

Abstract
The diagnosis of bacterial infections in hospital settings is currently performed using bacterial culture from sterile site, but they are lengthy and limited. Transcriptomic biomarkers are becoming promising tools for diagnosis with potential applicability in clinical settings. We evaluated a RT-qPCR assay for a 2-transcript host expression signature (FAM89A and IFI44L genes) inferred from microarray data that allow to differentiate between viral and bacterial infection in febrile children. This assay was able to discriminate viral from bacterial infections (P-value = 1.04 × 10−4; AUC = 92.2%; sensitivity = 90.9%; specificity = 85.7%) and showed very high reproducibility regardless of the reference gene(s) used to normalize the data. Unexpectedly, the monogenic IFI44L expression signature yielded better results than those obtained from the 2-transcript test (P-value = 3.59 × 10−5; AUC = 94.1%; sensitivity = 90.9%; specificity = 92.8%). We validated this IFI44L signature in previously published microarray and whole-transcriptome data from patients affected by different types of viral and bacterial infections, confirming that this gene alone differentiates between both groups, thus saving time, effort, and costs. Herein, we demonstrate that host expression microarray data can be successfully translated into a fast, highly accurate and relatively inexpensive in vitro assay that could be implemented in the clinical routine.
Subject terms: Transcriptomics, Bacterial infection, Gene expression
Introduction
A common practice in hospitals is to systematically administer antibiotics to febrile patients until the results from culture tests are available as preventive method, with the outcome that many viral infections are inadequately treated with anti-microbial drugs1,2. Moreover, it has been shown that excessive antibiotic administration may have led to an increase in bacterial resistance, not only at individual but also at more global levels3. Hence, misuse and overuse of the currently available therapeutic drugs is actively contributing to generate anti-microbial resistance4–6. This fact, together with the lack of new generation antibiotics, is making a “post-antibiotic era” a very real prospect in the near future; this is increasingly becoming a global health concern, as recently highlighted by the World Health Organization (WHO)7.
An early distinction between viral and bacterial patients might allow a more accurate diagnosis and treatment of the patient, reducing significantly the unnecessary use of antimicrobial drugs. In this context, the host transcriptome during the infection is proving to be a promising target to find out infectious disease-associated biomarkers8–12. Many studies have focused on the identification of host-specific transcriptomic biomarkers in viral and bacterial infections in both adults and children12–18. Out of all of these transcriptomic signatures, one in particular stands out owing to the very few mRNAs required16. By screening samples from febrile children using expression microarrays, Herberg et al.16 found two genes with different expression levels depending on the nature of the causative pathogen: Interferon Induced Protein 44 Like (IFI44L) gene is up-regulated in viral febrile children and, in contrast, Family with Sequence Similarity 89 Member A (FAM89A) gene has elevated expression in febrile children with bacterial infection. These biomarkers alone are capable of differentiating between bacterial and viral infections with high sensibility and specificity. These finding were subsequently replicated by Kaforou et al.15 in a cohort of febrile children under 60 days of age using the microarrays data from Mahajan et al.14. Most recently, Barral-Arca et al.19 validated this signature using whole transcriptome data from patients suffering from acute diarrhea with bacterial and viral etiology; they found that this signature clearly discriminated between viral and bacterial infections regardless of the pathogen, severity and ancestry of patients.
Even though expression microarrays are one of the most powerful screening approaches for the identification of disease-related biomarkers, findings need to be validated with highly sensitive techniques that allow the precise measurement of the expression levels of the candidate genes20,21. Moreover, their applicability as a diagnostic tool in clinical practice has been extensively questioned in the literature mainly due to problems related to standardization, reproducibility and reliability22–24. In contrast, real-time PCR (qPCR) has been considered the “gold standard” in genetic expression studies25, and it has been demonstrated to offer a sustainable approach to validate microarrays discovery results with high accuracy, time savings and a relatively low cost21,26,27. In general, there is a good correlation between microarray and qPCR expression results28, but this correlation is strongly affected by technical as well as analytical factors29–31. Owing to the lack of experimental details in many qPCR-based assays, some studies have emphasized the need of high quality qPCR laboratory performance32.
A rapid bedside test device development based on these two transcripts is desirable but difficult to achieve due to technology limitations33,34. Meanwhile, a well optimized, cost- and time-effective qPCR-based protocol to discriminate between viral and bacterial infections could be easily implemented in the hospital molecular diagnostics laboratory routine.
As a first step for the translational application of this host expression signature, we developed a two-step RT-qPCR assay to test and validate the 2-transcript signature of genes FAM89A (RefSeq mRNA: NM_198552.2) and IFI44L (RefSeq mRNA: NM_006820.3). We tested the expression of these two genes in a new cohort of febrile children with confirmed bacterial infection, confirmed viral infection, and healthy controls. In contrast to previous studies that only considered bacterial and viral patients, we introduced a control group in order to assess the test performance in healthy subjects. Our results are discussed in the light of previous findings15,16,19.
Results
Description of the cohort
A total of 25 samples were collected from febrile patients with confirmed viral (n = 11) and bacterial (n = 14) infection in acute phase of the disease. In addition, we collected 10 samples from healthy controls for the comparisons.
All patients were of self-reported South-European ancestry. Demographic as well as some clinical parameters of the cohort are described in Table 1 and Supplementary Table S2. There were not statistically significant differences of demographic features and white blood cell types between bacterial and viral groups (Table 1).
Table 1.
Demographic and clinical characteristics of the cohort.
| Characteristics | All | Bacterial | Viral | Control | Bacterial vs. Viral (P-value) |
|---|---|---|---|---|---|
| n | 35 | 14 | 11 | 10 | — |
| Age; median (IQR) | 2.2 (1.4–8.7) | 2.3 (1.2–8.7) | 3.5 (1.0–6.4) | 1.6 (1.5–8.5) | 0.809 |
| Sex (% Female/Male) | (43/57) | (36/64) | (45/ 55) | (50/50) | 0.697 |
| Admitted to hospital (%) | 84 | 93 | 72 | — | — |
| PICU (%) | 43 | 54 | 25 | — | — |
| Admission time (days); median (IQR) | 8.0 (6.0–13.0) | 10.0 (10.0–15.0) | 6.5 (5.5–7.0) | — | — |
| C-reactive protein (mg/L); median (IQR) | 46.0 (19.0–241.0) | 190.1 (71.25–198.8) | 9.6 (4.2–43.5) | — | — |
| Neutrophils; median (IQR) | 11.1 (7.5–14.6) | 11.6 (7.6–14.8) | 10.2 (6.2–13.3) | — | 0.250 |
| Lymphocytes; median (IQR) | 1.7 (0.8–4.0) | 1.4 (0.7–3.4) | 2.4 (1.0–3.8) | — | 0.654 |
| Monocytes; median (IQR) | 0.8 (0.4–1.2) | 1.1 (0.6–2.0) | 0.6 (0.3–1.0) | — | 0.072 |
| Days from symptoms (fever) to blood collection; mean | 2.36 | 2.64 | 2.00 | — | — |
| Maximum Tª before admission; median (IQR) | 39.0 (38.2–39.5) | 39.1 (38.6–39.9) | 39.0 (38.1–39.1) | — | — |
| Pathogens (%) | |||||
| Neisseria meningitidis | — | 42.9 | — | — | — |
| Streptococcus pneumoniae | — | 14.3 | — | — | — |
| Escherichia Coli | — | 21.4 | — | — | — |
| Staphylococcus aureus | — | 14.3 | — | — | — |
| Group A Streptococcus pyogenes | — | 7.1 | — | — | — |
| Enterovirus | — | — | 36.3 | — | — |
| Respiratory syncytial virus | — | — | 27.3 | — | — |
| Influenza A | — | — | 18.2 | — | — |
| Adenovirus | — | — | 18.2 | — | — |
The bacterial cohort consisted of six PICU admitted patients with sepsis and/or meningitis syndrome infected with Neisseria meningitidis (n = 5 meningococcus serogroup B and n = 1 meningococcus serogroup W135), three patients with focal pneumonia caused by Streptococcus pneumoniae (n = 2; one was a PICU patient) and GAS (Group A Streptococcus pyogenes; n = 1), three patients with Escherichia coli urinary tract infection, and two patients with bone infection and pyogenic infection with the detection of Staphylococcus aureus as causative pathogen.
