

Abstract
Pathogenic bacteria pose a serious public health threat. Rapid and cost effective detection of such bacteria remains a major challenge. Here, we present a DNAzyme-based fluorescent paper sensor for Klebsiella pneumoniae. The DNAzyme was generated by an in vitro selection technique to cleave a fluorogenic DNA-RNA chimeric substrate in the presence of K. pneumoniae. The DNAzyme was printed on a paper substrate in a 96 well format to serve as mix-and-read fluorescent assay which exhibited a limit of detection (LOD) 105 CFUs/mL. It was evaluated with 20 strains of clinical bacterial isolates and the DNAzyme produced the desired fluorescence signal with the samples of K. pneumoniae, regardless of their source or drug resistance. The assay is simple to use, rapid, inexpensive, avoids complex procedures of sample preparation and equipment. We believe, this DNAzyme-based fluorescent assay has potential for practical applications to identify K. pneumoniae.
Keywords: DNAzyme, Fluorogenic, Klebsiella pneumoniae, Paper sensor, Pathogenic bacteria
Graphical Abstract
Lighting up bacteria with DNAzyme.Infection of pathogenic bacteria remains a major health threat. Convenient detection of such pathogens plays a key role in protecting health and loss of economy. Herein, we present a DNAzyme that produce fluorescence signal in presence of K. pneumoniae.
Infectious diseases remain a major cause of morbidity and mortality throughout the world.[1] Besides the loss of lives, it also imposes a huge economic burden in our society.[1b,c,2] Additionally, the emergence of multidrug resistant bacteria has become a major public health concern. According to the Centers for Disease Control and Prevention (CDC), at least 2 million people in the United States alone are infected with drug resistant bacteria and 23,000 people die from these infections each year.[3] In 2017, the World Health Organization (WHO) published its first ever list of antibiotic-resistant “priority pathogens” that pose the greatest threat to human health and categorized them into critical, high and medium priority.[4] K. pneumoniae has been included in the critical group because of its rapid development of resistance towards multiple antibiotics.[4] K. pneumoniae is a gram negative bacteria and is a leading cause of respiratory tract, urinary tract, and blood stream infections.[5] Both carbapenemase and extended-spectrum beta-lactamase producing K. pneumoniae have created a critical health concern because of their resistance to a wide spectrum of beta lactam antibiotics.[5,6] Therefore, a rapid and selective assay for K. pneumoniae, in particular resistant isolates, can afford early intervention and thus play a vital role in the treatment and spread of these resistant strains.
The current standard methods for confirming the presence of carbapenemase producing K. pneumoniae (KPC) are culture followed by susceptibility testing.[7]Standard susceptibility testing can be further followed with molecular methods such as polymerase chain reaction (PCR) to confirm the presence of target genes. All these procedures are technically complex and require expensive equipment and highly trained personnel. On the other hand, immunoassays deliver relatively fast results. However, for the most part these methods are expensive, not sensitive or specific.[8] Therefore, there is still a need for new methods that can circumvent these limitations and can be used in resource deprived areas.
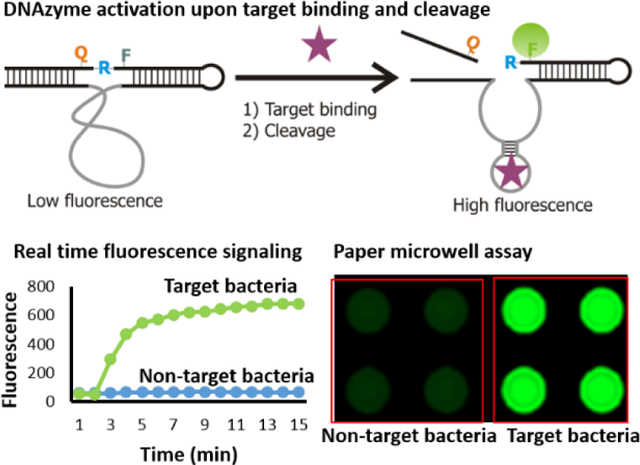
Recently, RNA-cleaving fluorogenic DNAzymes (RFDs) have emerged as potential candidates to develop bacterial bioassays.[9] These RFDs catalyze the cleavage of a DNA-RNA chimeric substrate at a single ribonucleotide junction embedded in a DNA sequence. The ribonucleotide is flanked by a fluorophore (F) and a quencher (Q), thus exhibits minimal fluorescence before the cleavage reaction because of the close proximity of F and Q. However, upon interaction with the target molecule in the complex crude extra/intra cellular mixture of the bacteria, the DNAzyme is activated and cleaves the substrate to separate F from Q and generates a high fluorescence signal (Figure 1A). RFDs are generated by an in vitro selection procedure using a random DNA library and crude extra/intra cellular mixtures of target bacteria including E. coli and C. difficile.[9] Once the DNAzymes are obtained, they can readily be used to formulate a mix-and-read fluorescent assay.[10] Advantageously, these RFDs can also be employed to develop colorimetric assays[11] including integration with isothermal secondary amplification.[12]
Figure 1.
