

SUMMARY
Exquisite regulation of energy homeostasis protects from nutrient deprivation but causes metabolic dysfunction upon nutrient excess. In human and murine adipose tissue, the accumulation of ligands of the receptor for advanced glycation end products (RAGE) accompanies obesity, implicating this receptor in energy metabolism. Here, we demonstrate that mice bearing global- or adipocyte-specific deletion of Ager, the gene encoding RAGE, display superior metabolic recovery after fasting, a cold challenge, or high-fat feeding. The RAGE-dependent mechanisms were traced to suppression of protein kinase A (PKA)-mediated phosphorylation of its key targets, hormone-sensitive lipase and p38 mitogen-activated protein kinase, upon β-adrenergic receptor stimulation—processes that dampen the expression and activity of uncoupling protein 1 (UCP1) and thermogenic programs. This work identifies the innate role of RAGE as a key node in the immunometabolic networks that control responses to nutrient supply and cold challenges, and it unveils opportunities to harness energy expenditure in environmental and metabolic stress.
Graphical Abstract
In Brief
Hurtado del Pozo et al. show that the deletion of adipocyte RAGE, whose ligands accumulate in metabolic stress, protects from obesity and cold challenges through the modulation of protein kinase A activities. This work adds RAGE to the immunometabolic networks that regulate energy expenditure in environmental and metabolic stress.
INTRODUCTION
The epidemic of obesity and its metabolic sequelae profoundly impact human health and longevity (Finkelstein et al., 2009; Flegal et al., 2012). Maintaining a precise balance between energy intake and expenditure is essential for an organism’s ability to store or utilize nutrients. Yet, hoarding energy is a double-edged sword, albeit, salutary in nutrient deprivation; in over-feeding, excess energy storage confers susceptibility to obesity and type 2 diabetes.
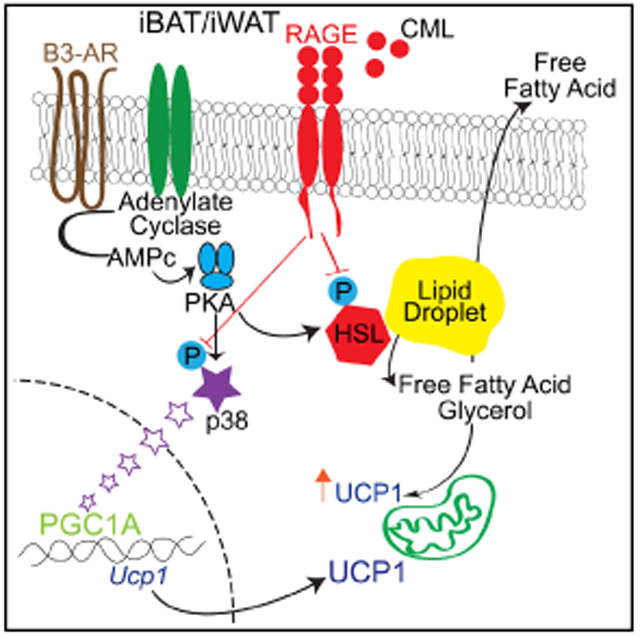
White adipose tissue (WAT) and brown adipose tissue (BAT) in mammals store energy in the form of triglycerides and release fatty acids and glycerol in response to catecholaminergic stimulation by sympathetic nerves (Bartness et al., 2010). Whereas WAT liberates fatty acids into the circulation, BAT preferentially oxidizes fatty acids and dissipates energy through uncoupled respiration and the production of heat (Cannon and Nedergaard, 2001; Collins, 2012). The recent identification of BAT in adult humans has rekindled an interest in harnessing brown fat to spur cellular energy expenditure (Nedergaard et al., 2007; van Marken Lichtenbelt et al., 2009; Cypess et al., 2009; Virtanen et al., 2009; Zingaretti et al., 2009). The physiologic process in which energy is dissipated in the form of heat in response to stressors such as cold challenges or over-feeding is termed “adaptive thermogenesis” and is dependent on the expression and activity of uncoupling protein 1 (UCP1) in brown adipocytes (Kopecky et al., 1995). Catecholamines stimulate β-adrenergic receptors (ARBs), culminating in increased activity of protein kinase A (PKA) and leading to phosphorylation of adipocyte lipases, such as hormone-sensitive lipase (HSL) and lipolysis, and of p38 mitogen-activated protein kinase (MAPK) and regulation of genes linked to lipid oxidation in brown adipocytes, including Ucp1 and Ppargc1a (peroxisome proliferator-activated receptor γ [PPARγ]-coactivator 1α) (Collins, 2012; Yehuda-Shnaidman et al., 2010). Though a process classically ascribed to BAT, during prolonged starvation or a cold challenge, white adipocytes undergo “beiging” or “browning,” in which WAT assumes increased UCP1-expressing adipocytes bearing thermogenic capacity (Kajimura et al., 2015; Lee et al., 2015b). Despite the profound importance of such metabolic plasticity, the natural “brakes” in adipose tissues—the best described of which is insulin (Burns et al., 1979; Jungas and Ball, 1963, 1964)—remain incompletely understood.
The immunoglobulin superfamily molecule, the receptor for advanced glycation end (RAGE) products, binds to a distinct repertoire of molecules, such as the carboxymethyllysine (CML)-advanced glycation end products (AGEs), high-mobility group box 1 (HMGB1), and S100/calgranulins, which accumulate in metabolic stress (López-Díez et al., 2016; Ramasamy et al., 2012). Although these ligands are classically linked to diabetes and inflammation, recent evidence places these molecules and RAGE itself in human and murine obese adipose tissue (Gaens et al., 2014; Song et al., 2014). RAGE localization in human obese adipose tissue aligns with our recent finding that mice globally devoid of Ager (the gene encoding RAGE) are protected from obesity and insulin resistance when fed a high-fat diet (HFD) compared to wild-type (WT) mice (Song et al., 2014). Yet, the mediating mechanisms have remained elusive.
Here, we show that mice bearing adipocyte-specific deletion of Ager display significant protection from HFD-induced obesity and insulin resistance and exhibit a superior ability to thermoregulate during a cold challenge, compared to mice in which adipocytes express Ager. WT mice that underwent a surgical transplantation of either interscapular BAT (iBAT) or subcutaneous inguinal WAT (iWAT) from mice with adipocyte-specific deletion of Ager demonstrate significant protection from HFD-induced obesity and insulin resistance, compared to WT mice transplanted with iBAT or iWAT in which adipocytes expressed Ager. We traced these adipocyte-intrinsic, RAGE-dependent mechanisms to RAGE ligand-mediated suppression of PKA-dependent phosphorylation of HSL and p38 MAPK—processes that, collectively, dampen UCP1 and thermogenic programs.
RESULTS
RAGE Is Expressed in Mature Adipocytes
We determined the expression patterns of RAGE in WAT and BAT in WT mice fed standard chow and found that Ager is expressed in BAT and WAT. Compared to epididymal adipose tissue (eWAT) or iWAT, a significantly higher expression of Ager mRNA was observed in iBAT (Figure 1A). In the iWAT and eWAT depots, a significantly higher expression of Ager mRNA transcripts was observed in the floating adipocytes than in the stromal vascular fraction (SVF) (Figure S1A). When preadipocytes from the SVF of iBAT, iWAT, and eWAT were differentiated into adipocytes, in each depot, a significantly higher expression of Ager mRNA was noted on days 3 or 8 of differentiation versus day 0, which paralleled time-dependent increases in Fasn, a marker for adipocyte differentiation (Figure 1B). There were no discernible differences in the morphology of iBAT-, iWAT-, or eWAT-derived primary adipocytes or in the neutral lipid content on day 7 of differentiation from eWAT-derived adipocytes (Figures S1B and S1C). Thus, increasing Ager expression accompanies, but is not required for, the differentiation of primary adipocytes from iBAT, iWAT, and eWAT.
Figure 1. RAGE Is Expressed in Adipose Tissue and Affects Thermogenic Gene Programs.

(A) qRT-PCR for relative Ager mRNA expression was performed in iBAT, iWAT, and eWAT retrieved from 12-week-old male wild-type (WT) C57BL/6 mice fed standard chow.
(B) Primary adipocytes from iBAT, iWAT, and eWAT of WT mice fed standard chow were subjected to qRT-PCR for detection of relative Ager and Fasn mRNA expression on day 0, 3, and 8 of differentiation.
(C and D) Core body temperature of 8-week-old male WT or Ager null mice determined in mice housed at (C) thermoneutrality (30°C) for 24 h and (D) at room temperature (~23°C).
(E) Oxygen consumption rates (OCRs) normalized to total protein were determined in primary adipocytes differentiated from iBAT from WT and Ager null mice. The means ± SEM are reported from five technical replicates with three biological replicates per group.
(F) iBAT and iWAT retrieved from 8-week-old WT or Ager null mice housed at room temperature were analyzed via qRT-PCR for detection of relative mRNA expression of Prdm16, Ppargc1a, Creb, Adrb3, Dio2, Cidea, Ucp1, Pnpla2, Fasn, Cebpa, Pparg, and Tfam.
Unless otherwise stated, the data are presented as mean ± SEM inN = 4–5 mice/group. Data analysis: (A and B) independent samples one-way ANOVA followed by a post hoc Tukey’s HSD test; (C and D) two-tailed Student’s t test; and (E, repeated measures, and F, independent samples) two-way ANOVA followed by post hoc Sidak test; *p < 0.05; **p < 0.01, and ***p < 0.001.
See also Figure S1.
Deletion of Ager Increases Adipocyte Thermogenic Gene Programs and Mitochondrial Activity
Although core body temperature at thermoneutrality (30°C) did not differ between the WT and Ager null mice (Figure 1C), at room temperature, mice devoid of Ager displayed a significantly higher core body temperature than the WT mice (Figure 1D). Body weight, adiposity, and plasma norepinephrine levels did not differ between the Ager null and the WT mice fed standard chow (Figures S1D-S1H).
As the highest expression of Ager in the adipose depots was in iBAT, we probed the effect of Ager deletion on mitochondrial function in iBAT-derived primary adipocytes from mice fed standard chow. Basal respiration rates and ATP production were significantly higher in adipocytes derived from Ager null than from WT mice (Figure 1E). The adipocytes derived from Ager null iBAT exhibited pronounced mitochondrial activity, as assessed by MitoTracker Red CMXRos (Koh et al., 2009) (Figure S1I). These findings suggest that RAGE contributes to the regulation of thermogenic programs in iBAT and to browning or beiging in iWAT.
To address this point, we retrieved adipose tissues from WT mice and mice globally devoid of Ager fed standard chow at room temperature. Ager deletion resulted in a significantly higher expression of thermogenic genes (Prdm16, Ppargc1a, Creb, Dio2, Cidea, and Ucp1) in iBAT and iWAT. Transcript levels of Adrb3 were significantly higher in iBAT, but not iWAT, of Ager null than of WT mice (Figure 1F). Regarding lipolysis (Pnpla2) and lipogenesis (Fasn, Cebpa, and Pparg), mRNA levels of these genes were significantly higher in Ager null than in WT iBAT and iWAT. In relation to mitochondrial properties, levels of Tfam in iBAT (Lidell et al., 2011), but not iWAT, were significantly higher in Ager null than in WT mice (Figure 1F), thus suggesting that mitochondrial biogenesis was higher in iBAT of Ager null than of WT mice.
Although eWAT is not primarily a thermogenic adipose tissue depot, we nevertheless examined the above markers. In eWAT, the levels of Creb, Dio2, Fasn, Cebpa, and Tfam were significantly higher in Ager null than in WT; there were no differences in Cidea, Ucp1, Pnpla, and Pparg; and the levels of Prdm16, Ppargc1a, and Adrb3 were significantly lower in the eWAT of Ager null than of WT mice (Figure S1J).
RAGE Dampens the Adaptive Response to Fasting and Cold Challenges
Our findings thus far indicate that the deletion of Ager upregulates adipocyte thermogenic gene programs in iBAT and iWAT. In adipocytes, β-adrenergic signals upregulate Ucp1 transcription, and, in part through lipolysis, the released fatty acids are proposed to enhance UCP1 activity in the mitochondria (Himms-Hagen et al., 2000; Rial et al., 1983). We tested these concepts in settings in which adaptive thermogenic programs are engaged.
First, we used a fasting-refeeding paradigm. After 24 h of fasting, WT mice displayed a significantly higher expression of Ager in iBAT, but not in iWAT. In iBAT, after 24 h of refeeding, the expression of Ager was significantly lower after fasting and returned to baseline (fed) levels (Figure 2A). Similar expression patterns were observed in eWAT under these conditions (Figure S2A). After 24 h of fasting, mice globally devoid of Ager displayed significantly less loss of body temperature than the WT mice did (Figure 2B). We examined the expression of Acox1 and Cpt2a, genes that are associated with fatty acid beta oxidation. In iBAT, levels of Acox1 and Cpt2a mRNA were significantly higher in the fasted Ager null than in the WT mice (Figure 2C), and in iWAT, significantly higher levels of Acox1 mRNA, but not Cpt2a mRNA, were observed in fasted mice devoid of Ager than in the WT mice (Figure 2D). In eWAT, there were no genotype-dependent differences in the fed or the fasted states (Figure S2B).
Figure 2. Effects of RAGE on Thermogenic Gene Expression during Fasting and Cold Challenge.

(A–D) Fasting and refeeding in 11-week-old male WT and Ager null mice.
(A) Ager expression in iBAT and iWAT from WT mice in a fed, fasted (24 h) or 24-h refed state.
(B) Change in core body temperature, [(Δ) temperature], between the fasted and the refed states was measured in WT or Ager null male mice housed at room temperature (RT).
(C and D) Relative mRNA expression of Acox1 and Cpt2a was measured by qRT-PCR in (C) iBAT and (D) iWAT from fed and fasted (24 h) WT and Ager null mice.
(E–I) Eight-week-old male WT or Ager null mice were exposed to a cold challenge (4°C).
(E and F) iBAT (E) and iWAT (F) from WT mice were subjected to qRT-PCR for relative Ager mRNA expression.
