
Figure 3.
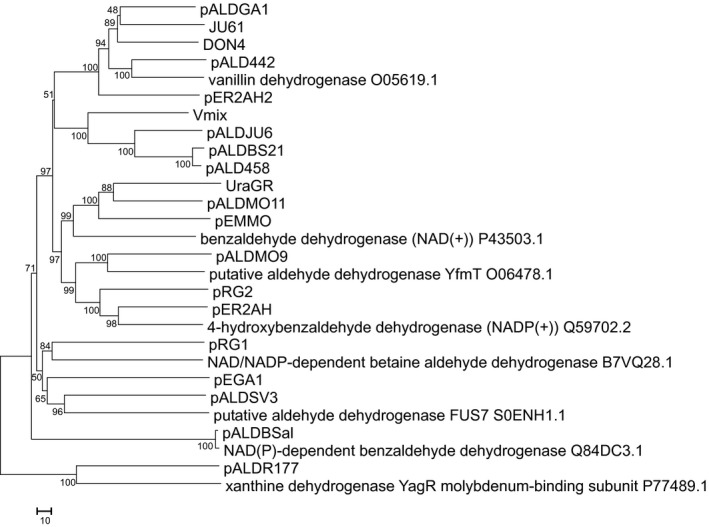
Sequence diversity of metagenomic aldehyde dehydrogenases. The evolutionary history was inferred using the Neighbor‐Joining method (Saitou & Nei, 1987), the evolutionary distances were computed using the number of differences method (Nei & Kumar, 2000) and are in the units of the number of amino acid differences per sequence. The percentage of replicate trees in which the associated taxa clustered together in the bootstrap test are shown next to the branches (Felsenstein, 1985). The tree is drawn to scale, with branch lengths in the same units as those of the evolutionary distances used to infer the phylogenetic tree. The optimal tree with the sum of branch length = 2,362.75195313 is shown. All positions containing gaps and missing data were eliminated. A total of 351 positions in the final dataset were used for data analysis