neurodevelopment and related cognitive deficits, aberrant behavior, and hyperphagia in PWS further complicated by growth hormone deficiency requiring growth hormone therapy which is now common in this disorder.
ACKNOWLEDGMENTS
We thank Carla Meister for preparation of the manuscript and Craig Lanio for technical support. Funding support was provided by the Clara Schiller Perpetual Charitable Trust, Prader-WilliSyndrome Association (USA), the National Institute of Child Health and Human Development (NICHD) HD02528 and from the Angelman, Rett and Prader-Willi Syndromes Consortium (U54 HD06122) which is part of the National Institute of Health (NIH) Rare Disease Clinical Research Network (RDCRN), supported through collaboration between the NIH Office of Rare Disease Research (ORDR) at the National Center of Advancing Translational Science (NCATS) and NICHD. The content is solely the responsibility of the authors and does not necessarily represent the office views of the National Institutes of Health.
Grant sponsor: Clara Schiller Perpetual Charitable Trust; Grant sponsor: Prader-Willi Syndrome Association (USA); Grant sponsor: The National Institute of Child Health and Human Development; Grant number: HD02528; Grant sponsor: Rett and Prader-Willi Syndromes Consortium; Grant number: U54 HD06122.
REFERENCES
- Abdallah MW, Larsen N, Grove J, Bonefeld-Jorgensen EC, Norgaard-Pedersen B, Hougaard DM, Mortensen EL. 2013. Neonatal chemokine levels and risk of autism spectrum disorders: Findings from a Danish historic birth cohort follow-up study. Cytokine. 61:370–376. [DOI] [PubMed] [Google Scholar]
- Abernethy TJ, Avery OT. 1941. The occurrence during acute infections of a protein not normally present in the blood: I. distribution of the reactive protein in patients’ sera and the effect of calcium on the flocculation reaction with c polysaccharide of pneumococcus. J Exp Med 73:173–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashwood P, Krakowiak P, Hertz-Picciotto I, Hansen R, Pessah I, Van de Water J. 2011. Elevated plasma cytokines in autism spectrum disorders provide evidence of immune dysfunction and are associated with impaired behavioral outcome. Brain Behav Immun 25:40–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bozzola M, De Benedetti F, De Amici M, Jouret B, Travaglino P, Pagani S, Conte F, Tauber M. 2003. Stimulating effect of growth hormone on cytokine release in children. Eur J Endocrinol 149:397–401. [DOI] [PubMed] [Google Scholar]
- Butler MG, Palmer CG. 1983Parental origin of chromosome 15deletionin Prader–Willi syndrome. Lancet 1:1285–1286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler MG, Meaney FJ, Palmer CG. 1986. Clinical and cytogenetic survey of 39 individuals with Prader-Labhart-Willi syndrome. Am J Med Genet 23:793–809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler MG. 1990. Prader–Willi syndrome: Current understanding of cause and diagnosis. Am J Med Genet 35:319–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler MG, Lee PDK, Whitman BY. 2006a. Management of Prader–Willi Syndrome. 3rd ed New York, NY:Springer-Verlag Publishers; p 1–550. [Google Scholar]
- Butler MG, Bittel DC, Kibiryeva N, Garg U. 2006b. C-reactive protein levels in subjects with Prader–Willi syndrome and obesity. Genet Med 8:243–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler MG. 2011. Prader–Willi syndrome: Obesity due to genomic imprinting. Curr Genomics 12:204–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cassidy SB, Schwartz S, Miller JL, Driscoll DJ. 2012. Prader–Willi syndrome. Genet Med 14:10–26. [DOI] [PubMed] [Google Scholar]
- Cocchi F, DeVico AL, Garzino-Demo A, Arya SK, Gallo RC, Lusso P. 1995. Identification of RANTES, MIP-1 alpha, and MIP-1 beta as the major HIV-suppressive factors produced by CD8+ T cells. Science 270:1811–1815. [DOI] [PubMed] [Google Scholar]