(G) (Δ) Temperature from baseline (room temperature) and after 48 h at 4°C is shown.
(H and I) WT or Ager null mice were housed at room temperature (~23°C) or exposed to a 4°C cold challenge for 6 or 12 h. iBAT (H) and iWAT (I) were dissected after and subjected to qRT-PCR for detection of relative Ucp1 mRNA expression.
In (B)–(D) and (G)–(I): WT (black bars) and Ager null (green bars) mice. Data are presented as mean ± SEM in N = 4–6 mice/group. Data analysis: (A, E, and F) one-way ANOVA with post hoc Tukey’s HSD test; (B and G) two-tailed Student’s t test; (C, D, H, and I) two-way ANOVA followed by a post hoc Sidak test or Tukey’s test, as appropriate; *p < 0.05; **p < 0.01, and ***p < 0.001.
See also Figure S2.
As an alternative paradigm to induce lipolysis and thermogenic programs, we performed a cold stress (Duncan et al., 2007). Compared to room temperature, the cold challenge resulted in a significantly increased expression of Ager in iBAT after 6 or 12 h at 4°C. (Figure 2E). In iWAT and eWAT, Ager expression was significantly higher after 12 h of exposure to 4°C, compared to room temperature (Figures 2F and S2C). To assess whether upregulation of Ager in iBAT and iWAT in mice subjected to the 4°C cold challenge affected thermoregulation in vivo, we assessed their core body temperature after 48 h of exposure to 4°C. Mice devoid of Ager exhibited significantly less loss of body temperature than did WT mice over 48 h (Figure 2G). In iBAT, at 6 or 12 h of exposure to 4°C, levels of Ucp1 mRNA did not differ between Ager null and WT mice; at room temperature, levels of Ucp1 mRNA transcripts were significantly higher in Ager null than in WT iBAT (Figure 2H). In iWAT, levels of Ucp1 mRNA were significantly higher in Ager null mice at room temperature and after 6 or 12 h in 4°C (Figure 2I). In eWAT, levels of Ucp1 were significantly lower in Ager null than in WT mice at room temperature and after 6 or 12 h at the 4° C cold challenge (Figure S2D). Thus, deletion of Ager protects against the loss of body temperature in mice subjected to the 4°C environment, which was accompanied by RAGE-dependent regulation of Ucp1 in iWAT.
Adipocyte-Specific Deletion of Ager Protects from Diet-Induced Obesity and Cold Challenge
In addition to adipocytes, immune and vascular cells and sympathetic fibers populate adipose tissue and might contribute to the impact of RAGE expression. To definitively probe for roles for RAGE in adipocyte-intrinsic responses to the HFD and cold challenge, we generated Ager floxed mice (Figures S3A and S3B). Agerflox/flox mice were bred with adiponectin (Adipoq) Cre-recombinase mice to yield Agerflox/flox Adipoq Cre (+) (adipocyte-specific Ager-deleted mice) and littermate Agerflox/flox Adipoq Cre (−) (Ager WT) mice. This resulted in the deletion of Ager from iWAT, eWAT, and iBAT, but not from the pancreas, muscle, and liver (Figure S3C). Hereafter, these animals will be referred to as Agerflox/flox Cre (+) and Agerflox/flox Cre (−) mice.
We phenotyped male littermate Agerflox/flox Cre (+) and Agerflox/flox Cre (−) mice at baseline while they were fed a standard chow diet and then after 6 weeks of HFD feeding (60% kcal/fat). At baseline, body weight and composition, average food intake, physical activity, and energy expenditure rates were indistinguishable between Agerflox/flox Cre (+) (green lines) and Agerflox/flox Cre (−) (red lines) mice (Figures 3A-3E). However, when fed a HFD, Agerflox/flox Cre (+) mice weighed significantly less than Cre (−) mice (Figure 3A). Whole body fat mass, measured using 1H-MRS, was significantly lower in the Agerflox/flox Cre (+) than in Cre (−) mice after 3 and 6 weeks of the HFD (Figure 3B). Average food consumption did not differ by genotype, except after 6 weeks of the HFD, in which lower food consumption was noted in Agerflox/flox Cre (+) than in Cre (−) mice (Figure 3C). No genotype-dependent differences in physical activity were noted at baseline or after 3 and 6 weeks of the HFD (Figure 3D). Agerflox/flox Cre (+) mice displayed significantly higher energy expenditure rates than Cre (−) mice after 3 weeks of the HFD, with trends in the same direction at 6 weeks of the HFD (Figure 3E). No differences in glucose-tolerance testing (GTT) were observed after 12 weeks of the HFD (Figure 3F). Agerflox/flox Cre (+) mice exhibited significantly greater insulin sensitivity than Cre (−) mice during an insulin-tolerance test (ITT) after 12 weeks of the HFD (Figures 3G and S3D).
Figure 3. Adipocyte-Specific Deletion of Ager Affects HFD-Induced Obesity and Thermoregulatory Responses to a Cold Challenge.

(A–G) Eight-week-old male Agerflox/flox Adipoq Cre (−) (red) (Agerflox/flox Cre (−)) and Agerflox/flox Adipoq Cre (+) (green) (Agerflox/flox Cre (+)) mice were fed a HFD (60% kcal/fat).
(A) Body weight gain after up to 6 weeks of high-fat feeding was recorded.
(B–E) Body composition (B), average food intake (C), physical activity (D), and energy expenditure (E) were assessed in mice fed standard chow (baseline) or after 3 or 6 weeks of a HFD. In (A)–(E), the mean ± SEM is reported in N = 8 mice/group.
(F and G) Glucose (F) and insulin tolerance (G) tests were performed after 12 weeks of a HFD. The mean ± SEM is reported in N = 4–5 mice/group.
(H–K) Eight-week-old male Agerflox/flox Cre (−) and Agerflox/flox Cre (+) mice were housed for 24 h at 4°C.
(H and I) (Δ) Temperature (body temperature at room temperature versus 24 h of 4°C) (H) and change in body weight (I) are reported.
(J and K) Relative mRNA expression of Ucp1, Dio2, Ppargc1a, and Cidea mRNA was assessed in (J) iBAT and (K) iWAT retrieved from mice housed at room temperature or after termination of a 24-h cold challenge (4°C).
In (F)–(K), the mean ± SEM is reported in N = 4–5 mice/group. In (A)–(K), Agerflox/flox Cre (−) shown as red and Agerflox/flox Cre (+) shown as green. Data analysis: (A–G, repeated measures, and J and K, independent samples) two-way ANOVA followed by a post hoc Bonferroni test; (H and I) two-tailed Student’s t test; *p < 0.05; **p < 0.01, ***p < 0.001, and ****p < 0.0001.
See also Figure S3.
We determined if a modulation of adipose inflammation accompanied the metabolic protection observed in Agerflox/flox Cre (+) mice after the HFD. In eWAT, but not iWAT, mRNA levels of Emr1 (F4/80) and Cc/2 were significantly lower in Agerflox/flox Cre (+) than in Cre (−) mice (Figure S3E). No differences in Tnfa or Il10 expression were observed in eWAT or iWAT between the genotypes (Figure S3E). Transcript levels for Irf4, which increases adipocyte lipolysis (Eguchi et al., 2011), were significantly higher in the eWAT, iWAT, and iBAT of Agerflox/flox Cre (+) mice than Cre (−) mice (Figure S3E). Tlr4, which has been linked to endotoxin-mediated stimulation of lipolysis in adipocytes (Zu et al., 2009), was significantly higher in eWAT, iWAT, and iBAT of Agerflox/flox Cre (+) than Cre (−) mice (Figure S3E), and levels of Tlr2 mRNA were significantly higher in eWAT and iWAT, not iBAT, of Agerflox/flox Cre (+) than of Cre (−) mice (Figure S3E). Thus, decreased adipose inflammation accompanied the observed protection in Agerflox/flox Cre (+) mice from diet-induced obesity and insulin intolerance.
We next exposed littermate Agerflox/flox Cre (+) and Cre (−) mice to a cold challenge. Compared to Agerflox/flox Cre (−) mice, Cre (+) mice exhibited significant protection from a loss of body temperature after 24 h of exposure to 4°C (Figure 3H), with no genotype-dependent differences observed in body weight changes (Figure 3I). We examined the expression of genes associated with thermogenic responses in iBAT and iWAT after cold exposure. In iBAT, there were no genotype-dependent differences in Ucp1, Dio2, Ppargc1a, or Cidea during the cold challenge, and only significantly lower Ucp1 was observed in Agerflox/flox Cre (+) at baseline (Figure 3J). In iWAT, after 24 h of 4°C, levels of Ucp1, Dio2, Ppargc1, and Cidea mRNA were significantly higher in Agerflox/flox Cre (+) than in Cre (−) mice (Figure 3K). In eWAT, levels of Ucp1 were significantly higher in Agerflox/flox Cre (+) than in Cre (−) mice at 4°C; no genotype-dependent differences were observed in Ppargc1a mRNA at 4°C but the levels of Ucp1 and Ppargc1a were significantly higher at room temperature in the Agerflox/flox Cre (+) than the Cre (−) mice (Figure S3F). These data support adipocyte-intrinsic roles for RAGE in adaptive thermogenic gene program responses after the HFD and 4°C cold challenge.
Transplantation of iBAT or iWAT from Agerflox/flox Cre (+) Mice into WT Mice Reduces Body Weight Gain and Upregulates Thermogenic Gene Programs in High-Fat Feeding
As the Adipoq Cre-recombinase strategy employed above affected levels of Ager in both brown and white adipocytes, the relative contribution of each depot could not be discerned. To address this point, we performed an adipose tissue surgical transplantation of iBAT bearing adipocyte-specific deleted Ager or adipocyte-expressing Ager into lean, young, WT mice. Figure S4A is a representative image demonstrating a well-vascularized, non-necrotic iBAT graft. After 4–5 days’ recovery from surgery, mice were switched to a HFD and were monitored over 20 weeks. Beginning at 4 weeks post-HFD, body weights in WT mice receiving Agerflox/flox Cre (+) iBAT were significantly lower than those of mice receiving Agerflox/flox Cre (−) iBAT, an observation that continued and was magnified throughout the HFD feeding period (Figures 4A and 4B). Fat mass was significantly lower in the WT mice receiving Agerflox/flox Cre (+) versus Cre (−) iBAT after 14 weeks of the HFD (Figure 4C). Although there were no differences in food intake or physical activity between the Agerflox/flox Cre (+) and Cre (−) mice iBAT donors (Figures 4D and 4E), energy expenditure was significantly higher in the mice receiving Agerflox/flox Cre (+) than those receiving Cre (−) iBAT after 16 weeks of the HFD (Figure 4F). After 12 weeks of the HFD, WT mice receiving Agerflox/flox Cre (+) iBAT were more glucose tolerant than mice receiving Cre (−) iBAT (Figure 4G). Insulin sensitivity was improved in mice receiving the Agerflox/flox Cre (+) than the Cre (−) iBAT (Figures 4H and S4B).
Figure 4. Surgical Transplantation of Adipocyte Ager-Deficient iBAT Affects HFD-Induced Metabolic Impairment in WT Mice.

(A) Representative photograph of mice surgically transplanted with iBAT from either Agerflox/flox Cre (−) (red, left) or Agerflox/flox Cre (+) iBAT (green, right) after 20 weeks on a HFD.
(B) Body weight curves through 12 weeks on a HFD.
(C) Body composition after 14 weeks of a HFD.
(D–F) Average daily food intake (D), physical activity (E), and energy expenditure (F) were assessed after 16 weeks on a HFD. For (B)–(F), the mean ± SEM is reported in N = 15 mice/group.
(G and H) Glucose (G) and insulin tolerance (H) tests were performed after 12 or 13 weeks on a HFD, respectively. The mean ± SEM is reported in N = 10–11 mice/group.
(I–M) Thermogenic programs.
(I, K, and M) The relative mRNA expression by qRT-PCR for Ucp1, Dio2, Ppargc1a, Cpt2a, and Pnpla2 for (I) native iBAT, (K) native iWAT, and (M) transplanted iBAT (tiBAT) retrieved from the WT recipient mice after 20 weeks on a HFD is reported in N = 4–6 mice/group.
(J and L) In (J, native iBAT) and (L, native iWAT), immunohistochemistry for the detection of UCP1 was performed and image analysis employed to quantify UCP1-positive area; representative images are shown at 20× magnification. The mean ± SEM is reported in N = 5 mice/group. Scale bar: 100 μm.
(A–M) Source of donor iBAT: Agerflox/flox Cre (−) shown as red and Agerflox/flox Cre (+) shown as green. Data analysis: (B, G, and H, repeated measures, and C, independent samples) two-way ANOVA followed by a post hoc Bonferroni test; (D and F–H, area under the curve, and I–M) two-tailed Student’s t test. Where group mean variances were statistically different (p < 0.05), data were analyzed post hoc using the non-parametric Mann-Whitney U test; *p < 0.05; **p < 0.01, ***p < 0.001, and ****p < 0.0001.
See also Figure S4.
We retrieved native iBAT and iWAT, transplanted iBAT (tiBAT) and eWAT, and assessed the expression of thermogenic genes after 20 weeks of a HFD. In native iBAT, the levels of Dio2, Cpt2a, and Pnpla2 mRNA levels were significantly higher in WT mice receiving Agerflox/flox Cre (+) than those receiving Cre (−) iBAT, while levels of Ucp1 and Ppargc1a mRNA were generally, but not significantly, higher (Figure 4I). Immunohistochemistry revealed that the native iBAT of mice receiving Agerflox/flox Cre (+) versus Cre (−) iBAT transplants displayed a significantly higher UCP1-positive area (Figure 4J). In native iWAT, there were no significant differences in levels of Ucp1, Dio2, Ppargc1a, Cpt2a, or Pnpla2 mRNA in WT mice receiving Agerflox/flox Cre (+) versus Cre (−) iBAT transplants (Figure 4K). However, immunostaining revealed that the native iWAT of mice that were transplanted with Agerflox/flox Cre (+) iBAT displayed significantly higher UCP1-positive areas than mice that had been transplanted with Cre (−) iBAT (Figure 4L). In the transplanted iBAT, levels of Ucp1, Dio2, Ppargc1a, Cpt2a, and Pnpla2 mRNA were significantly higher in transplanted iBAT originating from Agerflox/flox Cre (+) than from Cre (−) mice (Figure 4M). In native eWAT, levels of Ucp1, Ppargc1a, Cpt2a, and Pnpla2 mRNA transcripts were significantly higher in the mice receiving Agerflox/flox Cre (+) than those receiving Cre (−) iBAT, while levels of Dio2 mRNA transcripts were significantly lower (Figure S4C). Figure S4D is a representative image of the secondary antibody-alone control for the UCP1 immunostaining.