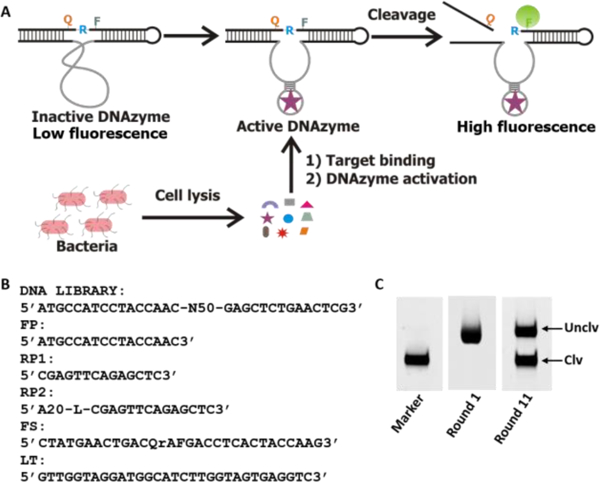
A) Schematic illustration of the mode of action of the DNAzyme. The inactive DNAzyme with low fluorescence becomes activated upon binding to the target molecule present in the cell lysate and cleaves the substrate at the designated cleavage site rendering fluorescence signal enhancement. B) DNA library and related oligonucleotide sequences used in the in vitro selection process. FP: forward primer, RP1: reverse primer that was used in the first PCR to enrich the selected pool, RP2: second reverse primer used in the second PCR for large scale production of the DNA pool in each round. A20 is 20 deoxyadenosine separated from the primer by a C18 glycol linker (Spacer 9 of IDT catalog). FQ30: the fluorogenic substrate wherein F is fluorescein-dT (fluorophore), rA is riboadenosine that serves as cleavage junction, Q is dabcyle-dT (quencher). LT: ligation template used for enzymatic ligation of the library with FQ30. C) Fluorescent gel images of the progress of selection. The marker was prepared by treating a portion of the FS ligated library with NaOH as discussed in the experimental section. Uclv: uncleaved full length sequence, Clv: cleaved sequence in the positive selection.
In this study, we aimed to generate a DNAzyme for K. pneumoniae and developed a paper-based assay in a 96 well format that could be used in point of care and low-resource settings. The DNAzyme was generated by our previously reported in vitro selection technique[9] using a random DNA library and a mixture of cell lysates composed of a mixture of carbapenam resistant K. pneumoniae which are listed in Table S1 in the supporting information (SI). The DNA library and the relevant sequences used for the in vitro selection are depicted in Figure 1B. We used a pool of cell lysates of K. pneumoniae strains as a complex target in the selection assuming that the DNAzymes obtained after the selection would respond to all K. pneumoniae rather than responding to a specific strain.
The DNA library covalently linked to the substrate (DL-FS) was mixed with cell lysates and incubated at room temperature for the cleavage reaction. The cleaved DNA molecules were purified by denaturing polyacrylamide gel electrophoresis (dPAGE) and amplified by PCR (See experimental section in the SI for details). This PCR enriched population was covalently attached to FS and applied to the next round of selection. The selection and enrichment process was repeated until a significant amount of cleaved product was obtained. A representative dPAGE image of the selection progress is shown in Figure 1C. To achieve selectivity, negative selection was applied in every two rounds of selection using a cell lysate mixture of carbapenem sensitive K. pneumoniae, E. coli and B. subtilis. After 11 rounds of selection, the enriched population was used in deep sequencing.