The viral cohort included four patients with Enterovirus viral meningitis; three patients with bronchiolitis produced by respiratory syncytial virus (RSV), two adenovirus positive patients with respiratory illness (one PICU patient) and two patients with Influenza A virus infection (one admitted to PICU). All patients classified as “viral” were negative in the blood culture test result.
Best reference gene selection
The expression levels of a reference gene should be virtually constant under different conditions and tissues. The raw threshold cycle (Ct; the PCR cycle at which a specific fluorescence becomes detectable) results obtained from each candidate (Supplementary Table S3) gene were represented in Fig. 1 and analyzed through descriptive statistics (mean, median, minimum, maximum and SD; Supplementary Table S4).
Figure 1.
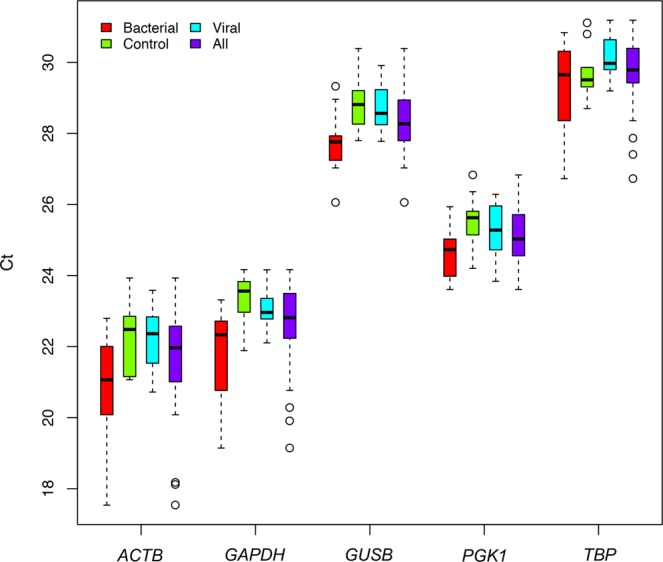
Boxplot of Ct raw data. Raw Ct data (y-axis) obtained from qPCR assay of candidate reference genes (x-axis) in viral, bacterial, and control cohorts. The box represents the interquartile range (25th to the 75th) containing the middle 50% of the data, the line in the box represents the median, and the whiskers represent the ranges for the bottom 25% and the top 25% of the data values, excluding outliers.
The mean Ct values of the candidate reference genes ranged from 21.6 to 29.7, indicating a moderate to relatively high expression of these genes in blood, with ACTB being the highest expressed gene and TBP the lowest. The highest gene expression stability between samples is represented by the reference genes PGK1 (SD = 0.843), GUSB (SD = 0.907) and TBP (SD = 0.997), while GAPDH (SD = 1.200) and ACTB (SD = 1.483) showed the highest instability values. Instability of the latter two genes seems to derive from the cohort of bacterial patients (SD = 1.310 for GAPDH, and SD = 1.665 for ACTB).
Determination of stability using available algorithms for best reference gene selection provided similar results considering the whole cohort (Fig. 2). Results from comprehensive ranking (Supplementary Table S5), which uses the stability scores obtained from Bestkeeper, ∆Ct method, NormFinder and geNorm tools to calculate an overall ranking, showed that the most stable candidates (from high stability to less stability) are GUSB (1.32), PGK1 (1.41) and TBP (2.91), while the genes with the worst stability values are GAPDH (3.72) and ACTB (5.00). This stability ranking is consistent when control, viral and bacterial patients are analyzed separately (with the exception of viral infected patients, in which GAPDH is ranked in second place, before TBP and PGK1 genes), with GUSB, PGK1 and TBP as the three most stable genes.
Figure 2.

Comprehensive stability ranking results of the different candidate reference genes in the different cohorts.
We additionally investigated the correlation between different algorithms by analyzing the correlation matrix obtained from the different stability rankings (Supplementary Fig. S1). The highest correlation was found to be between NormFinder vs. ∆Ct (Pearson r2 = 0.96; P-values range: 0.01–0.001) and RefFinder vs. ∆Ct (Pearson r2 = 0.96; P-values range: 0.01–0.001), whilst the lowest correlation was between NormFinder vs. Bestkeeper (Pearson r2 = 0.63; P-values range: 0.1–1). Despite the differences in stability rankings between the different methods, these results point to moderate to high correlation between them.
Fold change calculation
We calculated the fold change of bacterial and viral patients through the delta-delta Ct method or 2−∆∆Ct method35 using samples from healthy individuals as calibrator control (Supplementary Fig. S2). FAM89A gene yielded much lower expression values (mean Ct = 33.11) than IFI44L gene, and this was reflected in their fold change values with respect to control samples. Slight expression differences between bacterial and viral groups were recorded, with bacterial patients showing the highest FAM89A expression. Single gene normalization produced variable results, and reference genes with lower stability score (GAPDH and ACTB) yielded inverse expression pattern to FAM89A gene, pointing to problems derived from the use of only one gene to normalize the data, especially in genes with low expression values. This is also reflected in the correlation analysis using relative expression data (∆Ct) of viral, bacterial and control groups (Supplementary Fig. S3). In general, despite the overall good correlation values between normalizations, the worst correlation (Pearson r2 < 0.5) was generated by gene pairs in which GADPH and ACTB were involved. This was noted when analyzing all samples together or the viral-bacterial group alone (all groups: GAPDH–ACTB [Pearson r2 = 0.41, P-value range: 0.05–0.01], GAPDH–TBP [Pearson r2 = 0.43, P-value range: 0.01–0.001]; viral-bacterial group: GAPDH–ACTB [Pearson r2 = 0.38, P-value range: 0.1–0.05], GAPDH–TBP [Pearson r2 = 0.37, P-value range: 0.1–0.05], ACTB–PGK1 [Pearson r2 = 0.48, P-value range: 0.05–0.01]).
In addition, when we investigated the discrimination power of FAM89A using ∆Ct data we did not find consistent results among the different reference gene(s) used to normalize the expression (Supplementary Table S6). For the FAM89A gene, the use of multiple reference genes normalization produced more consistent results.
In the case of IFI44L gene, we recorded higher expression values (mean Ct = 27.67), and the fold change differences between viral and the other groups were clearly visible (Supplementary Fig. S2). Correlation between different normalization reference possibilities was very high in all cases (P-value = 0–0.001), including those pairs involving GADPH and ACTB (lower values: ACTB–GAPDH: Pearson r2 = 0.86; GAPDH–TBP: Pearson r2 = 0.85) (Supplementary Fig. S3).