We next transplanted iWAT expressing or devoid of adipocyte Ager into young, lean, WT mice and established the presence of vascularized non-necrotic grafts (representative image; Figure S5A). Four days after the transplantation, mice were switched to a HFD for an additional 20 weeks. Compared to recipients of Agerflox/flox Cre (−) iWAT, WT mice receiving Agerflox/flox Cre (+) iWAT displayed significantly less weight gain after 12 weeks of the HFD (Figures 5A and 5B). The WT mice recipients of Agerflox/flox Cre (+) iWAT displayed significantly less fat mass than recipients of Cre (−) iWAT (Figure 5C). No differences in food intake or physical activity were observed by iWAT donor genotype (Figures 5D and 5E). As with iBAT transplants, significantly higher energy expenditure was observed in WT recipients of Agerflox/flox Cre (+) than Cre (−) iWAT after 16 weeks of the HFD (Figure 5F). The WT mice receiving Agerflox/flox Cre (+) versus Cre (−) iWAT displayed superior glucose tolerance (Figure 5G) and insulin sensitivity while on the HFD (Figures 5H and S5B).
Figure 5. Surgical Transplantation of Adipocyte Ager-Deficient iWAT Affects HFD-Induced Metabolic Impairment in WT Mice.

(A) Representative photographs from mice receiving surgical transplantation of either Agerflox/flox Cre (−) (red, left) or Agerflox/flox Cre (+) iWAT (green, right) are shown after 20 weeks on a HFD.
(B) Body weights through 12 weeks of a HFD.
(C) Body composition after 14 weeks of a HFD.
(D–F) Average daily food intake (D), physical activity (E), and energy expenditure (F) were assessed after 16 weeks of a HFD. In (B)–(F), the mean ± SEM is reported in N = 7–9 mice/group.
(G and H) Glucose (G) and insulin tolerance (H) tests were performed after 12 and 13 weeks of a HFD, respectively. The mean ± SEM is reported in N = 5–6 mice/group.
(I–M) Thermogenic programs.
(I, K, and M) The relative mRNA expression by qRT-PCR for Ucp1, Dio2, Ppargc1a, Cpt2a, and Pnpla2 mRNA transcripts in (I) native iBAT, (K) native iWAT, and (M) transplanted iWAT (tiWAT) was examined, and the mean ± SEM is reported in N = 3–4 mice/group.
(J and L) In (J, native iBAT) and (L, native iWAT), immunohistochemistry for detection of UCP1 protein was performed and image analysis employed to quantify UCP1-positive area; representative images at 20× magnification are shown. The mean ± SEM is reported in N = 5 mice/group. Scale bar: 100 μm.
(A–M) Source of donor iWAT: Agerflox/flox Cre (−) shown as red and Agerflox/flox Cre (+) shown as green. Data analysis: (B, G, and H, repeated measures, and C, independent samples) two-way ANOVA followed by a post hoc Bonferroni test; (D–H, area under the curve, and I–M) two-tailed Student’s t test. Where group mean variances were statistically different (p < 0.05), data were analyzed post hoc using the non-parametric Mann-Whitney U test; *p < 0.05; **p < 0.01, ***p < 0.001, and ****p < 0.0001.
See also Figure S5.
After 20 weeks of the HFD, in native iBAT, WT mice receiving Agerflox/flox Cre (+) versus Cre (−) iWAT displayed a significantly higher expression of Ppargc1a and Pnpla2 mRNA transcripts, with no statistically significant differences in levels of Ucp1, Dio2, or Cpt2a mRNA transcripts (Figure 5I). Immunohistochemistry revealed that the native iBAT of mice that were transplanted with Agerflox/flox Cre (+) versus Cre (−) iWAT displayed a significantly higher UCP1-positive area (Figure 5J). In native iWAT, WT mice receiving Agerflox/flox Cre (+) versus Cre (−) iWAT displayed significantly higher levels of Ucp1, Dio2, and Pnpla2 mRNA transcripts; significantly lower levels of Ppargc1a mRNA; and no differences in Cpt2a mRNA transcripts (Figure 5K). Immunohistochemistry revealed that there were no significant differences in the UCP1-positive area in the iWAT of mice that had been transplanted with Agerflox/flox Cre (+) or Cre (−) iWAT (Figure 5L). In the tiWAT, the WT mice recipients of Agerflox/flox Cre (+) versus Cre (−) iWAT displayed significantly higher levels of Ucp1 and Pnpla2 mRNA transcripts, a trend to higher levels of Dio2 expression, lower levels of Ppargc1a, and no significant differences in Cpt2a mRNA transcripts (Figure 5M). In native eWAT, significantly higher levels of Ucp1, Ppargc1a, and Pnpla2 mRNA transcripts were observed in the recipients of Agerflox/flox Cre (+) than of Cre (−) iWAT, with no statistically significant differences in Dio2 or Cpt2a mRNA transcripts (Figure S5C). Thus, the deletion of Ager in iBAT or iWAT adipocytes transmits protection from diet-induced obesity and insulin and glucose intolerance and enhances expression of thermogenic genes.
To assess the effects of RAGE ligand sequestration, we previously treated WT mice fed a HFD for 18 weeks with soluble RAGE (sRAGE), the extracellular ligand-binding domains of RAGE, which act as a decoy receptor and thus suppress RAGE signaling. When treatment was given concurrently with a HFD, compared to vehicle-treated mice, mice treated with sRAGE displayed less body weight gain (Song et al., 2014). Compared to vehicle-treated mice, in the iWAT of sRAGE-treated mice, we noted the following: expression of Ucp1 mRNA was significantly higher; expression of Cidea was significantly higher in the eWAT, iWAT, and iBAT; expression of Ppargc1a mRNA was significantly higher in the eWAT and iBAT; and expression of Dio2 mRNA was significantly higher in eWAT (Figures S6A-S6D). Immunohistochemistry revealed more pronounced UCP1 immunoreactive epitopes in the eWAT, iWAT, and iBAT of sRAGE-treated mice (Figure S6E). Thus, both pharmacological and genetic approaches indicate that RAGE suppresses adipose tissue thermogenic programing.
RAGE Suppresses γ3-Adrenergic Stimulation of Lipolysis and Ucp1 mRNA in Adipocytes
Compared to thermoneutrality, exposure to 4°C results in significant increases in sympathetic nerve innervation to the eWAT and iWAT (Castro etal., 2017). As β-adrenergic stimulation of adipocytes is a primary mechanism for lipolysis and the generation of fatty acid products that regulate Ucp1 expression and activity (Cannon and Nedergaard, 2004; Collins and Surwit, 2001; Rial et al., 1983), we tested whether RAGE plays a role in this pathway in primary and cultured adipocytes.
We differentiated iBAT- and iWAT-derived adipocytes from WT and Ager null mice and stimulated them with CL316,243 (CL) (ARB3-specific agonist) to test the effects on lipolysis. In iBAT adipocytes, significantly higher CL316,243-stimulated levels of glycerol, and both basal and CL316,243-stimulated levels of non-esterified fatty acids (NEFAs), were observed in Ager null than in WT mice (Figure 6A). In iWAT adipocytes, basal and CL316,243-stimulated glycerol levels were significantly higher in adipocytes derived from Ager null mice, as was the CL316,243-mediated stimulation of the NEFA release (Figure 6B). Significantly higher oxygen consumption rates (OCRs) were observed in primary iBAT adipocytes from Ager null than from WT mice after treatment with isoproterenol (non-specific ARB agonist), which was minimally suppressed by oligomycin in the Ager null adipocytes, suggestive of maximum mitochondria uncoupling (Figure 6C). When primary iBAT adipocytes were treated with vehicle or norepinephrine (NE) + T3 (thyroid hormone), significantly higher Ucp1 mRNA transcripts were observed in adipocytes devoid of Ager versus WT at baseline and after NE/T3 (Figure 6D). Primary iBAT-derived adipocytes from Ager null versus WT mice displayed significantly higher levels of UCP1 (Figure 6E). These data point to adipocyte-intrinsic roles for RAGE in β-adrenergic stimulation of lipolysis and upregulation of Ucp1 expression and activity.
Figure 6. RAGE Affects β-Adrenergic-Stimulated Adipocyte Lipolysis and Thermogenic Programming.

Primary adipocytes were prepared from the SVF of 8-week-old WT and Ager null mice fed standard chow.
(A and B) iBAT- (A) and iWAT- (B) derived cells were incubated for 3 h in Krebs-Ringer bicarbonate buffer (KRB) and subjected to vehicle or CL316,243 (CL) (10 μM) for 3 h; glycerol and non-esterified fatty acids (NEFAs) in the media were measured. The means normalized to total protein per well ± SEM are reported from triplicate, independent experiments from cells derived from N = 5 mice/group.
(C) A representative OCR curve for iBAT-derived adipocytes during a mitochondrial stress test is shown under basal condition and following isoproterenol (10 μM). This experiment was performed three times using cells derived from N = 3 mice/group per experiment.
(D) Primary iBAT-derived adipocytes from iBAT of WT or Ager null mice were stimulated with norepinephrine (NE; 5 μM) and T3 (thyroid hormone, 2 nM) for 6 h, and relative Ucp1 mRNA expression was determined by qRT-PCR. The mean ± SEM is reported in N = 4 mice/group.
(E) Western blotting was performed for detection of UCP1/TUBULIN from primary iBAT adipocytes of 8-week-old WT and Ager null mice. The mean ± SEM is reported in N = 6 independent cell lysates per group along with representative blots.
(F and G) Primary adipocytes from (F) iBAT and (G) iWAT of WT mice were incubated with vehicle or the RAGE ligand CML-AGE (300 μg/ml) for 16 h alone or with NE (5 μM) for the final 6 h. qRT-PCR for detection of relative Ucp1 or Ppargc1a mRNA expression was performed. The mean ± SEM is reported from N = 5 mice/group.
(H) Cell lysates from primary adipocytes from iBAT of WT mice treated as in (F) and (G) were used for the detection of UCP1/TUBULIN. The mean ± SEM is reported in cell lysates from N = 3 mice/group.
(I) Schematic of Ucp1 promoter with enhancer elements. Undifferentiated C3H10T1/2 cells were transfected with mouse 3.1 kB Ucp1 promoter luciferase construct (Ucp1-luc) and stimulated for the last 6 h with NE(5 μM) and/or CML-AGE (300 μg/ml) for 16 h. Normalized luciferase activities are shown as fold-change compared to vehicle control.
(J) Ucp1 promoter luciferase constructs were generated and transfected into undifferentiated C3H10T1/2 cells and the cells cultured in medium containing NE (5 μM) for the last 6 h or CML-AGE (300 μg/ml) for 16 h. Normalized luciferase activities are shown relative to vehicle control. The mean ± SEM of three independent experiments is presented.
(K) PKA activity was determined in primary iBAT adipocytes from WT mice treated with CL316,243 (10 μM) for 15 min alone or after pre-incubation with the RAGE ligand CML-AGE (300 μg/ml) for 16 h. The mean ± SEM of N = 8 mice/group is presented.
In (A)–(E), WT (black) and Ager null (green). Data analysis: (A–D, repeated measures) two-way ANOVA followed by post hoc Bonferroni test; (E and H) two-tailed Student’s t test; (F, G, and I–K) one-way ANOVA followed by a post hoc Tukey’s HSD test; *p < 0.05, **p < 0.01, and ***p < 0.001.
See also Figure S6.
RAGE and its ligand, carboxymethyllysine (CML)-AGE, are highly expressed in obese human adipose tissue (Gaens et al., 2014). We previously showed that CML-AGEs were specific ligands of RAGE (Kislinger et al., 1999). WT primary adipocytes were treated with vehicle or NE, alone or with CML-AGE. In the basal state and upon treatment with NE, incubation with CML-AGE resulted in a significant suppression of Ucp1 and Ppargc1a mRNA expression in primary adipocytes from iBAT and iWAT (Figures 6F and 6G). In eWAT-derived adipocytes, CML-AGE treatment decreased Ucp1 and Ppargc1a at baseline and significantly suppressed the NE-stimulated induction of Ucp1 with a similar trend noted for Ppargc1a (Figure S6F). In iBAT-derived adipocytes, treatment with CML-AGE significantly decreased the expression of UCP1 (Figure 6H).
To determine if the effects of RAGE ligands were via direct action on Ucp1 transcription, we employed Ucp1 promoter luciferase constructs (Figure 6I) transfected into C3H10T1/2 cells. Compared to the vehicle, treatment with NE resulted in a significant increase in Ucp1 promoter luciferase activity, which was suppressed by CML-AGE (Figure 6I). We prepared mutants of the Ucp1 promoter in which only the proximal promoter, which possesses functional CRE-binding elements, or the enhancer elements, were expressed (Figure 6J). Treatment with CML-AGE exerted only modest effects on the suppression of basal but not NE-mediated promoter luciferase activities, indicating that the entire Ucp1 promoter was required to detect the effects of CML on transcriptional activity. On account of central roles for PKA in the β-adrenergic stimulation of thermogenic gene programs in adipocytes, we tested the effects of CML-AGE on PKA activity. In primary iBAT-derived adipocytes treated with CL316,243 and CML-AGE, a significant attenuation in PKA activity was observed (Figure 6K).