The top 10 sequences (Figure S2A in the SI) were chemically synthesized and tested for their cleavage performance with the reaction buffer (control) and cell lysate of carbapenem resistant K. pneumoniae (test). The cleavage results indicated that some DNAzyme candidates performed well without producing any background cleavage in the reaction buffer alone (Figure S2B in ESI). They cleave the substrate only in the presence of the K. pneumoniae cell lysate. Next, some DNAzymes with good cleavage activity were further tested for selectivity with E. coli (EC), B. subtilis (BS), L. monocytogenes (LM) and F. nucleatum (FN). The results presented in Figure 2A revealed that DNAzyme 6 (named RFD-KP6) exhibited the most specificity for K. pneumoniae although slight cleavage products appeared in presence of EC and BS. However, it did not discriminate the drug susceptible K. pneumoniae (Labeled as KPS in Figure 2B) from the carbapenem resistant K. pneumoniae (labeled as KPC in Figure 2B). These results suggest that, although it could not discriminate carbapenem susceptible from carbapenem resistant K. pneumoniae, the DNAzyme sensor has the potential to identify K. pneumoniae, regardless of susceptibility. The gel images indicated that, although the cleavage products were not significant, the band intensities of the uncleaved bands in presence of some not-target bacterial cell lysates also decreased. This is probably due to random degradation of the DNAzyme sequence by nuclease as the cell lysates that were used in the cleavage reaction contained high number of cells (~108 CFUs/mL). We further tested the fluorescence signaling ability of RFD-KP6 in real time using a fluorometer in the presence of KPC cell lysate. RFD-KP6 produced a high fluorescence signal in presence of the KPC cell lysate but no fluorescence signal enhancement was found in buffer alone (Figure 2C). A predicted secondary structure was obtained by mFold software and it has been presented in the SI (Figure S3).
Figure 2.
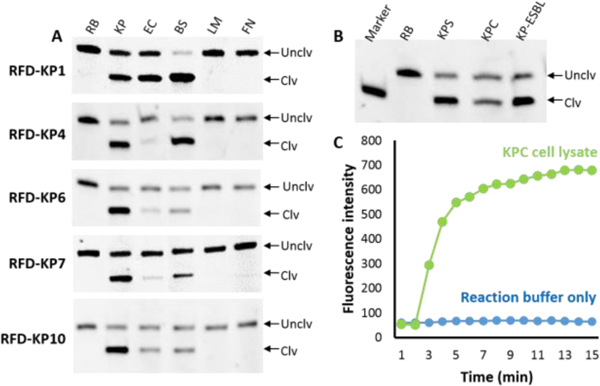
DNAzymes with select bacteria. RB: reaction buffer, KP: K. pneumoniae (carbapenem resistant), EC: E. coli, BS: B. subtilis, LM: L. monocytogenes, FN: F. nucleatum. B) Test of ability of discrimination of RFD-KP6 with drug susceptible K. pneumoniae (KPS), carbapenem resistant K. pneumoniae (KPC) and extended spectrum beta lactamase producing K. pneumoniae (KP-ESBL). C) Real time fluorescence signaling performance of RFD-KP6.
Next we designed a paper microzone device (PMD) as a mix-and-read assay which will allow multiple samples to be tested in a time convenient manner.[10c] In the first step, the 96-microzones were designed using Microsoft power point with black background. Then, it was wax printed in a plastic baked nitrocellulose paper as shown in Figure 3A (see details in the experimental section). The DNAzyme (RFD-KP6) along with 5% pullulan and 0.25M trehalose was printed on the microzones and air dried (Figure 3B). Pullulan is a natural sugar produced by a special fungus.[13] In our previous studies we have shown that dried pullulan films including trehalose can protect and stabilize labile biomolecules, affording longer storage at ambient temperature and maintenance of full activity.[14] Importantly, dried pullulan readily dissolves upon adding aqueous sample and does not interfere with biomolecules. Therefore, pullulan/trehalose was added to the DNAzyme during printing. The dried ready-to-use paper device was then tested for cleavage and signal generation. As shown in Figure 3C, the microzones treated with the K. pneumoniae cell lysate produced a strong fluorescent signal while the control microzones treated with reaction buffer alone did not. The fluorescence signal in the K. pneumoniae treated microzones was found to be at least 5-fold compared to that in the buffer treated microzones (Figure 3D).
Figure 3.
