
Figure 3. Cross-talk between the CDK4/6 and mitogenic signaling pathways in cancer.
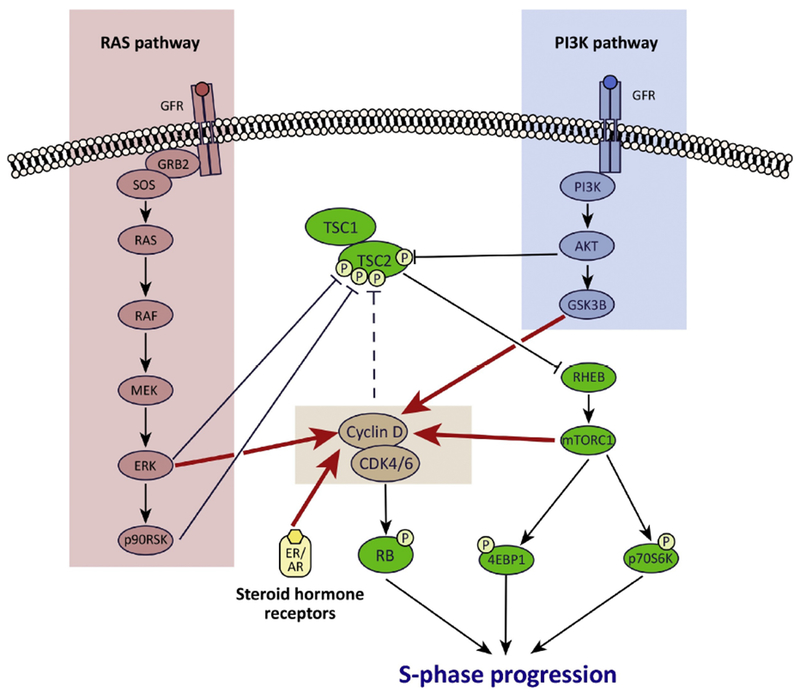
Extensive cross-talk exists between mitogenic signaling pathways and the CDK4/6 pathway in cancer cells. First, mitogenic signaling increases cyclin D1 levels to increase CDK4/6 activity via several mechanisms (red arrows): (a) PI3K pathway signaling reduces cyclin D1 turnover via GSK3β; (b) Ras pathway signaling promotes an ERK-dependent upregulation of transcription factors that drive cyclin D gene expression; (c) mTORC1 increases cyclin D protein translation; (d) CCND1 transcription is increased directly by activated ER (breast cancer) and AR (prostate cancer). In addition, these pathways also interact via their convergence on TSC2 and thus mTORCl: (a) ART, ERK, and p90RSK each directly phosphorylate TSC2 to activate mTORCl; (b) cyclin D-CDK4/6 also bind to and probably phosphorylate TSC2 to increase mTORCl activity (dashed line).
Rationale for synergistic combination therapy regimens: CDK4/6 inhibitors limit cell proliferation by reducing RB phosphorylation, but can also partially suppress TSC2 phosphorylation. Co-inhibiting the PI3K and/or Ras pathway not only reduces cyclin D1 levels (further enforcing RB activation), but also increases the suppression of TSC2 phosphorylation, maximizing mTORCl inhibition. Collectively, combined activation of RB and inhibition of mTORCl inhibition potently blocks progression of cells into S phase.