RAGE Ligands Suppress Phosphorylation of PKA Pathway Targets, Phosphorylated Hormone Sensitive Lipase (pHSL) Ser563 and p38 MAP Kinase, upon β3-Adrenergic Stimulation
To trace the fate of lipolysis and signaling pathways downstream of PKA that regulate adaptive thermogenesis, we focused on two PKA targets: HSL and p38 MAP kinase (MAPK) (Collins, 2012). Based on the discovery that the cytoplasmic domain of RAGE is essential for RAGE signaling, in part through its binding to DI-APH1 (Hudson et al., 2008; Touré et al., 2012), we recently described the small molecule antagonist, 2- [4- (acetylamino) phenyl] −4- Quinolinecarboxylic acid, methyl ester (“RAGE inhibitor”), which binds to the cytoplasmic domain of RAGE, blocks its interaction with DIAPH1, and suppresses RAGE ligand-stimulated signal transduction in cellular and murine models (Manigrasso et al., 2016). Significantly higher levels of glycerol release were observed in C3HT101/2 cells treated with CL316,243 and the RAGE inhibitor (RI) versus CL316,243 alone (Figure 7A). The RI had no independent effect on the numbers of lipid droplets, as assessed by staining with BODIPY (Figure 7B), nor did it affect the relative mitochondrial DNA content (Figure S7A).
Figure 7. Effects of RAGE Signal Transduction on the PKA Signaling Pathway, Adipocyte Lipolysis, and Phosphorylation of p38 MAP Kinase.

(A) Differentiated adipocytes from C3H10T1/2 cells were serum starved for 3 h and pretreated with the RAGE inhibitor (2- [4- (acetylamino) phenyl] −4- Quinolinecarboxylic acid, methyl ester) (1 μM) for 1 h prior to CL316,243 (10 μM) for 15 min. The mean glycerol content normalized to total protein per well ± SEM is reported from three independent experiments.
(B) C3H10T1/2 cells were differentiated to adipocytes and pre-incubated with a vehicle or the RAGE inhibitor (1 μM) for 90 min prior to CL316,243 (10 μM). Lipid droplets were stained using BODIPY and nuclei stained with DAPI. A representative fluorescence micrograph and mean ± SEM is shown. Scale bar: 100 μm. (C and D) C3H10T1/2 cells were differentiated and treated with a vehicle or CML-AGE (300 μg/ml) for 75 min alone or after pre-treatment with the RAGE inhibitor (1 μM) for 30 min followed by CL316,243 (10 μM) treatment for 15 min.
(C) Cells were lysed and western blotting performed for phosphorylated HSL Serine563, total HSL, phosphorylated p38 MAPK, total p38 MAPK, and GAPDH.
(D) Differentiated adipocytes from C3H10T1/2 cells were treated as in (C) with or without the addition of H89 (20 μM) for 30 min prior to CL316,243 (10 μM). Cells were lysed and western blotting performed for phosphorylated HSL Serine563, total HSL, phosphorylated p38 MAPK, total p38 MAPK, and GAPDH. Band intensities were normalized to the respective total HSL or total p38 MAPK, and the relative fold change is presented as mean ± SEM. Each experiment consisted of at least three independent studies with at least three technical replicates.
(E–H) Primary adipocytes from (E and G) iBAT or (F and H) iWAT of WT and Ager null mice were treated with the RAGE ligand CML-AGE (300 μg/ml) for 1 h, CL316,243 (10 μM) for 15 min, and with or without H89 (20 μM) for 30 min prior to CL. Cells were lysed and western blotting performed for the detection of phosphorylated HSL Serine563, total HSL, phosphorylated p38 MAPK, total p38 MAPK, and GAPDH. Band intensities were normalized to the respective total HSL or p38 MAPK, and the relative fold change is presented as mean ± SEM from N = 4 pooled mice/group in three independent experiments with at least 2–3 technical replicates per experiment.
Data analysis: (A–H) one-way ANOVA followed by a post hoc Tukey’s HSD test or Bonferroni test, as indicated; *p < 0.05; **p < 0.01, ***p < 0.001, and ****p < 0.0001.
See also Figure S7.
We assessed the effects of RAGE on phosphorylation of HSL Serine563 and p38 MAPK. Treatment of C3HT101/2 cells with CL316,243 resulted in significantly higher phospho/total pHSL Serine563 and phospho/total p38 MAPK compared to vehicle, which were both significantly reduced by treatment with CML-AGE. However, in cells treated with CL316,243 and CML-AGE, treatment with the RI resulted in significantly higher phospho-p38/total p38 MAPK and trended to higher phospho-HSL Serine563/total HSL, compared to treatment with CL316,243 and CML-AGE alone (Figure 7C). We hypothesized that if these effects of RAGE were through a blockade of phosphorylation of PKA targets, then direct catalytic inhibition of PKA should blunt the effects of the RI. First, we tested the efficacy of H89, a commonly used compound to inhibit PKA activity, on the suppression of the effects of CL316,243 (Chijiwa et al., 1990). In C3HT101/2 cells, treatment with H89 blocked the increased phosphorylated HSL Serine563/total HSL ratio and the increased phosphorylated p38/total p38 MAPK induced by CL316,243 (Figure S7B). We then tested if H89 blocked the effects of the RI. Compared to cells treated with CL316,243, CML-AGE, and the RI, the addition of H89 resulted in a significant reduction in phosphorylation of both HSL Serine563 and p38 MAPK (Figure 7D). Thus, direct catalytic inhibition of PKA blunted the effects of the RI on rescuing the suppressive effects of CML-AGE on the phosphorylation of pHSL Serine563 and p38 MAPK.
We next tested primary adipocytes from iBAT and iWAT. In WT, but not in Ager null primary adipocytes derived from iBAT (Figure 7E) and iWAT (Figure 7F), treatment with CML-AGE in CL316,243-treated cells resulted in significant suppression of phosphorylation of pHSL Serine563/total HSL and p38/total p38 MAPK. In primary iBAT- and iWAT-derived adipocytes, treatment with CL316,243 resulted in significantly increased phosphorylated HSL Serine563/total HSL and increased phosphorylated p38/total p38 MAPK, which was significantly attenuated by H89 (Figure S7C). Treatment of Ager null adipocytes from iBAT or iWAT with CL316,243, CML-AGE, and H89 resulted in significant decreases in phosphorylated HSL Serine563/total HSL and phosphorylated p38/total p38 MAP kinase, compared to cells treated with CL316,243 and CML-AGE alone (Figures 7G and 7H). Thus, direct catalytic inhibition of PKA significantly reduced the effects of deletion of Ager on rescuing the suppression of CML-AGE on phosphorylation of these two PKA targets. In vivo, expression of Lipe (gene encoding HSL), a target of activated PKA (Holm, 2003), was significantly higher in the floating adipocyte fraction retrieved from the iWAT of Ager null than from the WT mice (Figure S7D).
On account of the demonstrated roles for a PKA-independent, β3-adrenergic-mediated stimulation of extracellular signal-related kinase (ERK) MAPK on lipolysis in Robidoux et al. (2006), we tested the effects of the RAGE ligand, CML-AGE. In C3H10T1/2 cells, treatment with CL316,243 resulted in significantly increased phospho/total ERK MAPK, which was significantly suppressed by CML-AGE; the suppressive effects of CML-AGE were prevented by treatment with the RI (Figure S7E). However, in primary WT or Ager null adipocytes retrieved from iBAT or iWAT, no significant effects of CML-AGE on phospho/total ERK MAPK in CL316,243-treated cells were observed (Figure S7F).
There were no Ager-dependent differences in the protein levels of two other lipolytic molecules, adipose triglyceride lipase or monoacylglycerol lipase, upon treatment with CL316,243 (Figures S7G and S7H), and there were no genotype-dependent differences in the levels of cyclic AMP (cAMP) between WT and Ager null CL-316,243-treated primary adipocytes from iBAT, iWAT, or eWAT (Figure S7I).
DISCUSSION
The tethering of the biology of molecules implicated in inflammation to the regulation of metabolism underscores the significance of these two primal processes to drive and ensure organismal survival. Here, we demonstrate that the immunoglolubin superfamily molecule, RAGE, suppresses adaptive thermogenesis in both BAT and WAT, at least in part by reducing the effects of β-adrenergic signaling in adipocytes, through suppression of the phosphorylation of PKA targets. In contrast to the suppressive effects of RAGE on metabolic responses, distinct genes implicated in the inflammatory response positively regulate adipocyte lipolysis and expression of Ucp1. For example, IRF4 is induced in fasting, and experiments testing adipocyte-specific deletion of Irf4 (Lee et al., 2013) uncovered its roles in the positive regulation of adipocyte lipolysis (Eguchi et al., 2011; Kumari et al., 2016). Further, a role for toll-like receptor 4 (TLR4) in endotoxin-stimulated regulation of lipolysis in adipocytes was established (Zu et al., 2009) and shown to be independent of cAMP-PKA but dependent on the ERK1/2 signal transduction pathway. The observation that levels of Irf4 and Tlr4 were significantly higher in adipose tissues of HFD-fed mice bearing adipocyte-specific deletion of Ager versus animals expressing Ager in adipocytes suggests that RAGE may contribute, directly or indirectly, to the regulation of these genes in adipocytes.
What are the signals that trigger these events in the RAGE pathway? Many of the families of the RAGE ligands have “double lives,” in which their intracellular regulatory functions contrast with unique roles in the extracellular zones as “damage-associated molecular pattern (DAMP)” molecules (Bertheloot and Latz, 2017; Matzinger, 1994). In the intracellular space, the RAGE ligands S100/calgranulins and HMGB1 (Taguchi et al., 2000; Hofmann et al., 1999) modulate calcium homeostasis and are nonhistone DNA binding proteins, respectively. In the extracellular space, these RAGE ligands assume response-to-stress functions, such as driving cellular inflammation through activation of nuclear factor-κB and p38 MAPK (Gunasekaran et al., 2016; Fujiya et al., 2014). Direct roles for the RAGE ligands in obesity have been demonstrated, as blocking antibodies to HMGB1 reduced obesity and inflammation in a mouse model (Montes etal., 2015). As endogenous RAGE expression in adipocytes may be sufficient to quell thermogenic processes, it is possible that adipocytes generate and release RAGE ligands, thereby stringently regulating thermogenesis and energy expenditure. Indeed, our data in primary adipocytes from iBAT, iWAT, and eWAT revealed a time-dependent increase in Ager expression. In earlier work, ligand expression was linked to transcriptional upregulation of Ager in cultured cells (Li and Schmidt, 1997).
Our data indicate that in states of RAGE ligand enrichment, such as in high-fat feeding, the activity of the RAGE axis is heightened. Although iBAT from adipocyte Ager-deleted mice displays lower levels of Ucp1 and Dio2 at room temperature and in low-fat feeding (i.e., LOW RAGE LIGAND environment) when compared to iBAT from adipocyte Ager-expressing mice (Figure 3J), when these iBAT tissues are transplanted into environmentally-stressed WT mice fed a HFD (i.e, HIGH RAGE LIGAND environment), the absence of RAGE in adipocytes unrestrains thermogenic programming, as evidenced by the increased expression of Ucp1, Dio2, Ppargc1a, Cpt2a, and Pnpla2 mRNA in the transplanted iBAT of the adipocyte Ager-deleted versus Ager-expressing iBAT (Figure 4M). These considerations are supported by the work presented in Figures 6 and 7, in which the RAGE ligand CML-AGE suppresses lipolysis, phosphorylation of p38 MAPK, and thermogenic gene programs.
The activation of the β3-adrenergic receptor, which is highly expressed in adipocytes, results in the generation of cAMP and consequent PKA-mediated phosphorylation of HSL and perilipin (Soeder et al., 1999). The present studies illustrate that recently identified inhibitors of RAGE signal transduction (Manigrasso etal., 2016) block the RAGE ligand-mediated reduction of phosphorylation of PKA targets in adipocytes. Ager deletion, unlike the metabolic braking effects of KCNK3 (Chen et al., 2017), exerted no effects on cAMP levels in adipocytes at baseline or after CL316,243 stimulation. The observation that catalytic inhibition of PKA (H89) effectively blocked the effects of Ager deletion or the effects of the RI on adaptive thermogenesis in primary and cultured adipocytes supports a notion that the RAGE effects are mediated, at least in part, through the suppression of PKA-dependent phosphorylation of its downstream targets in adipocytes. In line with recent work that demonstrated that lipolysis in BAT is not required for cold-induced thermogenesis (Schreiber et al., 2017; Shin et al., 2017), our findings reveal broader effects for RAGE, in that beyond RAGE-dependent suppression of the β-adrenergic-mediated phosphorylation of PKA target HSL in adipocytes, fundamental signaling pathways that regulate Ucp1 and thermogenic genes, such as phosphorylation of p38 MAPK, are also downregulated by RAGE ligands and restored by Ager deletion or by treatment with RIs.
The present findings trace the impact of Ager deletion in the regulation of adaptive thermogenesis from the induction of lipolysis, particularly through the phosphorylation of HSL, to the generation of glycerol and free fatty acids and to the phosphorylation of p38 MAPK and regulation of Ucp1 mRNA transcription. It is established that circulating fatty acids from lipolysis serve as key cofactors facilitating the activation of UCP1 and the transport of protons across mitochondrial membranes, which is essential for mammalian regulation of heat (Garlid et al., 1996; Klingenberg and Winkler, 1985; Cannon and Nedergaard, 2004). In contrast to the suppressive effects of RAGE signaling on phosphorylation of p38 MAPK in primary adipocytes (iBAT and iWAT) and in C3H10T1/2 cells, our experiments revealed inconsistent findings regarding phosphorylation of ERK. Although the reasons for the differences in ERK MAPK phosphorylation vis-à-vis RAGE between cell lines and primary adipocytes remain to be identified, our data nevertheless point to clear roles for RAGE signaling-dependent regulation of HSL and p38 MAPK phosphorylation downstream of PKA.