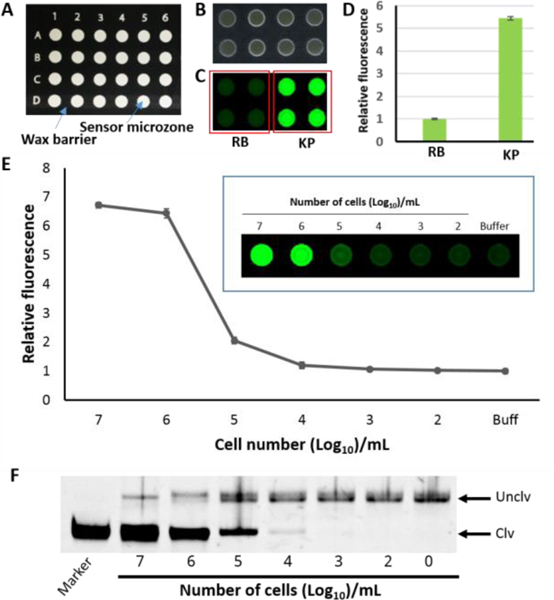
A) Design of paper sensor. B) Physical appearance of the paper device after printing of the DNAzyme. C) Cleavage and fluorescence signaling test of the paper device. D) Quantified fluorescence signal of C. E) Sensitivity test of RFD-KP6 with KPC with the paper microzone assay. Insert: fluorescent image of paper microzones with varying numbers of bacterial cells. The error bars are standard deviations of triplicate experiments. F) a representative denaturing gel
Next, we tested the sensitivity of RFD-KP6. For this purpose, samples of varying numbers of bacterial CFUs were prepared by serial dilution as described in the experimental section in the SI. Samples were applied on the DNAzyme printed microzones and incubated for the cleavage reactions. The fluorescent signal was quantified and plotted. The results showed that the paper device could produce detectable signal with 105 CFU/mL (Figure 3E). In order to confirm whether or not the fluorescent signals in the microzones were consistent with the cleavage products, we collected the reaction mixtures from each microzone from reactions with a different number of bacteria and ran them on dPAGE. The fluorescent signal matched with the amount of cleavage product (Figure 3F). Finally, we examined the performance of RFD-KP6 with clinical bacterial isolates. Twenty bacterial isolates blinded as to the identification (sample identification including name and information are given in Table S2 in ESI) were tested. The samples were applied to the paper microzone device and the fluorescent signal was detected by a fluorescent imager (see details in the experimental section). We found that some K. pneumoniae isolates produced the highest fluorescent signal compared to the other bacterial isolates. However, as seen in Figure 4, other bacterial isolates produced an equivalent fluorescence signal to the K. pneumoniae isolates, namely 441,443, 451 and 453. This may be due to other bacteria producing a high amount of nucleases which randomly degrade the DNAzyme rather than cleaving the substrate in the designated cleavage site. To verify this hypothesis, we collected the cleavage reaction mixtures from each microzone and analyzed them by dPAGE (Figure 4C). The dPAGE analysis produced consistet results with the fluorescence data. However, we noticed that some bacteria produced high amount of nucleases evidenced by the disappearance of the full length DNAzyme bands (Sample ID 438 and 453). Since nuclease activity is a major challenge in developing DNA based assays or therapeutics, attempts have been made to overcome this issue by incorporating modified nuecleotides.[15] Our future work will also explore this option when developing DNAzymes.
Figure 4.
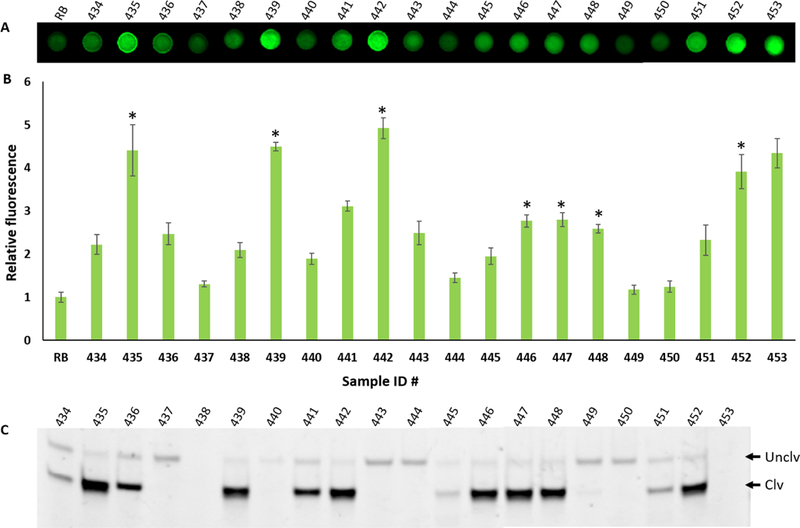
Evaluation of RFD-KP6 with bacterial isolates. A) Fluorescent gel image of the paper microzones after cleavage reaction with the samples (sample ID is shown above each microzone). B) Graph of the quantified fluorescent signal obtained from the paper microzones in A. The error bars represent standard deviation of triplicate experiments. C) A representative dPAGE gel image of the reaction mixture of each bacterial sample of the paper microzones of A. Unclv: Uncleaved intact full length DNAzyme sequence, Clv: cleaved product of the DNAzyme after the reaction. The name of the bacteria of each sample ID is provided in Table S2 in SI and the isolates of K. pneumoniae are designated by an asterisk.