Remarkably, the ability of IFI44L gene to separate viral group from bacterial and control groups is maintained constant regardless of the reference gene used to normalize the expression data (Supplementary Table S6; post-hoc analysis P-value range: 2.5 × 10−4–3.0 × 10−6 for viral vs. bacterial groups; P-values 8.0 × 10−4–3.6 × 10−6 for viral vs. control groups).
In both FAM89A and IFI44L genes we obtained very similar correlation results when control samples were removed from the analysis.
Disease risk score stability
We investigated if DRS is influenced by the use of different references gene(s) (Supplementary Fig. S4).
We observed a very high correlation between DRS values independently of the reference gene(s) (Pearson r2 ranging from 0.97 to 1.00; P-values range: 0–0.001; Supplementary Fig. S5) in both the whole cohort and the viral-bacterial sub-cohort. In addition, we only observed statistically significant differences in DRS (Supplementary Table S7), calculated using the different reference gene options, between the following pairs: ACTB vs. GUSB (P-value = 0.042), ACTB vs. TBP (P-value = 0.007) and GAPDH vs. TBP (P-value = 0.016) considering all dataset; and only in one pair (ACTB vs. TBP; P-value = 0.044) when we removed control samples from the analysis. Genes considered less stable according to the stability ranking (ACTB and GAPDH) were involved in all significantly different DRS values.
When we compared the ability of the DRS to distinguish between viral and bacterial infections using different normalization references, we found a statistically significant difference in the DRS of children affected by bacterial compared to those with viral infections, in all cases and independently of the normalization option employed (Table 2; P-values range: 7.03 × 10−5–2.37 × 10−4). These differences were also evident when we examined the ROC curves obtained considering different normalization references (Supplementary Fig. S6; AUC: 89–93%).
Table 2.
Statistical assessment of 2-transcript Disease Risk Score (DRS) assay to differentiate between viral and bacterial groups using different reference gene(s) to normalize the expression.
| RG | p-values | AUC (%) | |||
|---|---|---|---|---|---|
| Shapiro-Wilk test | f-test (variances) | t-test (groups) | |||
| Single RG normalization | GUSB | 0.286 | 0.618 | 7.03 × 10−5 | 92.2 |
| PGK1 | 0.222 | 0.253 | 2.37 × 10−4 | 89.0 | |
| TBP | 0.288 | 0.927 | 8.62 × 10−5 | 93.5 | |
| GAPDH | 0.185 | 0.260 | 1.66 × 10−4 | 90.3 | |
| ACTB | 0.210 | 0.258 | 7.41 × 10−5 | 92.2 | |
| Multiple RG normalization | GUSB-PGK1 | 0.265 | 0.364 | 1.37 × 10−4 | 91.0 |
| GUSB-PGK1-TBP | 0.296 | 0.495 | 1.04 × 10−4 | 92.2 | |
| GUSB-PGK1-TBP-GAPDH | 0.259 | 0.401 | 1.16 × 10−4 | 91.6 | |
AUC: area under the ROC curve; RF: reference gene.
All these data point to a good overall stability of the DRS, hence the test result is not affected by the use of different references to normalize the expression data.
Test performance
When we analyzed DRS data obtained from the most stable three-gene combination (GUSB–PGK1–TBP) we observed that the expression signature can properly discriminate between viral and bacterial infections (Fig. 3A; P-value = 1.04 × 10−4; AUC = 92.2% [95% CI: 81.9–100%]). Overall, bacterial patients yielded lower DRS values (median = 0.104; IQR = [0.025–0.215]) than viral patients (median = 0.315; IQR = [0.270–0.365]). The optimal threshold point value representing the maximum difference between sensitivity and specificity (the point on the ROC curve farthest from the equality, i.e., from the diagonal) was 0.248 (Fig. 3A,C; sensitivity = 90.9% [95% CI: 72.7–100%]; specificity = 85.7% [95% CI: 64.3–100%]). The proportion of positive results that are true positives (PPV; in our case positive means bacterial infection) was 83.3% whereas the proportion of negative results that are true negatives (NPV; in our case negative means viral infection) was 92.3%. The high performance of the test fits very well with previous findings15,16,19.
Figure 3.

Evaluation of DRS test performance. (A) Boxplot of DRS values from bacterial and viral infected samples using 1-transcript signature (IFI44L). Horizontal red dotted line indicates the optimal threshold value. (B) Boxplot of DRS values from bacterial and viral samples using 2-transcript signature. Horizontal blue dotted line indicates the optimal threshold value. (C) ROC curves generated from 2-transcript (blue) and 1-transcript (red) (IFI44L) DRS with AUC values and 95% CI. We used a t-test to evaluate statistical differences in DRS values between bacterial and viral patients in panels (A,B). The box represents the interquartile range (25th to the 75th) containing the middle 50% of the data, the line in the box represents the median, and the whiskers represent the ranges for the bottom 25% and the top 25% of the data values, excluding outliers.
DRS was not able to distinguish between bacterial and control samples (Supplementary Fig. S4). Despite this fact, control samples clearly show a narrower range of DRS values than the samples from bacterial infection, and they fall close to the upper DRS values of the bacterial patients.
Finally, we evaluated the ability of the assay to differentiate between the viral and bacterial group using only one of the two genes. Surprisingly, the results obtained using only expression data from IFI44L gene were similar to those derived from 2-gene DRS score (Fig. 3B,C; P-value = 3.59 × 10−5; AUC = 94.1% [95% CI: 85.3–100%]). In this case, the optimal threshold point value was −0.926, yielding sensitivity and specificity values of 90.9% and 92.8%, respectively (Fig. 3B).
To validate this unexpected result, we interrogated microarray data from Herberg et al.16 and similar values than those from the 2-transcripts were obtained for both definitively bacterial and viral samples (Fig. 4A, P-value = 6.61 × 10−17; Fig. 4B; P-value = 8.49 × 10−21; Fig. 4C; AUC = 0.91, 95% CI = [0.87–0.96]) and for probable bacterial and viral cohort (Supplementary Fig. S7A, P-value = 1.47 × 10−7; Supplementary Fig. S7B; P-value = 3.59 × 10−5; Supplementary Fig. S7C; AUC = 0.93, 95% CI = [0.85–1.00]).
Figure 4.

Evaluation of DRS test performance in microarray data from definitive bacterial and definitive viral infection in febrile children. (A) Boxplot of DRS values using 1-transcript signature (IFI44L). (B) Boxplot of DRS using 2-transcript signature. (C) ROC curves generated from 2-transcript (black) and 1-transcript ([IFI44L; blue], [FAM89A; yellow]) DRS with AUC values and 95% CI. We used a t-test to evaluate statistical differences in DRS values between bacterial and viral patients in panels (A,B). The box represents the interquartile range (25th to the 75th) containing the middle 50% of the data, the line in the box represents the median, and the whiskers represent the ranges for the bottom 25% and the top 25% of the data values, excluding outliers.
Likewise, monogenic expression signature was tested in whole transcriptome data (RNA-seq) from patients suffering bacterial and viral diarrhea (Fig. 5). Consistently, we observed that IFI44L expression signal alone can seamlessly separate patients with diarrhea of bacterial and viral etiologies; the results being comparable to the DRS values obtained using both FAM89A and IFI44L together (Fig. 5A, P-value = 7.00 × 10−11; Fig. 5B; P-value = 2.90 × 10−10; Fig. 5C; AUC = 0.80, 95% CI = [0.73–0.87]).