In the broader context of evolution and adaptive selection, then, what forces drive this metabolic braking role for Ager? Selective pressures in evolution favor phenotypes that safeguard survival in the face of paucities and extreme stresses (Hotamisligil, 2017). Hence, when nutrient supplies are unlimited and selective survival pressures are lost, the urge to hoard energy, if unchecked, may become a vulnerability. The gene AGER, located on chromosome 6 in the major histocompatibility complex (MHC) III humans (Wu et al., 2015), first appeared in Laurasiatheria (Sessa et al., 2014), a superorder of placental mammals that is part of the larger group of mammals classified as Eutheria. Eutheria are mammalian clades, and the oldest Eutherian species is believed to be Juramaia sinensis, which dates to 160 million years ago (mya) (Gerkema et al., 2013). A key property of the Eutherians is the expression of UCP1 in BAT, which imbues the capacity for non-shivering thermogenesis (Hughes et al., 2009). Mammals first appeared during the Mesozoic era, about 250 mya, which followed the period known as the “Great Dying,” in which a massive extinction of plants and land species ensued consequent to a period of intense global warming (Gerkema et al., 2013; Sun et al., 2012). Perhaps AGER evolved as a defense against starvation or swings in ambient temperatures through its ability to suppress adaptive thermogenesis.
These premises may indeed be consistent with hypotheses that reduced energy expenditure and obesity may be the unintended consequences of evolutionary pressures against severe starvation (“thrifty genotype”) (Neel, 1962; Prentice, 2005; Wang et al., 2014) or the need to protect from extreme temperature swings throughout human migration (Sellayah et al., 2014). At thermoneutrality, mice devoid of Ager bear body temperatures that are indistinguishable from those of WT animals, thereby implying that RAGE-dependent forces are not triggered during homeostasis, but are evoked under stress conditions in which the ligands of the receptor accumulate. Indeed, in the evolution of AGER, the major adaptively selected sites within the protein product RAGE are within the extracellular ligand binding domains, the intracellular adaptor binding regions, and the surfaces facilitating oligomerization and, thereby, signal transduction (Wu et al., 2015).
Notably, distinct pathways linking metabolism and inflammation, specifically TLR, insulin receptor, and tumor necrosis factor (TNF) receptor signaling, may be traced to orthologs in Drosophila (Hotamisligil, 2017). This is not the case for Ager. However, the RAGE ligands HMGB1 and S100/calgranulins, such as S100A8/A9, may also bind TLRs (Gunasekaran et al., 2016; Hiratsuka et al., 2008). TLRs play innate roles in host defense, but the role of Ager in response to infectious pathogens is assuredly more complex (van Zoelen et al., 2011). Whereas in cecal ligation and puncture (sepsis) or in Streptococcus pneumonia pneumonia, a deficiency of Ager in mice improved survival (Liliensiek et al., 2004; van Zoelen et al., 2009), the loss of Ager in Klebsiella pneumoniae pneumonia was detrimental (Achouiti et al., 2016). Together with the marked differences in appearance of TLR versus AGER in evolution and the lack of consistent roles for AGER in innate host defense to pathogens, it is plausible that RAGE’s ability to conserve energy underscores its primal endogenous function, which evolved, remarkably, at the expense of unremitting and chronic inflammation and the dampening of tissue repair on account of RAGE’s ability to usurp the functions of HMGB1 and S100/calgranulins ligands. Such a scenario frames a critical paradigm in the immunometabolic nexus of genes that integrate inflammatory and metabolic responses and identifies RAGE as an innate mechanism in the suppression of adipocyte metabolism and energy expenditure.
In summary, this work demonstrates that RAGE suppresses adaptive thermogenesis in both BAT and WAT during a physiological response to fasting, a cold challenge, or high-fat feeding; adds Ager to the cadre of genes that tether inflammation to the regulation of energy homeostasis; defines the endogenous function of RAGE in energy metabolism; and pinpoints tractable therapeutic targets to harness energy expenditure in metabolic disorders through the blockade of RAGE signal transduction.
STAR ★METHODS
LEAD CONTACT AND MATERIALS AVAILABILITY
Further information and request for resources and reagents should be directed to the lead Contact Ann Marie Schmidt (AnnMarie.Schmidt@nyulangone.org).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mouse Strains
All animal procedures were approved by the New York University, University of Massachusetts Medical School and Sanford Burnham Institute (Lake Nona, Orlando FLA) Institutional Animal Care and Use Committees and performed in accordance with the National Institutes of Health Animal Care Guidelines. Homozygous Ager null mice (Liliensiek et al., 2004) (C57BL/6 Ager null mice back-crossed > 20 generations into C57BL/6J [The Jackson Laboratory, Bar Harbor, ME]) and their littermate Ager-expressing controls or C57BL/6J were used. In order to generate adipocyte-specific Ager-deleted mice, we developed a floxed mouse line, suitable for the generation of a conditional deletion model for the Ager gene (GenOway, Lyon, France). Agerflox/flox mice were bred with Adiponectin (Adipoq) Cre-recombinase mice to yield Agerflox/flox Adipoq Cre (+) and Agerflox/flox Adipoq Cre (−) littermate mice, resulting in deletion of Ager from both white and brown adipose tissue depots. Adiponectin Cre-recombinase were obtained from the Jackson Laboratory (Stock No: 028020), which were in the C57BL/6J background. C57BL/6J (WT) mice were obtained from The Jackson Laboratory (Stock No: 00664). All mice studied were male, had free access to food and water, unless otherwise noted in specific fasting/refeeding or fasting state studies, and were subjected to 12 h light/dark cycles. Mice were bred, born and housed in a pathogen-free barrier environment, which was negative for Helicobacter or Norovirus pathogens. Mice were housed in standard caging up to 4 mice per cage and, where, indicated, were single housed for the indicated periods of time in order to perform specific studies such as indirect calorimetry and measurement of food intake that required the presence of only one mouse per cage. At 4–5 weeks of age, mice were anesthetized with 1% isoflurane, and tail snips were analyzed by PCR. To confirm adipose-specific deletion of Ager, fat depots (eWAT, iWAT, and iBAT) from offspring were isolated and purification of total genomic DNA (gDNA) was performed according to the manufacturer’s instructions (DNeasy Blood and Tissue, QIAGEN). Thereafter, gDNA was subjected to PCR analysis using primers spanning the deletion site (exon 6) in the Ager gene (GenBank Accession number NM_007425.3) forward: 5′-GGTACCCTGCACCCAACTAC-3′ and reverse: 5′-TTGATGGCCCTGGGATTGAC-3′). The following conditions with Platinum TM taq DNA polymerase from Invitrogen were used for PCR: 95°C for 30 s, 58°C for 30 s, and 72°C for 1 min (29 cycles). The resulting PCR products were separated using agarose gel electrophoresis, and the bands were visualized. The primary data illustrating the targeting vector construct and the characterization of these mice are shown in Figures S3A-S3C. In all cases, within genotypes, mice were randomly assigned to study groups upon review of a database, which indicates mouse date of birth, sex, identification number and body weight. Randomization and study group assignment was performed with the assistance of technical support staff in the laboratory who were not involved in the design or protocol of the specific experiments.
Cells Used in the Study
C3H10T1/2 cells, purchased from ATCC (Manassas, VA), catalog number ATCC CCL-226, were plated at low density and cultured in DMEM containing 10% (vol/vol) FBS and 1% (vol/vol) penicillin-streptomycin. Two days post-confluence (designated day 0), cells were induced to differentiate with DMEM containing 10% (vol/vol) FBS, 1 μg/ml insulin, 2 nM T3, 0.5 mM isobutylmethylxanthine, 1 μM dexamethasone and 1 μM Rosiglitazone until day 2-5. After this induction phase, complete medium was supplemented with insulin and T3 until day 8. Before any treatment, cells were starved for 3 h in 2% fatty acid free Krebs-Ringer bicarbonate (KRB) buffer. For treatments to stimulate RAGE, CML-AGE, 300 μg/ml, was added to the media for the indicated time periods.
Primary adipocytes were derived from eWAT, iWAT and iBAT of male mice from the lines of animals described above. Fat depots were retrieved and immediately washed with PBS, minced and digested with collagenase type II (1 mg/ml), 45 mins, 37°C) in DMEM containing 1% (wt/vol) BSA. Digested tissues were filtered through sterile 100 and 40 μm cell strainers and centrifuged (2000 rpm, 10 min). The floating fractions containing adipocytes were separated from the SVF and stored at −80°C with QIAzol (QIAgen) until further analysis. The cells were centrifuged at 2000 rpm for 5 mins, plated on a 12-well culture dish, and grown and differentiated to mature adipocytes. Briefly, cells were grown to confluence in DMEM, 10% (vol/vol) FBS and 1% (vol/vol) penicillin-streptomycin followed by standard adipogenic induction (1 μg/μl insulin, 2 nM T3, 0.5 mM isobutylmethylxanthine, 1 μM dexamethasone, and 1uM rosiglitazone). After 2-5 days induction phase, complete medium was supplemented with insulin and T3, until day 8. Before any treatment, cells were serum-starved for 1-3 h in either DMEM or KRB buffer containing 2% fatty acid free BSA.
METHOD DETAILS
Fasting experiments
Fasting and feeding studies were performed in mice fed a standard chow diet (5053 PicoLab Rodent Diet 20; LabDiet, Brentwood, MO). To determine the effect of nutritional status, 11-week-old male Ager null mice and WT mice were fed ad libitum, fasted for 24 h, or re-fed for 24 h after the 24 h fast.
Cold-induced thermogenesis and thermoneutrality studies
Cold-induced thermogenesis over 48 h was assessed at the Cardiometabolic Phenotyping Core of the Sanford Burnham Institute (Lake Nona, Orlando FL) where Ager null mice and WT mice were individually housed in home cages with ad libitum access to food and water. The cages were placed in an environmentally-controlled chamber (Powers Scientific, Inc.) with the temperature set at 23°C (standard housing temperature). The temperature was then gradually lowered to 4°C over a span of ~6 h. Mice remained in the chamber at 4°C for 48 h. During this 48 h period, core body temperature was measured three times per day (10:00,14:00 and 17:00) using a rectal probe attached to a digital thermometer. Short-term cold exposure (4°C for 24 h) thermogenesis studies were carried out at NYU Medical Center. Ager null, WT, Agerflox/flox Adipoq Cre (+) and Agerflox/flox Adipoq Cre (−) male mice 8 weeks old were exposed to 4°C for 24 h. Core body temperature was measured every 6 h using a rectal probe (ThermoWorks, Alpine UT). For thermoneutrality studies, male Ager null and WT mice, 8 weeks of age, were housed at 30°C for 24 h in individual cages. Core body temperature was measured every 6 h using a rectal probe (ThermoWorks, Alpine UT). At 24 h, mice were removed and immediately euthanized for tissue collection.
Diet-induced obesity and indirect calorimetry
For HFD studies Ageflox/flox Adipoq Cre (+) and Ageflox/flox Adipoq Cre (−) were fed a HFD with 60% of calories from lard (D12492; Research Diets, Inc., New Brunswick, NJ) or low-fat diet (LFD) with 13% of calories from fat (5053 PicoLab Rodent Diet 20; LabDiet, Brentwood, MO). Metabolic studies in Agerflox/flox Adipoq Cre (+) and (−) mice were performed at the National Mouse Metabolic Phenotyping Center (MMPC) at UMass Medical School (Worcester, MA) and metabolic studies in mice surgically transplanted with adipose were performed at NYU Medical Center. Metabolic cages (TSE Systems, Inc., Midland, MI) were used in conscious mice to simultaneously measure energy expenditure through indirect calorimetry analyses, food/water intake, and physical activity at before (baseline at 8 weeks of age) and after the indicated periods of HFD (Dagdeviren et al., 2016; Lee et al., 2015a).
Dual Energy X-Ray (DEXA) Absorptiometry
DEXA scans were employed to determine body composition. Before each scan session, the instrument was calibrated as per the manufacturer’s direction. Mice were then weighed, briefly anesthetized via isoflurane inhalation and placed on a scanning area of a Lunar PIXImus DEXA instrument (PIXImus, WI). Mice remained immobilized on this surface for the duration of the entire scan. At least 3 scans per mouse were performed and the average lean and fat mass were recorded.
Soluble (s) RAGE
Soluble RAGE (sRAGE) (human) was prepared, purified, and rendered free of endotoxin as previously described (Park et al., 1998) and 100 μg/day by intraperitoneal (IP) route was administered to mice beginning immediately at the time of HFD feeding at age 6 weeks. Control mice were treated with equal amounts of vehicle PBS.
Glucose and Insulin Tolerance Tests
Glucose (GTT) and insulin tolerance tests (ITT) were performed on mice after fasting for 5-6 h and acclimated to the testing room with access to water. Mice were injected intraperitoneally (IP) with either D-glucose (Sigma, 0.75-1.5 g per kg of body weight) or recombinant human regular insulin (0.5-1 U per kg of body weight); blood glucose levels were measured via tail sampling after fasting for a baseline measurement (t = 0) and again at t = 15, 30, 60, 90 and 120 mins post glucose or insulin bolus using a glucometer (Freestyle Freedom Line, Abbott Diabetes Care Inc, Alameda CA).
iBAT and iWAT transplantation
Following standard antiseptic surgical techniques, iBAT transplantation in the subcutaneous dorsal region was performed as described (Stanford et al., 2013) with the following modifications. iBAT pads were removed from 6-week old Agerflox/flox Adipoq Cre (+) and Agerflox/flox Adipoq Cre (−) male mice and transplanted into 6-week old WT recipient male mice. Specifically, after euthanasia of donor mice, bilateral iBAT was immediately excised, peripheral white fat was removed, and the remaining iBAT (~90 mg) washed in sterile PBS and transplanted into the subcutaneous anterior dorsal region of recipient mice as quickly as possible. With the same approach, bilateral iWAT (~250 mg) from two Agerflox/flox Adipoq Cre (+) or Agerflox/flox Adipoq Cre (−) donors was transplanted to a single recipient (2:1 donor:recipient) bilaterally into the posterior dorsal region of the recipient through small incisions of 2-3 mm in length beginning at the proximal end of the hind limb (Tran et al., 2008). Recipient mice were anesthetized with gas isoflurane and treated with buprenorphine during and after surgery for pain relief, mice were then monitored until recovered. In both cases, after 4 days of recovery from surgery, mice were fed a 60% HFD for the time course, as indicated in the figures. Mice were housed 4-5 per cage.