- Connolly AM, Chez M, Streif EM, Keeling RM, Golumbek PT, Kwon JM, Riviello JJ, Robinson RG, Neuman RJ, Deuel RM. 2006. Brain-derived neurotrophic factor and autoantibodies to neural antigens in sera of children with autistic spectrum disorders, Landau-Kleffner syndrome, and epilepsy. Biol Psychiatry 59:354–363. [DOI] [PubMed] [Google Scholar]
- Croonenberghs J, Bosmans E, Deboutte D, Kenis G, Maes M. 2002. Activation of the inflammatory response system in autism. Neuropsychobiology 45:1–6. [DOI] [PubMed] [Google Scholar]
- Danesh J, Wheeler JG, Hirschfield GM, Eda S, Eiriksdottir G, Rumley A, Lowe GD, Pepys MB, Gudnason V. 2004. C-reactive protein and other circulating markers of inflammation in the prediction of coronary heart disease. N Engl J Med 350:1387–1397. [DOI] [PubMed] [Google Scholar]
- Daugherty BL, Siciliano SJ, DeMartino JA, Malkowitz L, Sirotina A, Springer MS. 1996. Cloning, expression, and characterization of the human eosinophil eotaxin receptor. J Experimental Med 183:2349–2354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deal CL, Tony M, Hoybye C, Allen DB, Tauber M, Christiansen JS. 2013. GrowthHormone Research Society workshop summary: Consensus guidelines for recombinant human growth hormone therapy in Prader–Willi syndrome. J Clin Endocrinol Metab 98:E1072–E1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dykens EM, Lee E, Roof E. 2011. Prader–Willi syndrome and autism spectrum disorders: An evolving story. J Neurodev Disord 3:225–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garbett K, Ebert PJ, Mitchell A, Lintas C, Manzi B, Mirnics K, Persico AM. 2008. Immune transcriptome alterations in the temporal cortex of subjects with autism. Neurobiol Dis 30:303–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatton DD, Wheeler A, Sideris J, Sullivan K, Reichardt A, Roberts J, Clark R, Bailey DB. 2009. Developmental trajectories of young girls with fragile x syndrome. Am J Intellect Dev Disabil 114:161–171. [DOI] [PubMed] [Google Scholar]
- Ignatov A, Robert J, Gregory-Evans C, Schaller HC. 2006. RANTES stimulates Ca2+ mobilization and inositol trisphosphate (IP3) formation in cells transfected with G protein-coupled receptor 75. British J Pharmacol 149:490–497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeay S, Sonenshein GE, Postel-Vinay MC, Kelly PA, Baixeras E. 2002. Growth hormone can act as a cytokine controlling survival and proliferation of immune cells: New insights into signaling pathways. Mol Cell Endocrinol 188:1–7. [DOI] [PubMed] [Google Scholar]
- Kiess W, Gallaher B. 1998. Hormonal control of programmed cell death/ apoptosis. Eur J Endocrinol 138:482–491. [DOI] [PubMed] [Google Scholar]
- Kooijman R, Hooghe-Peters EL, Hooghe R. 1996. Prolactin, growth hormone, and insulin-like growth factor-I in the immune system. Adv Immunol 63:377–454. [DOI] [PubMed] [Google Scholar]
- Maghazachi AA, Al-Aoukaty A, Schall TJ. 1996. CC chemokines induce the generation of killer cells from CD56+ cells. Euro J Immunol 26:315–319. [DOI] [PubMed] [Google Scholar]
- Manzardo AM, Henkhaus R, Dhillon S, Butler MG. 2012. Plasma cytokine levels in children with autistic disorder and unrelated siblings. Int J Dev Neurosci 30:121–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masi A, Quintana DS, Glozier N, Lloyd AR, Hickie IB, Guastella AJ. 2014. Cytokine aberrations in autism spectrum disorder: A systematic review and meta-analysis. Mol Psychiatry. 2014. June 17. doi: 10.1038/mp.2014.59 [Epub ahead of print]. [DOI] [PubMed] [Google Scholar]
- Morel PA, Oriss TB. 1998. Crossregulation between Th1 and Th2 cells. Crit Rev Immunol 18:275–303. [DOI] [PubMed] [Google Scholar]
- Mustafa A, Nyberg F, Mustafa M, Bakhiet M, Mustafa E, Winblad B, Adem A. 1997. Growth hormone stimulates production of interferon-gamma by human peripheral mononuclear cells. Horm Res 48:11–15. [DOI] [PubMed] [Google Scholar]