It was also noticed that some K. pneumoniae strains (sample 446, 447 and 448) produced a somewhat lower fluorescent signal than others although the gel image showed almost similar cleavage products. We speculate that these strains may produce less target during culture thus causing slower cleavage reactions by the DNAzyme. It is important to note that the same samples were collected from the paper microzones to be analyzed by gel electrophoresis and it took significant time to collect the samples from the paper microzones. We assume that during the sample collection time the cleavage reactions continued to produce more cleavage products.
In summary we have generated a new fluorogenic DNAzyme by in vitro selection that catalyzes a fluorogenic substrate in the presence of K. pneumoniae. The DNAzyme is moderately specific in that it shows promise in being able to differentiate K. pneumoniae from other members of the Enterobacteriaceae, however, it was unable to differentiate susceptible isolates from carbapenem resistant isolates. With further improvement in specificity, this approach has the potential to provide a low cost, rapid method to identify these isolates once they are cultured.
Supplementary Material
Acknowledgements
The work was supported by National Institutes of Health (NIH)/ National Institute of Allergy and Infectious Diseases (NIAID) (1 R01 AI117061)
[**] Funding is provided by National Institutes of Health (NIH)/ National Institute of Allergy and Infectious Diseases (NIAID) (1 R01 AI117061)
References
- [1].a) Klevens RM, Edwards JR, Richards CL Jr., Horan TC, Gaynes RP, Pollock DA, Cardo DM, Public Health Rep. 2007,122,160–166; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Fonkwo PN, EMBO Rep., 2008, S13–17; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Suhrcke M, Stuckler D, Suk JE, Desai M, Senek M, McKee M, Tsolova S, Basu S, Abubakar I, Hunter P, Rechel B, Semenza JC, PLoS One. 2011, 6, e20724; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Osterholm MT, Engl N. J. Med. 2011, 364, 889–891; [DOI] [PubMed] [Google Scholar]; e) Scallan E, Hoekstra RM, Angulo FJ, Tauxe RV, Widdowson MA, Roy SL, Griffin PM, Emerg. Infect. Dis. 2011, 17, 7–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2]. http://www.who.int/healthinfo/global_burden_disease/estimates/en/index1.html.
- [3]. https://www.cdc.gov/drugresistance/index.html.