Protein extraction and western blot analysis
Protein was extracted from cells and tissue from eWAT, iWAT and iBAT or other cell or tissue lysates as indicated by using lysis buffer or RIPA (Cell Signaling) supplemented with complete protease inhibitor cocktail (Roche), and centrifuged at 13,000 g for 15 mins at 4°C. The supernatant was collected and protein concentration was measured using a bicinchoninic acid (BCA) protein quantification kit (Thermo Scientific, Waltham, MA). Samples were diluted to one constant protein concentration followed by addition of sample reducing agent and loading dye (Thermo Scientific, Waltham, MA). For western blot analyses, 20-50 μg of protein were separated in 4%−20% Mini-PROTEAN TGX gels (Bio-Rad, Hercules, CA) and blotted onto nitrocellulose membranes (Bio-Rad, Hercules, CA). Membranes were blocked at room temperature for 1 h with Odyssey LI-COR Blocking Buffer (LI-COR, Lincoln, NE) diluted 1:1 in Tris-buffered saline (TBS). Membranes were then incubated with primary antibodies (diluted 1:1000 in a 1:1 Blocking Buffer/TBS-T solution) overnight at 4°C. Primary antibodies detailed in the STAR methods key resource table were used. Membranes were washed consecutively 3 times for at least 5 mins each in TBS-T (0.1%). Blots were incubated with IRDye 680RD goat anti–mouse IgG and IRDye 800CW goat anti–rabbit IgG (LI-COR, Lincoln, NE) for 1 h at room temperature in blocking buffer containing 0.1% TBS-T and 0.1% SDS. Blots were washed 3 more times in TBS-T followed by a final wash in TBS, the blots were scanned with the LI-COR Odyssey Classic (LI-COR, Lincoln, NE) and quantified with Image Studio Lite software (LI-COR, Lincoln, NE) based on direct fluorescence measurement.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-UCP-1 (Western Blot) | Abcam | Cat# ab10983; RRID:AB_2241462 |
| Anti-UCP-1 (Immunohistochemistry) | Abcam | Cat# Ab23841; RRID:AB_2213764 |
| Anti-phospho HSL (Ser563) | Cell Signaling | Cat# 4139S; RRID:AB_2135495 |
| Anti-total HSL | Cell Signaling | Cat# 4107S; RRID:AB_2296900 |
| Anti-Tubulin | Cell Signaling | Cat# 3873S; RRID:AB_1904178 |
| Anti-phospho ERK | Cell Signaling | Cat# 9101L; RRID:AB_331646 |
| Anti-total ERK | Cell Signaling | Cat # 9102; RRID:AB_330744 |
| Anti-ATGL | Cell Signaling | Cat # 2439S; RRID:AB_2167953 |
| Phospho-p38 MAPK (Thr180/Tyr182) | Cell Signaling | Cat #9211; RRID:AB_331641 |
| Anti-p38 MAPK | Cell Signaling | Cat # 9212; RRID:AB_330713 |
| Anti-Beta Actin | Santa Cruz | Cat# sc-81178; RRID:AB_2223230 |
| Anti-Monoacylglycerol Lipase (MGL) | Abcam | Cat# ab180016 |
| IRDye 680RD goat anti–mouse IgG | Li-Cor | Cat# 925-68070; RRID:AB_2651128 |
| Dylight 800-conjugated goat anti–mouse IgG | Li-Cor | Cat# 925-32211; RRID:AB_2651127 |
| Anti-GAPDH | Santa Cruz | Cat# sc-47724; RRID:AB_627678 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Hoeschst 33342 | Invitrogen | H3570 |
| Seahorse XF Cell Mito Stress Test | Agilent | 103015-100 |
| Seahorse XFe24 FluxPak | Agilent | 100850-001 |
| iScript cDNA Synthesis Kit, 500 × 20 μl rxns | Biorad | 1708891BUN |
| 10x Tris/Glycine Buffer | Bio-Rad | 1610771EDU |
| 10x Tris/Glycine/SDS | Bio-Rad | 1610732EDU |
| 4–15% Mini-PROTEAN® TGX Precast Protein Gel | Bio-Rad | 4561086 |
| Cell Lysis Buffer (10X) | Cell Signaling | 9803 |
| RIPA Buffer (10X) | Cell Signaling | 9806 |
| 4-20% Mini-PROTEAN TGX gels | Bio-Rad | 456-1096 |
| Trans-Blot® Turbo Midi Nitrocellulose Transfer | Bio-Rad | 1704159 |
| 10x Tris/Glycine/SDS Buffer | Bio-Rad | 161-0772 |
| 10x Tris/Glycine Buffer | Bio-Rad | 161-0771 |
| Sybr Green | Applied Biosystems | 4385618 |
| Odyssey® Blocking Buffer (PBS) | Li-Cor | 927-40003 |
| MitoTracker® Red CMXRos | Life Technologies | M7512 |
| DMEM, high glucose, HEPES, no phenol red | Life Technologies | 21063-029 |
| Phusion® High-Fidelity PCR Kit | New England Biolabs, Inc | E0553L |
| Dual-Glo® Luciferase Assay System | Promega | E2940 |
| Glo Lysis Buffer, 1X | Promega | E2661 |
| QIAzol Lysis Reagent | QIAGEN | 7906 |
| RNeasy Mini Kit (250) | QIAGEN | 79306 |
| Paraformaldehyde solution 4% in PBS | Santa Cruz Biotechnology | CAS 30525-89-4 |
| (−)-Norepinephrine | Sigma | A7257 |
| 3,3′,5-Triiodo-L-thyronine sodium salt | Sigma | T6397 |
| 3-Isobutyl-1-methylxanthine | Sigma | I5879 |
| CL 316243, | Sigma | C5976 |
| Collagenase from Clostridium histolyticum | Sigma | C6885-1G |
| Dexamethasone | Sigma | D4902 |
| Free Glycerol Reagent | Sigma | F6428 |
| Glucose | Sigma | G8270 |
| Glycerol Standard | Sigma | G1394 |
| H-89 dihydrochloride hydrate | Sigma | B1427-5MG |
| Insulin from bovine pancreas | Sigma | I6634 |
| Isoproterenol | Sigma | I5627 |
| Rosiglitazone | Sigma | R2408 |
| DMEM | ThermoFisher Scientific | 11995065 |
| Lipofectamine® 3000 Transfection Reagent | ThermoFisher Scientific | L3000001 |
| Optimem | ThermoFisher Scientific | 31985047 |
| Bovine Serum Albumin - Fatty Acid Free | Roche | 3117057001 |
| FBS | Corning | 35-010-CV |
| Glucose strips - FreeStyle Lite Blood Glucose Test Strips | Abbott | 70819-70 |
| D-Glucose | Alfa Aesar | A16828 |
| Magnesium Chloride | Sigma | M8266 |
| Sodium Chloride | Fisher Scientific | BP318-212 |
| Potassium Chloride | Fisher Scientific | P217-500 |
| Sodium Phosphate Dibasic | Sigma | S3264 |
| LipidTox | ThermoFisher Reagent | H34476 |
| BODIPY | ThermoFisher Reagent | D3922 |
| DAPI | Life Technologies | D1306 |
| Sodium Phosphate Monobasic | Sigma | S8282 |
| Sodium Bicarbonate | Fisher Scientific | S3233-500 |
| Insulin (Human) - Humulin R | Lilly | 0002-8215-01 |
| CML-AGE | Synthesized in Laboratory per Kislinger et al., 1999 |
N/A |
| 2- [4- (acetylamino) phenyl] −4- Quinolinecarboxylic acid, methyl ester | Synthesized Per the Methods/Source outlined in Manigrasso et al., 2016 | N/A |
| Critical Commercial Assays | ||
| 2-CAT (A-N) Research ELISA | Rocky Mountain Diagnostics | BA E-5400 |
| cAMP Direct Immunoassay Kit | Abcam | Ab65355 |
| Glycerol/Triglyceride Determination Kit | Sigma | TR0100-1KT |
| NEFA HR(2) color reagent A | Wako Diagnostics | 99934691 |
| NEFA-HR(2) color reagent B | Wako Diagnostics | 99134891 |
| NEFA HR(2) Solvent A | Wako Diagnostics | 99534791 |
| NEFA HR(2) Solvent B | Wako Diagnostics | 99335191 |
| cAMP Assay Kit | Abcam | ab65355 |
| Pierce BCA Protein Assay Kit | ThermoFisher | 23225 |
| PKA Kinase Activity Assay Kit | Abcam | ab139435 |
| Experimental Models: Cell Lines | ||
| C3H/10T1/2, Clone 8 (ATCC® CCL-226) | ATCC American Type Culture Collection | ATCC CCL-226 |
| Experimental Models: Organisms/Strains | ||
| C57BL/6J mice | Jackson Laboratory | JAX:000664 |
| Ager Flox/ Flox mice | N/A | N/A |
| Adipoq Cre Recombinase mice | Jackson Laboratory | 28020 |
| Ager null mice | N/A | N/A |
| Recombinant DNA | ||
| pGL3- UCP1 | Koza Laboratory | N/A |
| PGL3 Basic | Promega | E1741 |
| Software and Algorithms | ||
| Graphpad Prism 8.02 | GraphPad Software | https://www.graphpad.com |
| Leica Digital Image Hub | Leica Biosystems | https://www.leicabiosystems.com |
| ImageJ | NIH | https://imagej.nih.gov |
| Fiji (distribution of ImageJ) | Schindelin et al., 2012 | https://fiji.sc |
| WAVE Seahorse XFe24 Analyzer | Agilent | https://www.agilent.com |
| LI-COR Odyssey Classic | Li-Cor | https://www.licor.com |
| Trans-Blot Turbo Transfer System | Bio-Rad | http://www.bio-rad.com |
| Image Studio Lite | Li-Cor | https://www.licor.com |
RNA analysis and Real-time quantitative PCR
Total RNA was extracted from cells and tissue samples using TRIzol and RNeasy kits (QIAGEN) followed by cDNA synthesis using iScript Reverse Transcription Supermix for RT-PCR (Bio-Rad). qPCR was performed using the TaqMan method (50°C for 2 mins, 95°C for 10 mins, and 40 cycles of 95°C for 15 s and 60°C for 1 min) with premade primer sets (Applied Biosystems). The relative abundance of transcripts was normalized according to the expression of Actb using the ΔΔCt method. Primer sequences are listed in Table S1.
Mitochondrial DNA content
Genomic DNA was isolated from C3H10T1/2 cells using the DNeasy Blood & Tissue Kit (QIAGEN) following manufacturer’s instructions. Quantitative real-time PCR assessed genomic expression of mitochondrial NADH dehydrogenase subunit 1 (Nd1) Fwd 5′ GGCCACATGGCTACCATTTG 3′ and Rev 5′ CCGACCACTCACATTCACC 3′ normalized to Hbb (β-globin) Fwd 5′ ACACAAGGGACAAGAGGAATCC 3′ and Rev 5′ GGGTCAGGTCTTTGCTTCCA 3′ in genomic extracts from adipocytes differentiated from C3H10T1/2 cells treated with vehicle or CL316243 (Sigma) with or without the small molecule RAGE inhibitor (RI); 2- [4- (acetylamino) phenyl] −4- Quinolinecarboxylic acid, methyl ester. MitoTracker® Red CMXRos (Thermofisher) (75nM) was used to check the function of mitochondria in live iBAT-derived primary adipocytes from WT and Ager null mice at 37°C for 30 min to stain functional mitochondria, according to manufacturer’s protocol.
XF24 Extracellular Flux Analysis (Seahorse)
SVF cells isolated from the indicated adipose tissue depots from WT and Ager null mice were seeded in 24-well Seahorse plates and differentiated, as described above. The oxygen consumption rate (OCR) of 8 days differentiated cells was determined with an XF24 Extracellular Flux Analyzer (Seahorse Bioscience, Proteigene, Saint Marcel, France). Isoproterenol (10 μM) was used to characterize β-adrenergic inducible respiration and injected before oligomycin was indicated. Uncoupled and maximum OCR were determined with oligomycin (1 μM) and FCCP (1.5 μM). Rotenone (1 μM) and antimycin A (1 μM) were used to inhibit complex I-and III-dependent respiration, respectively. OCR was determined by plotting the oxygen tension and acidification of the medium in the chamber as a function of time and normalized by protein concentration (picomoles per min per milligram), respectively, as per the manufacturer’s instructions.
Assessment of Lipolysis
Differentiated adipocytes from iWAT or iBAT or differentiated C3H10T1/2 cells were starved for 3 h. Fresh medium (DMEM without Red Phenol) with 1% fatty acid free-BSA was added and the cells were immediately subjected to the indicated treatment (CL316243) for the indicated times. Sampled medium was used to measure glycerol release with a free glycerol reagent (Sigma Aldrich), according to the manufacturer’s instructions. Results were normalized to total sample protein content. For free fatty acid measurement, fully differentiated iWAT and iBAT-derived primary adipocytes were starved for the indicated times in KRB buffer containing 2% fatty acid free BSA (Roche) followed by treatment with either vehicle or CL316243 (Sigma Aldrich). Media and cell samples were collected at the indicated times and stored at −80°C until analyzed. Non-esterified free fatty acids (NEFA) from media were measured using a colormetric NEFA assay (WAKO) per the manufacturer’s protocol and normalized to total sample protein content.
Cell transfection and reporter assay
Undifferentiated C3H10T1/2 cells, clone 8 (ATCC® CCL-226) were transfected with pGL3 (Addgene) or complete Ucp1 promoter luciferase reporter constructs (generously shared by Dr. R. Koza) and 220-bp BAT-specific enhancer and proximal promoter of the mouse Ucp1 gene obtained by PCR amplification as described (Rim and Kozak, 2002) using lipofectamine 3000 (Thermofisher). At 24 h after transfection, cells were stimulated with NE (5 μM) and CML-AGE (300 μg/ml) as indicated. Cell lysates were harvested 48 h after transfection, and luciferase activity was quantified by standard luminometer assay and normalized to Renilla signal (Renilla Luciferase Assay System, PROMEGA).