Figure 5.

Evaluation of DRS test performance in whole transcript data from bacterial and viral diarrhea patients. (A) Boxplot of DRS values using 1-transcript signature (IFI44L). (B) Boxplot of DRS using 2-transcript signature. Dots indicate disease severity. Dots indicate disease severity. (C) ROC curves generated from 2-transcript (blue) and 1-transcript ([IFI44L; green], [FAM89A; red]) DRS with AUC values and 95% CI. We used a t-test to evaluate statistical differences in DRS values between bacterial and viral patients in panels (A,B). The box represents the interquartile range (25th to the 75th) containing the middle 50% of the data, the line in the box represents the median, and the whiskers represent the ranges for the bottom 25% and the top 25% of the data values, excluding outliers.
Therefore, test performance is not compromised by the use of IFI44L expression alone.
Discussion
Disease-associated biomarkers are currently a major research focus in the medical field, as their study and translational applications may contribute to understand, diagnose, prognosticate and treat patients in more efficient ways. Several biomarkers (proteins or mRNA) have been evaluated in the context of a broad range of infectious diseases12,16,18,36 and non-infectious complex diseases37–40 so far. Here, we have shown that a RT-qPCR assay for a 2-transcript host expression signature (FAM89A and IFI44L genes) inferred from microarray data is able to efficiently separate viral from bacterial infections. In addition, IFI44L expression signature alone seems to differentiate both groups, thus saving time, effort, and costs over the two genes signature. Blood-based host transcriptomics in pediatric patients constitute a promising tool for disease biomarker identification. A test based on host expression data offers additional benefits compared to the typical routine testing. Bacterial culture tests from sterile sites, routinely implemented in hospitals, usually cover the most common pathogens, often leading to false negative results, particularly when the infection resides in a non-accessible site, the patient was previously treated with antibiotics or if the causative pathogen is not interrogated16,41,42. Moreover, the time from admission to test results usually takes more than 24 hours. A rapid host-based expression assay could enhance the diagnosis of febrile children with viral or bacterial etiologies and will reduce the need of clinicians prescribing antibiotics as a preventive tool. Thus, an early differentiation between viral and bacterial patients will help improve triage in emergency departments, decrease the misuse of antibiotics, and guide the clinics to a more precise diagnosis. This will prevent the onset of antibiotics resistance as well as more broadly improving the quality of the health system.
As a first step towards establishing a host-based test in hospital routine, we have investigated two mRNA biomarkers, previously identified in microarray expression experiments16, in a custom relative expression RT-qPCR assay using commercially available Taqman probes. One of the most critical issues in relative expression experiments based on qPCR can be the selection of appropriate reference genes. It has been reported that the accuracy of the results obtained is strongly dependent on the selection of candidate reference genes. Validations of housekeeping genes must be specific of a particular experimental condition, and are a key component in assessing any new condition32. Because of this, important efforts have been devoted in the present study to the evaluation and selection of the most convenient reference genes. Our experiments indicate that the three most stable reference genes, from higher to lower stability, are GUSB, PGK1 and TBP (Fig. 2). Discrepancies between stability rankings were previously reported and may be attributed to different algorithms implemented in the available methods43. Our data indicate that the well-known genes GAPDH and ACTB are too unstable; this could be due to their known variable expression patterns in different tissues and conditions44,45. In addition, it has been reported that the expression of these two genes can be modified during infection46,47. For routine applications, it is reasonable to strive for a balance between the benefits of adding a new reference gene to achieve extra precision during the normalization process, and the cost of including additional reference genes. Keeping this in mind and considering that the accepted recommendation is to use at least two reference genes to normalize qPCR data48, we chose to employ the three most stable reference candidates to normalize the qPCR expression of FAM89A and IFI44L genes.
We found that a RT-qPCR test based on DRS displays very good performance at distinguishing between viral and bacterial infections in febrile children (P-value = 1.04 × 10−4; AUC = 92.2% [95% CI: 81.9–100%]; Fig. 3A,C), with a potential clinical use for both “ruling in” (specificity >85%) and “ruling out” (sensitivity >90%) viral infections. These results are in good agreement with previous findings15,16,19. Contrary to microarrays and RNA-seq studies, DRS based on the RT-qPCR assay is higher in patients with viral infection and lower in patients affected by bacterial infection. This difference is due to the methodological differences between the microarray and qPCR techniques for measuring gene expression. Microarrays data work with expression intensity values (higher expression is related to higher intensity); hence, calculation of DRS from these data usually returns higher DRS values when FAM89A gene is highly expressed with respect to IFI44L gene, i.e., in bacterial samples. However, lower DRS values are expected in viral patients due to the higher expression of gene IFI44L compared to gene FAM89A. In contrast, qPCR uses Ct values and, therefore, higher expression is represented by lower Ct values. When the DRS formula is applied to qPCR data, high expression of gene IFI44L and gene FAM89A results in low ∆Ct(IFI44L) and ∆Ct(FAM89A), respectively. Hence, samples with higher expression of gene IFI44L and lower expression of gene FAM89A (viral patients) will give a higher DRS value than samples with lower expression of gene IFI44L and higher expression of gene FAM89A (bacterial samples). Notwithstanding this difference between DRS scales obtained from microarray-based and qPCR-based studies, this does not interfere with the test performance.
The DRS 2-transcript test was not able to differentiate between control and bacterial patients but it clearly distinguished the viral group from bacterial and control groups. Note, however, that in the clinical practice the utility of this test lies in its ability to efficiently separate viral from bacterial children with fever; in such contexts, controls do not necessarily play a role.
The expression signals produced by FAM89A gene showed low expression level, low expression differences between viral and bacterial cohort and high variability when different normalization controls are employed. In contrast, IFI44L gene produces a more robust and reliable expression signal and it is less prone to variability derived from the use of different normalization references. Surprisingly, our results indicate that a test based on a single expression signal generated by the IFI44L gene not only suffices to differentiate accurately between viral and bacterial children with fever, but it also performs slightly better than the 2-transcript signature (Fig. 3B,C; P-value = 3.59 × 10−5; AUC = 94.1% [95% CI: 85.3–100%]; sensitivity >90% and specificity >92%). In addition, we succeeded in validating this finding in two independent cohorts composed by microarray data from febrile children with definitively bacterial and viral infection16 (Fig. 4A,C; P-value = 6.61 × 10−17; AUC = 0.91, 95% CI = [0.87–0.96]) and whole transcriptome data from acute diarrhea of viral and bacterial aetiology (Fig. 5A,C; P-value = 7.00 × 10−11; AUC = 0.80; 95% CI: 0.73–0.87). Both microarray and RNA-seq data also allowed to confirm the limited informativeness of the FAM89A signature. This simplification of the test to a single gene expression provides the opportunity to save time, effort and costs.
Some interesting issues remain to be investigated with regards to this transcriptomic signature, namely: (i) the evolution of gene expression over time from the onset of fever, and (ii) variations in its expression patterns with regards to the causative pathogen, co-infections and/or mixed viral/bacterial infections, (iii) non-infectious febrile illness (i.e. rheumatoid arthritis) and (iv) adult patients.
