

HIV-1 particle production and infection are highly ordered processes. Viral Gag proteins play a central role in the assembly and disassembly of viral molecules. Of these, capsid protein (CA) is a major contributor to the Gag-Gag interactions. CA consists of two structured domains, i.e., N-terminal (NTD) and C-terminal (CTD) domains, connected by an unstructured domain named the interdomain linker. While multiple regions in the NTD and CTD are reported to play roles in virion morphogenesis and infectivity, the roles of the linker region in Gag assembly and virus particle formation remain elusive. In this study, we showed by biological and molecular analyses that the linker region functions as an intramolecular modulator to tune Gag assembly, virion production, and viral infectivity. Our study thus illustrates a hitherto-unrecognized mechanism, an allosteric regulation of CA structure by the disordered protein element, for HIV-1 replication.
KEYWORDS: Gag-CA, HIV-1, assembly, interdomain linker, virus particle production
ABSTRACT
The retroviral Gag capsid (Gag-CA) interdomain linker is an unstructured peptide segment connecting structured N-terminal and C-terminal domains. Although the region is reported to play roles in virion morphogenesis and infectivity, underlying molecular mechanisms remain unexplored. To address this issue, we determined biological and molecular phenotypes of HIV-1 CA linker mutants by experimental and in silico approaches. Among the nine linker mutants tested, eight exhibited attenuation of viral particle production to various extents mostly in parallel with a reduction in viral infectivity. Sucrose density gradient, confocal microscopy, and live-cell protein interaction analyses indicated that the defect is accompanied by attenuation of Gag-Gag interactions following Gag plasma membrane targeting in the cells. In silico analyses revealed distinct distributions of interaction-prone hydrophobic patches between immature and mature CA proteins. Molecular dynamics simulations predicted that the linker mutations can allosterically alter structural fluctuations, including the interaction surfaces apart from the mutation sites in both the immature and mature CA proteins. These results suggest that the HIV-1 CA interdomain linker is a cis-modulator of the CA interaction surfaces to optimize efficiency of Gag assembly, virion production, and viral infectivity.
IMPORTANCE HIV-1 particle production and infection are highly ordered processes. Viral Gag proteins play a central role in the assembly and disassembly of viral molecules. Of these, capsid protein (CA) is a major contributor to the Gag-Gag interactions. CA consists of two structured domains, i.e., N-terminal (NTD) and C-terminal (CTD) domains, connected by an unstructured domain named the interdomain linker. While multiple regions in the NTD and CTD are reported to play roles in virion morphogenesis and infectivity, the roles of the linker region in Gag assembly and virus particle formation remain elusive. In this study, we showed by biological and molecular analyses that the linker region functions as an intramolecular modulator to tune Gag assembly, virion production, and viral infectivity. Our study thus illustrates a hitherto-unrecognized mechanism, an allosteric regulation of CA structure by the disordered protein element, for HIV-1 replication.
INTRODUCTION
Gag precursor protein of retroviruses, including human immunodeficiency virus type 1 (HIV-1), is a major viral structural protein and plays pivotal roles in both early and late phases of replication (1, 2). Correct assembly of immature viral particles by Gag proteins, followed by the steps of budding and maturation, leads to proper formation of the viral core, which is essential for infectious virion production and virus replication. HIV-1 Gag protein consists of four major domains, matrix (Gag-MA), capsid (Gag-CA), nucleocapsid (Gag-NC), and p6 (Gag-p6), and two spacer peptides, SP1 and SP2. Each domain of HIV-1 Gag protein has an independent role during HIV-1 particle assembly: Gag-MA anchors Gag proteins to plasma membrane (PM) through its N-terminal myristoylation; Gag-NC is involved in packaging of viral genome RNA (gRNA), and the interaction of Gag-NC with gRNA also has been suggested to be important for Gag oligomerization; and Gag-p6 participates in the release of viral particles by utilizing host’s ESCRT (endosomal sorting complex required for transport) machinery. Of the four Gag domains, Gag-CA is a main driver of Gag-Gag interactions required for HIV-1 particle assembly (1–7).
HIV-1 Gag-CA is composed of two independent structured domains, the N-terminal domain (CA-NTD; residues 1 to 145) and the C-terminal domain (CA-CTD; residues 151 to 231), and a short flexible peptide (interdomain linker; residues 146 to 150) that links these domains (Fig. 1A) (1, 5, 7). The HIV-1 CA-NTD and CA-CTD contain seven and five α-helices (H1 to H7 and H8 to H12), respectively. Another helix, designated 310 helix (310H; residues 150 to 152), is located downstream of the linker region (Fig. 1B) (8, 9). Immediately after 310H, a major homology region (MHR; residues 153 to 172) has been mapped; this region is highly conserved among retroviruses and has been shown to be critical for the assembly of retroviruses, including HIV-1 (Fig. 1) (10–13). Many studies showed that mutations in Gag-CA have pleiotropic effects on various viral replication steps (14–19). The Gag-CA mutants that decrease virion production or form aberrant core displayed simultaneous drastic reduction in viral infectivity as determined by single-cycle infection assays using various indicator cell lines. The mutations that cause defects in virion production were identified in H4 to H6 of the CA-NTD and in MHR and H9 to H10 of the CA-CTD (19–22). Recent structural analyses of immature retroviral capsids of HIV-1 and Mason-Pfizer monkey virus (M-PMV) by cryo-electron microscopy (cryo-EM) revealed the residues that are predicted to be involved in interhexameric NTD-NTD contacts, interhexameric CTD-CTD contacts, and intrahexameric CTD-CTD contacts formed by Gag-CA (23–25). Interestingly, virion production-defective mutations described above were located within and adjacent to the Gag-CA contact sites revealed by structural analyses, suggesting that the residues/regions in both the CA-NTD and CA-CTD are required for Gag assembly and subsequent virus particle production (Fig. 1) (21).