Measurement of cAMP levels
Differentiated adipocytes from iBAT iWAT and eWAT from WT and Ager null mice were grown with complete medium supplemented with insulin and T3. At day 7 of differentiation, complete medium was replaced with DMEM + 0.2% BSA for 16 h. The cells were then treated with serum free DMEN ± 10 μM CL316,243 for 10 mins. Cells were collected, lysed, and intracellular cAMP levels were measured with cAMP Assay Kit (Competitive ELISA) (ab65355). cAMP levels were normalized to protein content.
Protein Kinase A Activity Assay
Differentiated primary brown adipocytes from WT mice were treated overnight with vehicle or RAGE ligand, CML-AGE (300 μmg/ml) followed by stimulation with CL316,243 for 15 mins. Cells were washed twice with RIPA buffer. Fifteen μg of cell lysate protein were used for the kinase reaction according to the manufacturer’s instructions (Enzo Life Science, NY). For the negative control, only the RIPA buffer was used in the reaction mixture. Data were normalized to total protein content.
Histology and Immunohistochemistry
For staining of adipose tissue for UCP1 antigen, iBAT, iWAT and eWAT samples were dissected and fixed in 4% paraformaldehyde (PFA) overnight or up to 48 h at 4°C. Samples were then washed at least three times in PBS and processed for paraffin embedding. Embedded blocks were sectioned at 5 μm thickness and placed onto histology slides. Slides were deparaffinized (Leica AR9222 dewax) and stained on a Leica BondRX® autostainer. Prior to the incubation with the primary antibody, sections underwent heat retrieval with Bond Epitope Retrieval Buffer 2 (Leica ER2, AR9640), and incubation with hydrogen peroxidase to eliminate endogenous peroxidase activity. To achieve an appropriate range of staining with different sample subsets, the primary antibody, UCP1 (Abcam ab23841) was diluted ((Leica diluent, ARD1001EA) 1:100 and 1:500 for iWAT and iBAT, respectively) and stained for 12 h. Sections were subsequently incubated with Leica anti-rabbit polymer conjugated to horse radish peroxidase (DAB KIT, Leica DS9800) and developed with diaminobenzidine. After counterstaining with hematoxylin, the sections were imaged on a Leica SCN400 whole slide scanner up to 40x magnification. Using the Slidepath Digital Imaging Hub software (Leica Biosystems), 3 randomly-selected section images per animal were captured at 20x magnification and used for quantification of UCP1 by an experimenter naive to the identification of the experimental groups.
Immunofluorescence microscopy
C3H/10T1/2 cells or SVF-derived adipocytes isolated either from WT or Ager null mice were grown on coverslips to obtain fully differentiated primary adipocytes. Cells were fixed with 4% PFA for 20 mins. After 5 mins of Triton X-100 0.2% permeabilization and 15 mins of blocking (PBS-BSA 3%), lipid droplets were stained using BODIPY or LipidTox for the indicated times. After washing 10 times with 1x PBS, cells were incubated with DAPI (Life Technologies) for 5 mins at room temperature. Cells were then washed extensively with 1x PBS and mounted on glass slides with Prolong-Gold (Life Technologies). All gains for the acquisition of comparable images were maintained constant and the Leica 5500B microscope was used to capture the images. Analysis of the images was performed using ImageJ (NIH) and Adobe Photoshop CC 2015.
Image analysis and quantification
MacBiophotonics ImageJ was used for image quantification and analysis of LipidTOX/BODIPY staining. To quantify the staining of lipid droplets with LipidTOX/BODIPY, the immunofluorescence of this dye was recorded as a digital, 8-bit gray-scale, 1024×1024 resolution images. The lipid droplet labeling image threshold was set at 90 on a 0–255 black to white scale (all pixels with a value under 90 were excluded from the quantification) to remove background pixels from measurement. The number of individual cells in every image was counted by DAPI staining. Quantification of LipidTOX/BODIPY staining in each condition is represented as mean intensity value divided by number of nuclei.
The Fiji distribution of ImageJ was used for image quantification and analysis of UCP1 immunostaining. To quantify the area of UCP1 staining, the images were first de-convoluted to separate the DAB (3,3′-diaminobenzidine) stain from the hematoxylin counter-stain using the Fiji plug-in Color De-convolution 1.7 (vector-H DAB). The resulting image was then subjected to automated thresholding using the Otsu method (Otsu, 1979; Ruifrok and Johnston, 2001; Schindelin et al., 2012). Quantification of UCP1-positive area was calculated as μm2 and the average of three images per animal was used for statistical analysis.
QUANTIFICATION AND STATISTICAL ANALYSES
The Figure Legends indicate all of the subject numbers/replicates per study; the mean ± standard error of the mean (SEM) is reported. The of details the specific statistical analyses based on the conditions and comparisons are listed in each figure legend and employed methods such as Student’s t test or ANOVA followed by the appropriate and indicated post hoc tests. In the event that variances were statistically different and thus not meeting the requirement of normal distribution, a non-parametric Mann-Whitney U test was employed. P values of < 0.05 were determined a priori as the threshold for statistical significance. Data analysis was performed using GraphPad Prism Software Version 8.02 (GraphPad, San Diego, CA).
DATA AND CODE AVAILABILITY
We have not generated software and datasets in this manuscript.
ADDITIONAL RESOURCES
We have not generated a new website or forum.
Supplementary Material
Highlights.
Ligands of RAGE accumulate in obese murine and human adipose tissues
Adipocyte-specific RAGE deletion protects mice from obesity and cold stress
RAGE suppresses key protein kinase A activities to restrain adaptive thermogenesis
Transplantation of adipocyte-RAGE-deleted adipose tissue into mice prevents obesity
ACKNOWLEDGMENTS
The authors gratefully acknowledge the expert assistance of Ms. Latoya Woods in the preparation of the manuscript. We thank Branka Dabovic and the Experimental Pathology Research Laboratory staff for their expert advice and immunohistochemistry support. This work was supported by grants from the United States Public Health Service (1R01DK109675 to A.M.S. and R.R.; 1PO1HL131481 to A.M.S.; 5T32HL098129-10 to H.H.R.; and 1F31AG054129-01 to J.D.); the American Diabetes Association (1-15-MI-14 to C.H.d.P.); and the American Heart Association (17SFRN33520045 to A.M.S. and H.H.R.). Part of this work was funded by Research Funds of the Diabetes Research Program at NYU (to A.M.S. and R.R.). The Experimental Pathology Research Laboratory is partially funded by the Cancer Center Support grant P30CA016087 at NYU Langone’s Cancer Center. Part of this work was funded by the National Mouse Metabolic Phenotyping Center at UMass funded by NIH grant 5U2C-DK093000 (to J.K.K.).
Footnotes
DECLARATION OF INTERESTS
A.M.S. and R.R. are co-inventors of patent applications related to the work in this manuscript.
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2019.06.061.
REFERENCES
- Achouiti A, de Vos AF, van’t Veer C, Florquin S, Tanck MW, Nawroth PP, Bierhaus A, van der Poll T, and van Zoelen MA (2016). Receptor for Advanced Glycation End Products (RAGE) Serves a Protective Role during Klebsiella pneumoniae - Induced Pneumonia. PLoS ONE 11, e0141000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartness TJ, Vaughan CH, and Song CK (2010). Sympathetic and sensory innervation of brown adipose tissue. Int. J. Obesity 34, S36–S42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertheloot D, and Latz E (2017). HMGB1, IL-1α, IL-33 and S100 proteins: dual-function alarmins. Cell. Mol. Immunol. 14, 43–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burns TW, Terry BE, Langley PE, and Robison GA (1979). Insulin inhibition of lipolysis of human adipocytes: the role of cyclic adenosine monophosphate. Diabetes 28, 957–961. [DOI] [PubMed] [Google Scholar]
- Cannon B, and Nedergaard J (2001). Respiratory and thermogenic capacities of cells and mitochondria from brown and white adipose tissue. Methods Mol. Biol. 155, 295–303. [DOI] [PubMed] [Google Scholar]
- Cannon B, and Nedergaard J (2004). Brown adipose tissue: function and physiological significance. Physiol. Rev. 84, 277–359. [DOI] [PubMed] [Google Scholar]
- Castro É, Silva TEO, and Festuccia WT (2017). Critical review of beige adipocyte thermogenic activation and contribution to whole-body energy expenditure. Horm. Mol. Biol. Clin. Investig 31(2). [DOI] [PubMed] [Google Scholar]
- Chen Y, Zeng X, Huang X, Serag S, Woolf CJ, and Spiegelman BM (2017). Crosstalk between KCNK3-Mediated Ion Current and Adrenergic Signaling Regulates Adipose Thermogenesis and Obesity. Cell 171, 836–848.e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chijiwa T, Mishima A, Hagiwara M, Sano M, Hayashi K, Inoue T, Naito K, Toshioka T, and Hidaka H (1990). Inhibition of forskolin-induced neurite outgrowth and protein phosphorylation by a newly synthesized selective inhibitor of cyclic AMP-dependent protein kinase, N-[2-(p-bromocinnamylamino) ethyl]-5-isoquinolinesulfonamide (H-89), of PC12D pheochromocytoma cells. J. Biol. Chem. 265, 5267–5272. [PubMed] [Google Scholar]
- Collins S (2012). β-Adrenoceptor Signaling Networks in Adipocytes for Recruiting Stored Fat and Energy Expenditure. Front. Endocrinol. (Lausanne) 2, 102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins S, and Surwit RS (2001). The beta-adrenergic receptors and the control of adipose tissue metabolism and thermogenesis. Recent Prog. Horm. Res. 56, 309–328. [DOI] [PubMed] [Google Scholar]
- Cypess AM, Lehman S, Williams G, Tal I, Rodman D, Goldfine AB, Kuo FC, Palmer EL, Tseng YH, Doria A, et al. (2009). Identification and importance of brown adipose tissue in adult humans. N. Engl. J. Med. 360, 1509–1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dagdeviren S, Jung DY, Lee E, Friedline RH, Noh HL, Kim JH, Patel PR, Tsitsilianos N, Tsitsilianos AV, Tran DA, et al. (2016). Altered Interleukin-10 Signaling in Skeletal Muscle Regulates Obesity-Mediated Inflammation and Insulin Resistance. Mol. Cell. Biol. 36, 2956–2966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duncan RE, Ahmadian M, Jaworski K, Sarkadi-Nagy E, and Sul HS (2007). Regulation of lipolysis in adipocytes. Annu. Rev. Nutr. 27, 79–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eguchi J, Wang X, Yu S, Kershaw EE, Chiu PC, Dushay J, Estall JL, Klein U, Maratos-Flier E, and Rosen ED (2011). Transcriptional control of adipose lipid handling by IRF4. Cell Metab. 13, 249–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finkelstein EA, Trogdon JG, Cohen JW, and Dietz W (2009). Annual medical spending attributable to obesity: payer-and service-specific estimates. Health Aff. (Millwood) 28, w822–w831. [DOI] [PubMed] [Google Scholar]
- Flegal KM, Carroll MD, Kit BK, and Ogden CL (2012). Prevalence of obesity and trends in the distribution of body mass index among US adults, 1999–2010. JAMA 307, 491–497. [DOI] [PubMed] [Google Scholar]
- Fujiya A, Nagasaki H, Seino Y, Okawa T, Kato J, Fukami A, Himeno T, Uenishi E, Tsunekawa S, Kamiya H, et al. (2014). The role of S100B in the interaction between adipocytes and macrophages. Obesity (Silver Spring) 22, 371–379. [DOI] [PubMed] [Google Scholar]
- Gaens KH, Goossens GH, Niessen PM, van Greevenbroek MM, van der Kallen CJ, Niessen HW, Rensen SS, Buurman WA, Greve JW, Blaak EE, et al. (2014). Nε-(carboxymethyl)lysine-receptor for advanced glycation end product axis is a key modulator of obesity-induced dysregulation of adipokine expression and insulin resistance. Arterioscler. Thromb. Vasc. Biol. 34, 1199–1208. [DOI] [PubMed] [Google Scholar]
- Garlid KD, Orosz DE, Modrianský M, Vassanelli S, and Jezek P (1996). On the mechanism of fatty acid-induced proton transport by mitochondrial uncoupling protein. J. Biol. Chem. 271, 2615–2620. [DOI] [PubMed] [Google Scholar]
- Gerkema MP, Davies WI, Foster RG, Menaker M, and Hut RA (2013). The nocturnal bottleneck and the evolution of activity patterns in mammals. Proc. Biol. Sci. 280, 20130508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunasekaran MK, Virama-Latchoumy AL, Girard AC, Planesse C, Guérin-Dubourg A, Ottosson L, Andersson U, Césari M, Roche R, and Hoareau L (2016). TLR4-dependant pro-inflammatory effects of HMGB1 on human adipocyte. Adipocyte 5, 384–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Himms-Hagen J, Melnyk A, Zingaretti MC, Ceresi E, Barbatelli G, and Cinti S (2000). Multilocular fat cells in WAT of CL-316243-treated rats derive directly from white adipocytes. Am. J. Physiol. Cell Physiol. 279, C670–C681. [DOI] [PubMed] [Google Scholar]
- Hiratsuka S, Watanabe A, Sakurai Y, Akashi-Takamura S, Ishibashi S, Miyake K, Shibuya M, Akira S, Aburatani H, and Maru Y (2008). The S100A8-serum amyloid A3-TLR4 paracrine cascade establishes a pre-metastatic phase. Nat. Cell Biol. 10, 1349–1355. [DOI] [PubMed] [Google Scholar]