The present study represents an important step towards the implementation of a functional host transcriptomic-based test capable of distinguishing between viral and bacterial infections in febrile children, thus contributing to a more accurate diagnosis and treatment of pediatric patients.
Methods
Sample size and statistical power calculation
In order to estimate the minimum sample size required to detect differences between viral and bacterial groups with high probability (0.8–0.9) we performed a post-hoc statistical power analysis based on these two transcripts data (IFI44L and FAM89A) previously published by Herberg et al.16 retrieved from GEO database (Gene Expression Omnibus; https://www.ncbi.nlm.nih.gov/geo/; accession number: GSE72829). We calculated the Disease Risk Score (DRS; see Data analysis in M&M section for more details about DRS calculation) mean from definitively bacterial (n = 52) and definitively viral (n = 92) samples to obtain delta parameter (mean difference) of each group as well as standard deviation (SD) from both groups together. We used pwr R package49 to carry out the analysis with a significance level of 0.05 and a statistical power of 80% and 90%.
This analysis indicates that a sample size of n = 8.4 samples per group is needed to discriminate between viral and bacterial group with a statistical power of 80% (delta = 0.69; SD = 0.47). When we raised the power to 90% we obtained a sample size of n = 10.8 samples per group. Therefore, we need a minimum of 8 samples per group to have enough statistical power to detect true differences between both groups.
Sample collection
The study was approved by the Ethical Committee of Clinical Investigation of Galicia (CEIC ref. 2016/331) and was conducted according to the principles of the Declaration of Helsinki and in accordance with all applicable Spanish normative, namely, Law for Biomedical Research (Law 14/2007-3 of July), Law 41/2002 of Autonomy of the Patient, Decree SAS/3470/2009 for Observational Studies and Law 15/1999 of Data Protection. Written informed consent was obtained from a parent or legal guardian for each subject before study inclusion.
PAXgeneTM tubes were used to collect 2.5 ml blood samples from 25 febrile children admitted to Hospital Clínico Universitario from Santiago de Compostela (Spain) with confirmed bacterial (n = 14) and confirmed viral infections (n = 11). The clinical selection and classification of the patients was carried out according to the decision scheme previously published by Herberg et al.16. Additionally, we collected healthy control samples (n = 10) for the comparisons. Demographic and clinical data from these patients and controls are summarized in Table 1.
PAXgeneTM RNA samples were collected within 24 hours of admission to hospital following the manufacturer guidelines, stored at −20 °C for 24 hours and then stored at −80 °C until the RNA extraction.
RNA extraction and quantification
Samples were thawed and left at room temperature overnight before extraction in order to increase the RNA yields. RNA extraction was performed using PAXgene Boold RNA kit (Qiagen). Samples were treated with RNase-free DNase I during the extraction process to destroy all DNA present in the sample that could interfere in downstream applications. After extraction, RNA amount and purity of the samples was measured using NanoDrop 1000 (ThermoFisher) (Supplementary Table S1).
RT-qPCR reactions
RT-PCR reaction was carried out to obtain cDNA from RNA samples. Some preliminary assays were performed in order to check and select an adequate RNA and cDNA input concentration and to test the expression level of the genes in our samples. High level of Ct variation is indicative of poor precision and consequently low fold changes are more difficult to quantify accurately. Therefore, we normalized RNA input for RT-PCR reaction to 100 ng/μl using treated ultrapure DEPC-H2O. A volume of 10 μl of this dilution was used in the reaction (total RNA template was 1 μg). Firstly, 1 μl of Random primers p(dN)6 (Roche) and 1 μl of DNTPs mix (Invitrogen) were added to the template. Random primers are hexamers of random sequences that hybridize to random sites to perform conversion of the whole RNA to cDNA. The mixture was then heated to 65 °C for 5 minutes and immediately placed in ice. After a brief centrifugation, 4 μl of 5× First-Strand Buffer, 2 μl of DTT (0.1 M) and 1 μl of Recombinant RNasin® Ribonuclease Inhibitor (Promega) were added and the mixture was incubated at 25 °C for 2 minutes. Finally, 1 μl (200 units) of SuperScript™ II Reverse Transcriptase (Invitrogen) was introduced to the mix for a final reaction volume of 20 μl. The RT-PCR reaction was carried out under the following conditions: 15 minutes at 25 °C, 50 minutes at 42 °C, 15 minutes at 70 °C.
For the qPCR assay we used 10 μl of Master mix (2×) Kapa Probe Fast qPCR Kit, 7 μl of MiliQ water, 1 μl of the TaqMan assay mix and 2 μl of the cDNA (1:5 dilution). In order to test the assay precision, three replicates of each gene per sample were run. Replicates with standard deviations over 0.5 were repeated. qPCR reaction was carried out in a StepOnePlus Real-Time PCR System (Applied Biosystems) with the following parameters: 2 minutes at 50 °C, 10 minutes at 95 °C, 40 cycles of 15 seconds at 95 °C, and 1 minute at 60 °C.
Selection of best reference gene candidates
The selection of the best reference gene(s) for the assay was conducted through testing five of the most commonly used human reference genes: PGK1 (Phosphoglycerate Kinase 1), ACTB (beta-actin), GUSB (beta-glucuronidase), TBP (TATA-binding protein) and GAPDH (glyceraldehyde 3-phosphate dehydrogenase) (TaqMan probes IDs: Hs00943178_g1, Hs99999903_m1, Hs00984230_m1, Hs00939627_m1, Hs00427621_m1, Hs03929097_g1, respectively) (Table 3). We selected best reference gene candidates using different software packages available: geNorm50, NormFinder51, BestKeeper52, and the ∆Ct method53. BestKeeper and ∆Ct method use raw Ct data for calculations, while geNorm and NormFinder work on relative quantities. Finally, we calculated the overall final ranking using the web-based tool RefFinder54, which considers the ranking of the above-mentioned tools to assign a final weight to each candidate gene, and calculates the geometric mean of their weights to build the final ranking.
Table 3.
List of candidate reference genes and genes of interest evaluated in the present study. RG: reference gene; GOI: gene of interest. Chromosome locations are referred to the reference genome GRCh38.
| RG/GOI | Symbol | Protein name | Accession number | Chromosome location | Taqman probe |
|---|---|---|---|---|---|
| RG | ACTB | β-actin | NM_001101 | Chr.7: 5527148–5530601 | Hs99999903_m1 |
| RG | GAPDH | Glyceraldehyde 3-phosphate dehydrogenase | NM_002046 | Chr.12: 6534405–6538375 | Hs03929097_g1 |
| RG | GUSB | β-glucuronidase | NM_000181 | Chr.7: 65960684–65982314 | Hs00939627_m1 |
| RT | PGK1 | Phosphoglycerate kinase 1 | NM_000291 | Chr.X: 78104169–78126827 | Hs00943178_g1 |
| RG | TBP | TATA box-binding protein | NM_003194 | Chr.6: 170554333–170572870 | Hs00427621_m1 |
| GOI | IFI44L | Interferon induced protein 44 like | NM_006820 | Chr.1: 78620382–78646145 | Hs00915294_g1 |
| GOI | FAM89A | Family with sequence similarity 89 member A | NM_198552 | Chr.1: 231018958–231040249 | Hs00293357_m1 |
We constructed the correlation matrix between rankings using R software55.