- [4]. http://www.who.int/mediacentre/news/releases/2017/bacteria-antibiotics-needed/en/
- [5].a) Khaertynov KS, Anokhin VA, Rizvanov AA, Davidyuk YN, Semyenova DR, Lubin SA, Skvortsova NN, Front. Med., 2018, 5, 225; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Jiao Y, Qin Y, Liu J, Li Q, Dong Y, Shang Y, Huang Y, Liu R, Pathog. Glob. Health. 2015, 109, 68–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].a) Kurupati P, Chow C, Kumarasinghe G, Poh CL, J. Clinic. Microbiol. 2004, 42, 1337–1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].a) Hirsch EB, Tam VH, J. Antimicrob. Chemother. 2010, 65, 1119–1125; [DOI] [PubMed] [Google Scholar]; b) Doern CD, Dunne WM Jr., C-A D Burnham, 2011, 49, 1143–1147, [DOI] [PMC free article] [PubMed] [Google Scholar]; c) https://www.cdc.gov/labtraining/training-courses/master/antimicrobial-susceptibility-testing-methods-ast.html [Google Scholar]
- [8].a) Baker CA, Rubinelli PM, Park SH, Ricke SC, Crit Rev Microbiol. 2016, 42, 656–675; [DOI] [PubMed] [Google Scholar]; b) Vidic J, Manzano M, Chang CM, Jaffrezic-Renault N, Vet Res. 2017, 48,11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].a) Ali MM, Aguirre SD, Lazim H, Li Y Angew. Chem. Int. Ed. Engl., 2011, 50, 3751–3754; [DOI] [PubMed] [Google Scholar]; b) Shen Z, Wu Z, Chang D, Zhang W, Tram K, Lee C, Kim P, Salena BJ, Li Y, Angew. Chem. Int. Ed. Engl., 2016, 55, 2431–2434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].a) Aguirre SD, Ali MM, Salena BJ, Li Y, Biomolecules, 2013, 3, 563–577; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Aguirre SD, Ali MM, Kanda P, Li Y, J. Vis. Exp., 2012, 63, e3961; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Ali MM, Brown CL, Jahanshahi-Anbuhi S, Li Y, Brennan JD, Sceintific Reports, 2017, 7, 12335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Tram K, Kanda P, Salena BJ, Huan S, Li Y, Angew. Chem. Int. Ed. Engl., 2014, 53, 12799–12802; [DOI] [PubMed] [Google Scholar]; e) Tram K, Manochehry S, Feng Q, Chang D, Salena BJ, Li Y, J. Vis. Exp., 2016, 115, e54546; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Zheng L, Qia P, Zhang D, Sensors & Actuators: B. Chemical, 2018, 27642–27647 [Google Scholar]
- [12].a) Liu M, Zhang Q, Li Z, Gu J, Brennan JD, Li Y, Nat Commun., 2016, 7, 12074; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Liu M, Zhang Q, Chang D, Gu J, Brennan JD, Li Y, Angew. Chem. Int. Ed. Engl. 2017, 56, 6142–6146 [DOI] [PubMed] [Google Scholar]
- [13].a) Bernier B, Canadian J Microbiol., 1958, 4 195–204, [DOI] [PubMed] [Google Scholar]; b) Bender H, Lechmann J, Wallenfells K, Biochemica Biophysica Acta, 1959, 36, 309–316 [DOI] [PubMed] [Google Scholar]
- [14].a) Jahanshahi-Anbuhi S, Pennings K, Leung V, Liu M, Carrasquilla C, Kannan B, Li Y, Pelton R, Brennan JD, Filipe CD. Angew. Chem. Int. Ed. Engl., 2014, 53, 6155–6158; [DOI] [PubMed] [Google Scholar]; b) Hsieh PY, Ali MM, Tram K, Jahanshahi-Anbuhi S, Brown C, Brennan JD, Filipe CD, Li Y, ChemBioChem, 2017, 18, 502–505; [DOI] [PubMed] [Google Scholar]; c) Kannan CB, Jahanshahi-Anbuhi S, Pelton RH, Li Y, Filipe CD, Brennan JD, Anal. Chem., 2015, 87, 9288–9293; [DOI] [PubMed] [Google Scholar]; c) Liu M, Hui CY, Zhang Q, Gu J, Kannan B, Jahanshahi-Anbuhi S, Filipe CD, Brennan JD, Li Y, Angew. Chem. Int. Ed. Engl., 2016, 55, 2709–2713 [DOI] [PubMed] [Google Scholar]
- [15].a) Schubert S, Gül DC, Grunert H-P, Zeichhardt H, Erdmann VA, Kurreck J, Nucleic Acids Res. 2003, 31, 5982–5992; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Chakravarthy M, Aung-Htut MT, Le BT, Veedu RN, Sci Rep. 2017, 7, 1613; [DOI] [PMC free article] [PubMed] [Google Scholar]; C) Ni S, Yao H, Wang L, Lu J, Jiang F, Lu A, Zhang G, Int J Mol Sci. 2017, 18, 1683; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Gawande BN, Rohloff JC, Carter JD, Carlowitz IV, Zhang C, Schneider DJ, Janjic N, Proc Natl Acad Sci U S A, 2017. 114, 2898–2903; [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Kimoto M, Yamashige R, Matsunaga K, Yokoyama S, Hirao I, Nat Biotechnol. 2013, 31, 453–457 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.