- Hofmann MA, Drury S, Fu C, Qu W, Taguchi A, Lu Y, Avila C, Kambham N, Bierhaus A, Nawroth P, et al. (1999). RAGE mediates a novel proinflammatory axis: a central cell surface receptor for S100/calgranulin polypeptides. Cell 97, 889–901. [DOI] [PubMed] [Google Scholar]
- Holm C (2003). Molecular mechanisms regulating hormone-sensitive lipase and lipolysis. Biochem. Soc. Trans. 31, 1120–1124. [DOI] [PubMed] [Google Scholar]
- Hotamisligil GS (2017). Inflammation, metaflammation and immunometabolic disorders. Nature 542, 177–185. [DOI] [PubMed] [Google Scholar]
- Hudson BI, Kalea AZ, Del Mar Arriero M, Harja E, Boulanger E, D’Agati V, and Schmidt AM (2008). Interaction of the RAGE cytoplasmic domain with diaphanous-1 is required for ligand-stimulated cellular migration through activation of Rac1 and Cdc42. J. Biol. Chem. 283, 34457–34468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes DA, Jastroch M, Stoneking M, and Klingenspor M (2009). Molecular evolution of UCP1 and the evolutionary history of mammalian non-shivering thermogenesis. BMC Evol. Biol. 9, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jungas RL, and Ball EG (1963). Studies on the metabolism of adipose tissue. XII. The effects of insulin and epinephrine on free fatty acid and glycerol production in the presence and absence of glucose. Biochemistry 2, 383–388. [DOI] [PubMed] [Google Scholar]
- Jungas RL, and Ball EG (1964). Studies on the metabolism of adipose tissue. XVII. In vitro effects of insulin upon the metabolism of the carbohydrate and triglyceride stores of adipose tissue from fasted-refed rats. Biochemistry 3, 1696–1702. [DOI] [PubMed] [Google Scholar]
- Kajimura S, Spiegelman BM, and Seale P (2015). Brown and Beige Fat: Physiological Roles beyond Heat Generation. Cell Metab. 22, 546–559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kislinger T, Fu C, Huber B, Qu W, Taguchi A, Du Yan S, Hofmann M, Yan SF, Pischetsrieder M, Stern D, and Schmidt AM (1999). N(epsilon)-(carboxymethyl)lysine adducts of proteins are ligands for receptor for advanced glycation end products that activate cell signaling pathways and modulate gene expression. J. Biol. Chem. 274, 31740–31749. [DOI] [PubMed] [Google Scholar]
- Klingenberg M, and Winkler E (1985). The reconstituted isolated uncoupling protein is a membrane potential driven H+ translocator. EMBO J. 4, 3087–3092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koh YJ, Park BH, Park JH, Han J, Lee IK, Park JW, and Koh GY (2009). Activation of PPAR gamma induces profound multilocularization of adipocytes in adult mouse white adipose tissues. Exp. Mol. Med. 41, 880–895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopecky J, Clarke G, Enerbäck S, Spiegelman B, and Kozak LP (1995). Expression of the mitochondrial uncoupling protein gene from the aP2 gene promoter prevents genetic obesity. J. Clin. Invest. 96, 2914–2923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumari M, Wang X, Lantier L, Lyubetskaya A, Eguchi J, Kang S, Tenen D, Roh HC, Kong X, Kazak L, et al. (2016). IRF3 promotes adipose inflammation and insulin resistance and represses browning. J. Clin. Invest. 126, 2839–2854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee KY, Russell SJ, Ussar S, Boucher J, Vernochet C, Mori MA, Smyth G, Rourk M, Cederquist C, Rosen ED, et al. (2013). Lessons on conditional gene targeting in mouse adipose tissue. Diabetes 62, 864–874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee E, Jung DY, Kim JH, Patel PR, Hu X, Lee Y, Azuma Y, Wang HF, Tsitsilianos N, Shafiq U, et al. (2015a). Transient receptor potential vanilloid type-1 channel regulates diet-induced obesity, insulin resistance, and leptin resistance. FASEB J. 29, 3182–3192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee MW, Odegaard JI, Mukundan L, Qiu Y, Molofsky AB, Nussbaum JC, Yun K, Locksley RM, and Chawla A (2015b). Activated type 2 innate lymphoid cells regulate beige fat biogenesis. Cell 160, 74–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, and Schmidt AM (1997). Characterization and functional analysis of the promoter of RAGE, the receptor for advanced glycation end products. J. Biol. Chem. 272, 16498–16506. [DOI] [PubMed] [Google Scholar]
- Lidell ME, Seifert EL, Westergren R, Heglind M, Gowing A, Sukonina V, Arani Z, Itkonen P, Wallin S, Westberg F, et al. (2011). The adipocyte-expressed forkhead transcription factor Foxc2 regulates metabolism through altered mitochondrial function. Diabetes 60, 427–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liliensiek B, Weigand MA, Bierhaus A, Nicklas W, Kasper M, Hofer S, Plachky J, Gröne HJ, Kurschus FC, Schmidt AM, et al. (2004). Receptor for advanced glycation end products (RAGE) regulates sepsis but not the adaptive immune response. J. Clin. Invest. 113, 1641–1650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- López-Díez R, Shekhtman A, Ramasamy R, and Schmidt AM (2016). Cellular mechanisms and consequences of glycation in atherosclerosis and obesity. Biochim. Biophys. Acta 1862, 2244–2252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manigrasso MB, Pan J, Rai V, Zhang J, Reverdatto S, Quadri N, De-Vita RJ, Ramasamy R, Shekhtman A, and Schmidt AM (2016). Small Molecule Inhibition of Ligand-Stimulated RAGE-DIAPH1 Signal Transduction. Sci. Rep. 6, 22450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matzinger P (1994). Tolerance, danger, and the extended family. Annu. Rev. Immunol. 12, 991–1045. [DOI] [PubMed] [Google Scholar]
- Montes VN, Subramanian S, Goodspeed L, Wang SA, Omer M, Bobik A, Teshigawara K, Nishibori M, and Chait A (2015). Anti-HMGB1 antibody reduces weight gain in mice fed a high-fat diet. Nutr. Diabetes 5, e161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nedergaard J, Bengtsson T, and Cannon B (2007). Unexpected evidence for active brown adipose tissue in adult humans. Am. J. Physiol. Endocrinol. Metab 293, E444–E452. [DOI] [PubMed] [Google Scholar]
- Neel JV (1962). Diabetes mellitus: a “thrifty” genotype rendered detrimental by “progress”? Am. J. Hum. Genet. 14, 353–362. [PMC free article] [PubMed] [Google Scholar]
- Otsu N (1979). A Threshold Selection Method from Gray-Level Histograms. IEEE Trans. Syst. Man Cybern. 9, 62–66. [Google Scholar]
- Park L, Raman KG, Lee KJ, Lu Y, Ferran LJ Jr., Chow WS, Stern D, and Schmidt AM (1998). Suppression of accelerated diabetic atherosclerosis by the soluble receptor for advanced glycation endproducts. Nat. Med. 4, 1025–1031. [DOI] [PubMed] [Google Scholar]
- Prentice AM (2005). Early influences on human energy regulation: thrifty genotypes and thrifty phenotypes. Physiol. Behav. 86, 640–645. [DOI] [PubMed] [Google Scholar]
- Ramasamy R, Yan SF, and Schmidt AM (2012). The diverse ligand repertoire of the receptor for advanced glycation endproducts and pathways to the complications of diabetes. Vascul. Pharmacol. 57, 160–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rial E, Poustie A, and Nicholls DG (1983). Brown-adipose-tissue mitochondria: the regulation of the 32000-Mr uncoupling protein by fatty acids and purine nucleotides. Eur. J. Biochem. 137, 197–203. [DOI] [PubMed] [Google Scholar]
- Rim JS, and Kozak LP (2002). Regulatory motifs for CREB-binding protein and Nfe2l2 transcription factors in the upstream enhancer of the mitochondrial uncoupling protein 1 gene. J. Biol. Chem. 277, 34589–34600. [DOI] [PubMed] [Google Scholar]
- Robidoux J, Kumar N, Daniel KW, Moukdar F, Cyr M, Medvedev AV, and Collins S (2006). Maximal beta3-adrenergic regulation of lipolysis involves Src and epidermal growth factor receptor-dependent ERK1/2 activation. J. Biol. Chem. 281, 37794–37802. [DOI] [PubMed] [Google Scholar]
- Ruifrok AC, and Johnston DA (2001). Quantification of histochemical staining by color deconvolution. Anal. Quant. Cytol. Histol. 23, 291–299. [PubMed] [Google Scholar]
- Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, et al. (2012). Fiji: an open-source platform for biological-image analysis. Nat. Methods 9, 676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schreiber R, Diwoky C, Schoiswohl G, Feiler U, Wongsiriroj N, Abdellatif M, Kolb D, Hoeks J, Kershaw EE, Sedej S, et al. (2017). Cold-Induced Thermogenesis Depends on ATGL-Mediated Lipolysis in Cardiac Muscle, but Not Brown Adipose Tissue. Cell Metab. 26, 753–763.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sellayah D, Cagampang FR, and Cox RD (2014). On the evolutionary origins of obesity: a new hypothesis. Endocrinology 155, 1573–1588. [DOI] [PubMed] [Google Scholar]
- Sessa L, Gatti E, Zeni F, Antonelli A, Catucci A, Koch M, Pompilio G, Fritz G, Raucci A, and Bianchi ME (2014). The receptor for advanced glycation end-products (RAGE) is only present in mammals, and belongs to a family of cell adhesion molecules (CAMs). PLoS ONE 9, e86903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin H, Ma Y, Chanturiya T, Cao Q, Wang Y, Kadegowda AKG, Jackson R, Rumore D, Xue B, Shi H, et al. (2017). Lipolysis in Brown Adipocytes Is Not Essential for Cold-Induced Thermogenesis in Mice. Cell Metab. 26, 764–777.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soeder KJ, Snedden SK, Cao W, Della Rocca GJ, Daniel KW, Luttrell LM, and Collins S (1999). The beta3-adrenergic receptor activates mitogen-activated protein kinase in adipocytes through a Gi-dependent mechanism. J. Biol. Chem. 274, 12017–12022. [DOI] [PubMed] [Google Scholar]
- Song F, Hurtado del Pozo C, Rosario R, Zou YS, Ananthakrishnan R, Xu X, Patel PR, Benoit VM, Yan SF, Li H, et al. (2014). RAGE regulates the metabolic and inflammatory response to high-fat feeding in mice. Diabetes 63, 1948–1965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanford KI, Middelbeek RJ, Townsend KL, An D, Nygaard EB, Hitchcox KM, Markan KR, Nakano K, Hirshman MF, Tseng YH, and Goodyear LJ (2013). Brown adipose tissue regulates glucose homeostasis and insulin sensitivity. J. Clin. Invest. 123, 215–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Y, Joachimski MM, Wignall PB, Yan C, Chen Y, Jiang H, Wang L, and Lai X (2012). Lethally hot temperatures during the Early Triassic green-house. Science 338, 366–370. [DOI] [PubMed] [Google Scholar]
- Taguchi A, Blood DC, del Toro G, Canet A, Lee DC, Qu W, Tanji N, Lu Y, Lalla E, Fu C, et al. (2000). Blockade of RAGE-amphoterin signalling suppresses tumour growth and metastases. Nature 405, 354–360. [DOI] [PubMed] [Google Scholar]
- Touré F, Fritz G, Li Q, Rai V, Daffu G, Zou YS, Rosario R, Ramasamy R, Alberts AS, Yan SF, and Schmidt AM (2012). Formin mDia1 mediates vascular remodeling via integration of oxidative and signal transduction pathways. Circ. Res. 110, 1279–1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran TT, Yamamoto Y, Gesta S, and Kahn CR (2008). Beneficial effects of subcutaneous fat transplantation on metabolism. Cell Metab. 7, 410–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Marken Lichtenbelt WD, Vanhommerig JW, Smulders NM, Drossaerts JM, Kemerink GJ, Bouvy ND, Schrauwen P, and Teule GJ (2009). Cold-activated brown adipose tissue in healthy men. N. Engl. J. Med. 360, 1500–1508. [DOI] [PubMed] [Google Scholar]
- van Zoelen MA, Schouten M, de Vos AF, Florquin S, Meijers JC, Nawroth PP, Bierhaus A, and van der Poll T (2009). The receptor for advanced glycation end products impairs host defense in pneumococcal pneumonia. J. Immunol. 182, 4349–4356. [DOI] [PubMed] [Google Scholar]
- van Zoelen MA, Achouiti A, and van der Poll T (2011). RAGE during infectious diseases. Front. Biosci. (Schol. Ed.) 3, 1119–1132. [DOI] [PubMed] [Google Scholar]
- Virtanen KA, Lidell ME, Orava J, Heglind M, Westergren R, Niemi T, Taittonen M, Laine J, Savisto NJ, Enerbäck S, and Nuutila P (2009). Functional brown adipose tissue in healthy adults. N. Engl. J. Med. 360, 1518–1525. [DOI] [PubMed] [Google Scholar]
- Wang SP, Yang H, Wu JW, Gauthier N, Fukao T, and Mitchell GA (2014). Metabolism as a tool for understanding human brain evolution: lipid energy metabolism as an example. J. Hum. Evol. 77, 41–49. [DOI] [PubMed] [Google Scholar]
- Wu X, Wu J, Thompson CW, and Li Y (2015). Adaptive evolution of the MHC class III-encoded receptor RAGE in primates and murine rodents. Int. J. Immunogenet. 42, 461–468. [DOI] [PubMed] [Google Scholar]
- Yehuda-Shnaidman E, Buehrer B, Pi J, Kumar N, and Collins S (2010). Acute stimulation of white adipocyte respiration by PKA-induced lipolysis. Diabetes 59, 2474–2483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zingaretti MC, Crosta F, Vitali A, Guerrieri M, Frontini A, Cannon B, Nedergaard J, and Cinti S (2009). The presence of UCP1 demonstrates that metabolically active adipose tissue in the neck of adult humans truly represents brown adipose tissue. FASEB J. 23, 3113–3120. [DOI] [PubMed] [Google Scholar]
- Zu L, He J, Jiang H, Xu C, Pu S, and Xu G (2009). Bacterial endotoxin stimulates adipose lipolysis via toll-like receptor 4 and extracellular signal-regulated kinase pathway. J. Biol. Chem. 284, 5915–5926. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
We have not generated software and datasets in this manuscript.