Gene expression of FAM89A and IFI44L
We followed a qPCR TaqMan-based approach to test the host expression balance between these two transcripts in febrile pediatric patients. Probe IDs corresponding to FAM89A and IFI44L transcripts were first retrieved from Chip GeneChipTM HG-U133 Plus 2.0 (Affymetrix Inc; 226448_at and 204439_at, respectively). TaqManTM (ThermoFisher Scientific) probes exactly matching the array probes from GeneChipTM were then selected (assay IDs: Hs00293357_m1 for the FAM89A transcript and Hs00915294_g1 for the transcript IFI44L) in order to avoid potential variations due to the use of different probe sequences29.
Data analysis
A relative quantification method 2−∆∆Ct 35 was used to normalize the samples with respect to reference genes and obtain fold change values of viral and bacterial cohorts using control samples as calibrator. TaqMan probes are extensively checked, and therefore an efficiency close to 100% can be assumed (see details: https://assets.thermofisher.com/TFS-Assets/LSG/Application-Notes/cms_040377.pdf; last access 29/01/2019).
We evaluated the statistical significance in demographics and cell types between viral and bacterial cohorts using one-factor ANOVA or Wilcoxon test for numeric variables and depending on the data (normality, equality of variances and homoscedasticity), and Fisher’s exact test for categorical variables.
Different statistical tests were carried out on relative expression data from FAM89A and IFI44L genes depending on the assumptions of normality (Shapiro-Wilk) and homoscedasticity (Breusch-Pagan): one factor ANOVA, robust one-way ANOVA and Kruskal-Wallis. Post-hoc tests analyses were performed using Tukey test and Games-Howell test, as appropriate.
We followed Kaforou et al.56 to compute the Disease Risk Score (DRS) for each sample. We first obtained individual DRS values using normalized values (∆Ct). We then incremented the scale of the ∆Ct values by a factor of 10 in order to avoid negative values when the log-scale is applied. The final DRS formula was as follows:
After DRS calculation we studied the impact of using different single and multiple reference genes on DRS stability. Firstly, we performed a correlation analysis to check the extent of correlation between DRS calculated from different reference gene combinations. Secondly, we investigated if there were significant differences in DRS using different reference genes as normalizers. Due to the fact that the data did not meet the normal distribution (Shapiro-Wilk test), we used a non-parametric Kruskal-Wallis and a post-hoc Games-Howell tests to carry out the analysis. Finally, we used a t-test for independent samples to evaluate the statistical significance of differences in DRS values between bacterial vs. viral patients and how the use of different reference gene to normalize the data affects the capacity of DRS to differentiate between these two groups. The latter was done after testing that the data followed a normal distribution (Shapiro-Wilk test) and met the assumption of equality of variances (f-test).
We investigated the discrimination power of the individual IFI44L gene signature using the formula:
We maintained the negative sign to avoid points of the ROC (Receiver Operating Characteristic) curve being located below the diagonal, meaning an inverse relationship between the measure and the event probability (i.e., the larger the measure, the less likely the event).
We used the ROCR package57 to evaluate the predictive accuracy of the 2-transcript signature, by creating a ROC curve to asses test performance; this represents the sensitivity (true positive rate) vs. 1 − specificity (false positive rate) for all possible values of the cut-point between two status. In addition, we calculated the area under the ROC curve (AUC) and the confidence interval as well as the positive predictive value (PPV) and negative predictive value (NPV) to measure the diagnostic potential of the expression signature. The optimal cut-point value, defined as the point on the ROC curve that classifies most of the individuals correctly (maximizing sensitivity and specificity), was calculated using the Youden method included in the OptimalCutPoints R package58. The calculation of the confidence intervals for sensitivity and the specificity was based on a stratified bootstrap resampling.
Graphical representations and statistical tests were carried out using R software55.
Monogenic signature validation in microarray and whole transcriptome data
We evaluated the discrimination power of the 1-transcript signature (IFI44L) in microarray data from Herberg et al.16 retrieved from GEO database (accession number: GSE72829) consisting of n = 52 definitively bacterial and n = 92 definitively viral samples from febrile children. Furthermore, we also checked the performance in probable viral and probable bacterial cohorts included from the same study (n = 42 probable bacterial; n = 5 probable viral). Samples were processed and analyzed using same methodology described in16. Normalization of raw data was carried using the same method as in16 (lumi R package59).
We also tested the IFI44L signature in whole transcriptome data (RNA-Seq) previously analyzed in Barral-Arca et al.19 and also available in GEO database (accession number: GSE69529). RNA expression dataset includes Mexican children (n = 174) younger than 10 years of age with acute diarrhea associated with a single viral or bacterial pathogen [dataset]60. Processing of RNA-seq data was carried out as described previously19. Normalization of the raw whole transcriptome data was performed through the same procedure used in19 (DESeq2R package61).
Graphical representations and statistical tests were carried out using R software55.
Supplementary information
Acknowledgements
This study received support from the Instituto de Salud Carlos III (Proyecto de Investigación en Salud, Acción Estratégica en Salud): project GePEM ISCIII/PI16/01478/Cofinanciado FEDER) and project DTS19/00049/Cofinanciado FEDER (A.S.) and project ReSVinext ISCIII/PI16/01569/Cofinanciado FEDER (F.M.-T.); Consellería de Sanidade, Xunta de Galicia (RHI07/2-intensificación actividad investigadora, PS09749 and 10PXIB918184PR), Instituto de Salud Carlos III (Intensificación de la actividad investigadora 2007–2012, PI16/01569), Fondo de Investigación Sanitaria (FIS; PI070069/PI1000540) del plan nacional de I + D + I and ‘fondos FEDER’ (F.M.-T.), and 2016-PG071 Consolidación e Estructuración REDES 2016GI-1344 G3VIP (Grupo Gallego de Genética Vacunas Infecciones y Pediatría, ED341D R2016/021) (A.S. and F.M.-T). We would like to thank also the collaboration of the Translational Research Network in Pediatric Infectious Diseases (RITIP – www.ritip.org) and their members. We extend our gratitude to the children and parents/guardians who participated in this study and to the nursery and laboratory services from the Hospital Clínico Universitario de Santiago de Compostela for their effort.
Author Contributions
A.S., F.M.-T. and A.G.-C. conceived and designed the study. M.C.-L., I.R.-C., M.J.-C., J.G.-R. and S.P. were involved in sample recruitment and organization of RNA collections and clinical data. A.G.-C. and F.B. performed the laboratory work. A.G.-C., J.P.-S. and R.B.-A., analyzed the data. A.G.-C. wrote the first draft of the manuscript and it was revised by F.M.-T. and A.S. All the authors contributed to the final version of the manuscript.
Competing Interests
The authors declare no competing interests.
Footnotes
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary information accompanies this paper at 10.1038/s41598-019-48162-9.