- Najjar S, Pearlman DM. 2014. Neuroinflammation and white matter pathology in schizophrenia: systematic review. Schizophr Res 171: 1117–1118. [DOI] [PubMed] [Google Scholar]
- Nicholls RD, Knoll JH, Butler MG, Karam S, Lalande M. 1989. Genetic imprinting suggested by maternal heterodisomy in nondeletion Prader–Willi syndrome. Nature 342:281–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parmely MJ. 2006. USMLE Road Map: Immunology. NewYork, NY: Lange Medical Books/ McGraw-Hill Companies. [Google Scholar]
- Prader A, Labhart A, Willi H. 1956. Ein syndrome von adipositas, kleinwuchs, kryptorchismus und oligophrenie nach myatonieartigem zustand im neugeborenenalter. Schweizerishe Med Wochen 86: 1260–1261. [Google Scholar]
- Reuschenbach M, Dorre J, Waterboer T, Kopitz J, Schneider M, Hooger-brugge N, Jager E, Kloor M, Von Knebel Doeberitz M. 2014. A multiplex method for the detection of serum antibodies against in silico-predicted tumor antigens. Cancer Immunol Immunother. 63:1251–1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skogstrand K, Thorsen P, Norgaard-Pedersen B, Schendel DE, Sorensen LC, Hougaard DM. 2005. Simultaneous measurement of 25 inflammatory markers and neurotrophins in neonatal dried blood spots by immunoassay with xMAP technology. Clin Chem 51:1854–1866. [DOI] [PubMed] [Google Scholar]
- Sodhi A, Tripathi A. 2008a. Prolactin and growth hormone induce differential cytokine and chemokine profile in murine peritoneal macrophages in vitro: Involvement of p-38 MAP kinase. STAT3 and NF-kB . Cytokine 41:162–173. [DOI] [PubMed] [Google Scholar]
- Sodhi A, Tripathi A. 2008b. Prolactin induced production of cytokines in macrophages involves Ca + + and p42/44 MAP kinase signaling pathway. Growth Factors 26:212–219. [DOI] [PubMed] [Google Scholar]
- Stuart MJ, Baune BT. 2014. Chemokines and chemokine receptors in mood disorders, schizophrenia, and cognitive impairment: A systematic review of biomarker studies. Neurosci Biobehav Rev 42C:93–115. [DOI] [PubMed] [Google Scholar]
- Tripathi A, Sodhi A. 2009. Growth hormone-induced production of cytokines in murine peritoneal macrophages in vitro: Role of JAK/STAT, PI3K, PKC and MAP kinases. Immunobiology 214:430–440. [DOI] [PubMed] [Google Scholar]
- Uronen-Hansson H, Allen ML, Lichtarowicz-Krynska E, Aynsley-Green A, Cole TJ, Hoiden-Guthenberg, Fryklund I, Klein L. 2003. Growth hormone enhances proinflammatory cytokine production by monocytes in whole blood. Growth Horm IGF Res 13:282–286. [DOI] [PubMed] [Google Scholar]
- Valekova I, Kupcova Skalnikova H, Jarkovska K, Motlik J, Kovarova H. 2014. Multiplex immunoassays for quantification of cytokines, growth factors, and other proteins in stem cell communication. Methods Mol Biol. 2014. July 26 [Epub ahead of print]. [DOI] [PubMed] [Google Scholar]
- Viardot A, Sze L, Purtell L, Sainsbury A, Loughnan G, Smith E, Herzog H, Steinbeck K, Campbell LV. 2010. Prader–Willi syndrome is associated with activation of the innate immune system independently of central adiposity and insulin resistance. J Clin Endocrinol Metab 95:3392–3399. [DOI] [PubMed] [Google Scholar]
- Visser M, Bouter LM, McQuillan GM, Wener MH, Harris TB. 1999. Elevated C-reactive protein levels in overweight and obese adults. JAMA 282:2131–2135. [DOI] [PubMed] [Google Scholar]
- Warwick-Davies J, Lowrie DB, Cole PJ. 1995. Growth hormone is a human macrophage activating factor. Priming of human monocytes for enhanced release of H2O2. J Immunol 154:1909. [PubMed] [Google Scholar]
- Zerbo O, Yoshida C, Grether JK, Van de Water J, Ashwood P, Delorenze GN. 2014. Neonatal cytokines and chemokines and risk of Autism Spectrum Disorder: The Early Markers for Autism (EMA) study: a case-control study. J Neuroinflammation 11:113. [DOI] [PMC free article] [PubMed] [Google Scholar]