References
- 1.Hersh AL, Shapiro DJ, Pavia AT, Shah SS. Antibiotic prescribing in ambulatory pediatrics in the United States. Pediatrics. 2011;128:1053–1061. doi: 10.1542/peds.2011-1337. [DOI] [PubMed] [Google Scholar]
- 2.Gaur AH, Hare ME, Shorr RI. Provider and practice characteristics associated with antibiotic use in children with presumed viral respiratory tract infections. Pediatrics. 2005;115:635–641. doi: 10.1542/peds.2004-0670. [DOI] [PubMed] [Google Scholar]
- 3.Bell BG, Schellevis F, Stobberingh E, Goossens H, Pringle M. A systematic review and meta-analysis of the effects of antibiotic consumption on antibiotic resistance. BMC infectious diseases. 2014;14:13. doi: 10.1186/1471-2334-14-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fauci AS, Marston lD The perpetual challenge of antimicrobial resistance. JAMA. 2014;311:1853–1854. doi: 10.1001/jama.2014.2465. [DOI] [PubMed] [Google Scholar]
- 5.Lee GC, et al. Outpatient antibiotic prescribing in the United States: 2000 to 2010. BMC Med. 2014;12:96. doi: 10.1186/1741-7015-12-96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hicks LA, et al. Outpatient antibiotic prescribing and nonsusceptible Streptococcus pneumoniae in the United States, 1996–2003. Clin Infect Dis. 2011;53:631–639. doi: 10.1093/cid/cir443. [DOI] [PubMed] [Google Scholar]
- 7.WHO. Antimicrobial resistance: global report on surveillance., (WHO Library Cataloguing-in-Publication Data, 2014).
- 8.Sweeney TE, Braviak L, Tato CM, Khatri P. Genome-wide expression for diagnosis of pulmonary tuberculosis: a multicohort analysis. Lancet Respir Med. 2016;4:213–224. doi: 10.1016/S2213-2600(16)00048-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mejias A, et al. Whole blood gene expression profiles to assess pathogenesis and disease severity in infants with respiratory syncytial virus infection. PLoS Med. 2013;10:e1001549. doi: 10.1371/journal.pmed.1001549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Berry MP, et al. An interferon-inducible neutrophil-driven blood transcriptional signature in human tuberculosis. Nature. 2010;466:973–977. doi: 10.1038/nature09247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zak DE, et al. A blood RNA signature for tuberculosis disease risk: a prospective cohort study. Lancet. 2016;387:2312–2322. doi: 10.1016/S0140-6736(15)01316-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.van Houten CB, et al. A host-protein based assay to differentiate between bacterial and viral infections in preschool children (OPPORTUNITY): a double-blind, multicentre, validation study. Lancet Infect Dis. 2017;17:431–440. doi: 10.1016/S1473-3099(16)30519-9. [DOI] [PubMed] [Google Scholar]
- 13.Srugo Isaac, Klein Adi, Stein Michal, Golan-Shany Orit, Kerem Nogah, Chistyakov Irina, Genizi Jacob, Glazer Oded, Yaniv Liat, German Alina, Miron Dan, Shachor-Meyouhas Yael, Bamberger Ellen, Oved Kfir, Gottlieb Tanya M., Navon Roy, Paz Meital, Etshtein Liat, Boico Olga, Kronenfeld Gali, Eden Eran, Cohen Robert, Chappuy Helène, Angoulvant François, Lacroix Laurence, Gervaix Alain. Validation of a Novel Assay to Distinguish Bacterial and Viral Infections. Pediatrics. 2017;140(4):e20163453. doi: 10.1542/peds.2016-3453. [DOI] [PubMed] [Google Scholar]
- 14.Mahajan P, et al. Association of RNA Biosignatures With Bacterial Infections in Febrile Infants Aged 60 Days or Younger. JAMA. 2016;316:846–857. doi: 10.1001/jama.2016.9207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kaforou M, Herberg JA, Wright VJ, Coin LJM, Levin M. Diagnosis of Bacterial Infection Using a 2-Transcript Host RNA Signature in Febrile Infants 60 Days or Younger. JAMA. 2017;317:1577–1578. doi: 10.1001/jama.2017.1365. [DOI] [PubMed] [Google Scholar]
- 16.Herberg JA, et al. Diagnostic Test Accuracy of a 2-Transcript Host RNA Signature for Discriminating Bacterial vs Viral Infection in Febrile Children. JAMA. 2016;316:835–845. doi: 10.1001/jama.2016.11236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bhattacharya S, et al. Transcriptomic Biomarkers to Discriminate Bacterial from Nonbacterial Infection in Adults Hospitalized with Respiratory Illness. Scientific reports. 2017;7:6548. doi: 10.1038/s41598-017-06738-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sampson DL, et al. A Four-Biomarker Blood Signature Discriminates Systemic Inflammation Due to Viral Infection Versus Other Etiologies. Scientific reports. 2017;7:2914. doi: 10.1038/s41598-017-02325-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Barral-Arca R, Pardo-Seco J, Martinon-Torres F, Salas A. A 2-transcript host cell signature distinguishes viral from bacterial diarrhea and it is influenced by the severity of symptoms. Scientific reports. 2018;8:8043. doi: 10.1038/s41598-018-26239-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chuaqui RF, et al. Post-analysis follow-up and validation of microarray experiments. Nat Genet. 2002;32(Suppl):509–514. doi: 10.1038/ng1034. [DOI] [PubMed] [Google Scholar]
- 21.Wang Y, et al. Large scale real-time PCR validation on gene expression measurements from two commercial long-oligonucleotide microarrays. BMC genomics. 2006;7:59. doi: 10.1186/1471-2164-7-59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hackett JL, Lesko LJ. Microarray data–the US FDA, industry and academia. Nat Biotechnol. 2003;21:742–743. doi: 10.1038/nbt0703-742. [DOI] [PubMed] [Google Scholar]
- 23.Petricoin EF, 3rd, et al. Medical applications of microarray technologies: a regulatory science perspective. Nat Genet. 2002;32(Suppl):474–479. doi: 10.1038/ng1029. [DOI] [PubMed] [Google Scholar]
- 24.Shi L, et al. The MicroArray Quality Control (MAQC)-II study of common practices for the development and validation of microarray-based predictive models. Nat Biotechnol. 2010;28:827–838. doi: 10.1038/nbt.1665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shi L, et al. The MicroArray Quality Control (MAQC) project shows inter- and intraplatform reproducibility of gene expression measurements. Nat Biotechnol. 2006;24:1151–1161. doi: 10.1038/nbt1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Provenzano, M. & Mocellin, S. Complementary Techniques. In: Mocellin, S. (eds) Microarray Technology and Cancer Gene Profiling. Advances in Experimental Medicine and Biology 593. Springer, New York, NY (2007). [DOI] [PubMed]
- 27.Dallas PB, et al. Gene expression levels assessed by oligonucleotide microarray analysis and quantitative real-time RT-PCR–how well do they correlate? BMC genomics. 2005;6:59. doi: 10.1186/1471-2164-6-59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Arikawa E, et al. Cross-platform comparison of SYBR Green real-time PCR with TaqMan PCR, microarrays and other gene expression measurement technologies evaluated in the MicroArray Quality Control (MAQC) study. BMC genomics. 2008;9:328. doi: 10.1186/1471-2164-9-328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Etienne Wiguins, Meyer Martha H., Peppers Johnny, Meyer Ralph A. Comparison of mRNA gene expression by RT-PCR and DNA microarray. BioTechniques. 2004;36(4):618–626. doi: 10.2144/04364ST02. [DOI] [PubMed] [Google Scholar]
- 30.Morey JS, Ryan JC, Van Dolah FM. Microarray validation: factors influencing correlation between oligonucleotide microarrays and real-time PCR. Biol Proced Online. 2006;8:175–193. doi: 10.1251/bpo126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Park WD, Stegall MD. A meta-analysis of kidney microarray datasets: investigation of cytokine gene detection and correlation with rt-PCR and detection thresholds. BMC genomics. 2007;8:88. doi: 10.1186/1471-2164-8-88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bustin SA, et al. The MIQE guidelines: minimum information for publication of quantitative real-time PCR experiments. Clin Chem. 2009;55:611–622. doi: 10.1373/clinchem.2008.112797. [DOI] [PubMed] [Google Scholar]
- 33.Gliddon HD, Herberg JA, Levin M, Kaforou M. Genome-wide host RNA signatures of infectious diseases: discovery and clinical translation. Immunology. 2018;153:171–178. doi: 10.1111/imm.12841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Vashist SK, Luppa PB, Yeo LY, Ozcan A, Luong JHT. Emerging Technologies for Next-Generation Point-of-Care Testing. Trends Biotechnol. 2015;33:692–705. doi: 10.1016/j.tibtech.2015.09.001. [DOI] [PubMed] [Google Scholar]
- 35.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 36.Jansen AFM, et al. CXCL9, a promising biomarker in the diagnosis of chronic Q fever. BMC infectious diseases. 2017;17:556. doi: 10.1186/s12879-017-2656-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wagener AH, et al. Toward composite molecular signatures in the phenotyping of asthma. Ann Am Thorac Soc. 2013;10(Suppl):S197–205. doi: 10.1513/AnnalsATS.201302-035AW. [DOI] [PubMed] [Google Scholar]
- 38.Morin PJ. Claudin proteins in human cancer: promising new targets for diagnosis and therapy. Cancer Res. 2005;65:9603–9606. doi: 10.1158/0008-5472.CAN-05-2782. [DOI] [PubMed] [Google Scholar]
- 39.Li LM, et al. Serum microRNA profiles serve as novel biomarkers for HBV infection and diagnosis of HBV-positive hepatocarcinoma. Cancer Res. 2010;70:9798–9807. doi: 10.1158/0008-5472.CAN-10-1001. [DOI] [PubMed] [Google Scholar]
- 40.Gorostidi A, et al. Alphalpha-synuclein levels in blood plasma from LRRK2 mutation carriers. PLoS One. 2012;7:e52312. doi: 10.1371/journal.pone.0052312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Iroh Tam PY, Bernstein E, Ma X, Ferrieri P. Blood Culture in Evaluation of Pediatric Community-Acquired Pneumonia: A Systematic Review and Meta-analysis. Hosp Pediatr. 2015;5:324–336. doi: 10.1542/hpeds.2014-0138. [DOI] [PubMed] [Google Scholar]
- 42.Martin NG, Sadarangani M, Pollard AJ, Goldacre MJ. Hospital admission rates for meningitis and septicaemia caused by Haemophilus influenzae, Neisseria meningitidis, and Streptococcus pneumoniae in children in England over five decades: a population-based observational study. Lancet Infect Dis. 2014;14:397–405. doi: 10.1016/S1473-3099(14)70027-1. [DOI] [PubMed] [Google Scholar]
- 43.Wang Q, et al. Stability of endogenous reference genes in postmortem human brains for normalization of quantitative real-time PCR data: comprehensive evaluation using geNorm, NormFinder, and BestKeeper. Int J Legal Med. 2012;126:943–952. doi: 10.1007/s00414-012-0774-7. [DOI] [PubMed] [Google Scholar]
- 44.Huggett J, Dheda K, Bustin S, Zumla A. Real-time RT-PCR normalisation; strategies and considerations. Genes Immun. 2005;6:279–284. doi: 10.1038/sj.gene.6364190. [DOI] [PubMed] [Google Scholar]
- 45.Lin J, Redies C. Histological evidence: housekeeping genes beta-actin and GAPDH are of limited value for normalization of gene expression. Dev Genes Evol. 2012;222:369–376. doi: 10.1007/s00427-012-0420-x. [DOI] [PubMed] [Google Scholar]
- 46.Dheda Keertan, Huggett Jim F., Bustin Stephen A., Johnson Margaret A., Rook Graham, Zumla Alimuddin. Validation of housekeeping genes for normalizing RNA expression in real-time PCR. BioTechniques. 2004;37(1):112–119. doi: 10.2144/04371RR03. [DOI] [PubMed] [Google Scholar]
- 47.Kuchipudi SV, et al. 18S rRNA is a reliable normalisation gene for real time PCR based on influenza virus infected cells. Virol J. 2012;9:230. doi: 10.1186/1743-422X-9-230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Nicot N, Hausman JF, Hoffmann L, Evers D. Housekeeping gene selection for real-time RT-PCR normalization in potato during biotic and abiotic stress. J Exp Bot. 2005;56:2907–2914. doi: 10.1093/jxb/eri285. [DOI] [PubMed] [Google Scholar]
- 49.Champely, S. Package: Pwr. Retrieved from, http://cran.r-project.org/packagepwr (2013).
- 50.Vandesompele J, et al. Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol. 2002;3:RESEARCH0034. doi: 10.1186/gb-2002-3-7-research0034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Andersen CL, Jensen JL, Orntoft TF. Normalization of real-time quantitative reverse transcription-PCR data: a model-based variance estimation approach to identify genes suited for normalization, applied to bladder and colon cancer data sets. Cancer Res. 2004;64:5245–5250. doi: 10.1158/0008-5472.CAN-04-0496. [DOI] [PubMed] [Google Scholar]
- 52.Pfaffl MW, Tichopad A, Prgomet C, Neuvians TP. Determination of stable housekeeping genes, differentially regulated target genes and sample integrity: BestKeeper–Excel-based tool using pair-wise correlations. Biotechnol Lett. 2004;26:509–515. doi: 10.1023/B:BILE.0000019559.84305.47. [DOI] [PubMed] [Google Scholar]
- 53.Silver N, Best S, Jiang J, Thein SL. Selection of housekeeping genes for gene expression studies in human reticulocytes using real-time PCR. BMC Mol Biol. 2006;7:33. doi: 10.1186/1471-2199-7-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Xie Fuliang, Xiao Peng, Chen Dongliang, Xu Lei, Zhang Baohong. miRDeepFinder: a miRNA analysis tool for deep sequencing of plant small RNAs. Plant Molecular Biology. 2012;80(1):75–84. doi: 10.1007/s11103-012-9885-2. [DOI] [PubMed] [Google Scholar]
- 55.R Core Team. R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria (2014).
- 56.Kaforou M, et al. Detection of tuberculosis in HIV-infected and -uninfected African adults using whole blood RNA expression signatures: a case-control study. PLoS Med. 2013;10:e1001538. doi: 10.1371/journal.pmed.1001538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sing T, Sander O, Beerenwinkel N, Lengauer T. ROCR: visualizing classifier performance in R. Bioinformatics. 2005;21:3940–3941. doi: 10.1093/bioinformatics/bti623. [DOI] [PubMed] [Google Scholar]
- 58.Lopez-Raton M, Rodriguez-Alvarez MX, Cadarso-Suarez C, Gude-Sampedro F. OptimalCutpoints: An R Package for Selecting Optimal Cutpoints in Diagnostic Tests. Journal of Statistical Software. 2014;61:1–36. doi: 10.18637/jss.v061.i08. [DOI] [Google Scholar]
- 59.Du P, Kibbe WA, Lin SM. lumi: a pipeline for processing Illumina microarray. Bioinformatics. 2008;24:1547–1548. doi: 10.1093/bioinformatics/btn224. [DOI] [PubMed] [Google Scholar]
- 60.Khaenam, P. et al. Expression profiling by high throughput sequencing (NCBI GEO (GSE69529), 2105).
- 61.Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.