FIG 1.

Structure and amino acid sequence of HIV-1 Gag-CA illustrating the positions of the interdomain linker region and residues used for mutagenesis. (A) Three-dimensional (3D) locations of amino acid residues analyzed in the present mutational study. The Gag-CA monomeric structure is derived from PDB code 4USN (25). A red ribbon indicates five residues in the linker region (residues 146 to 150). Residues E75/E76 in helix 4 (H4) and K158/Y164 in the MHR, which are known to be important for Gag assembly (11, 19, 20), are also shown. These sites were used as references for the present mutational analyses. (B) Alignment of HIV-1 and SIVmac Gag-CA sequences. Amino acid sequences in Gag-CA of HIV-1NL4-3 (GenBank accession no. AF324493) and SIVmacMA239 (GenBank accession no. M33262) were aligned by Genetyx version 14. Locations of helices (H) in this alignment are as described previously (21), and the CypA binding loop, interdomain linker, 310 helix (310H) (8, 9), and MHR are indicated. Amino acid sites to which mutations were introduced in this study are indicated by red letters.
Cryo-EM analyses have provided a highly ordered structural model for the Gag-CA layer in retroviral immature particles that shows various interactions between the CA-NTD and CA-CTD (24, 25). However, the cryo-EM density corresponding to the CA linker region is partly visible for immature M-PMV capsid and can be seen only at lower isosurface thresholds for immature HIV-1 particles. These results have suggested an unstructured nature for the domain, and the observed flexibility appears to work for ensuring the correct arrangement of the CA-NTD and CA-CTD in HIV-1 mature capsids (9, 26, 27). Because the linker region of HIV-1 Gag-CA is inherently unstructured and flexible and is not located at the capsid surface, studies on its significance in Gag-Gag interactions and HIV-1 particle production have been limited. A previous study has shown that although mutants of the HIV-1 Gag-CA linker region exhibit wild-type Gag expression, processing, and progeny production, they are noninfectious, having defects in conical core assembly and viral DNA synthesis (28). Based on these results, the linker region of the HIV-1 Gag-CA has been concluded to be critical for proper core assembly and stability (28). In addition, one of the serine residues within the domain (at position 149) has been shown to be a major phosphorylation site in HIV-1 Gag-CA (29–32). The alanine substitution of S149 led to aberrant core assembly or impaired core stability (28, 29, 31, 32), whereas it did not appear to affect viral particle production (29, 30). In contrast, for Moloney murine leukemia virus, insertion of amino acid residues into the Gag-CA linker region, or fusion with HIV-1 sequence at the linker region, reduced viral particle production (33). Thus, it remains unclear how mutations in the retroviral Gag-CA linker region, including that of HIV-1, influence Gag assembly and progeny production.
In this study, we mainly focused on defining the role of the HIV-1 Gag-CA linker region in Gag assembly and viral particle production at the late viral replication phase. To better understand molecular events associated with the interdomain linker, we performed in vitro experimental and in silico structural analyses. Here we provide evidence for critical involvement of the HIV-1 Gag-CA linker region in Gag-Gag interaction/Gag assembly, virus particle production, and viral infectivity.
RESULTS
Mutations in HIV-1 Gag-CA linker region can affect both early and late replication phases.
To elucidate how the HIV-1 CA linker region acts at early and late replication stages, we conducted site-directed mutagenesis of the linker region by regular alanine substitutions as previously described (28) (Table 1). A CA-NTD mutant (EE75/76AA) and two MHR mutants (K158A and Y164A), which were reported to display a marked reduction in virus particle production and viral infectivity, were constructed and used as controls (19, 21, 22). Viral growth ability overall and viral replication abilities at early and late phases were monitored by multicycle replication ability in human T cell line H9, single-cycle infectivity in reporter cell line TZM-bl (early), and virus production assay in transfected H9 cells (late), respectively. For infection experiments, input viruses were normalized by virion-associated reverse transcriptase (RT) activity (RT units) to allow comparison of relative infectivity.
TABLE 1.
Characterization of HIV-1 CA mutants in this study
| Parent and mutant clones | Location of substitutions | Growth in H9 cellsa,b | Infectivity in TZM-bl cellsb,c | Virus particle production in H9 cellsb,c |
|---|---|---|---|---|
| NL4-3 | None | ++ | 1.00 ± 0.00 | 1.00 ± 0.00 |
| EE75/76AA | NTD helix 4 | – | 0.08 ± 0.04 | 0.10 ± 0.06 |
| K158A | CTD MHR | – | 0.07 ± 0.04 | 0.12 ± 0.02 |
| Y164A | CTD MHR | – | 0.07 ± 0.04 | 0.04 ± 0.02 |
| S146A | Linker | + | 0.67 ± 0.03 | 1.02 ± 0.10 |
| P147A | Linker | + | 0.66 ± 0.06 | 0.45 ± 0.08 |
| T148A | Linker | + | 0.62 ± 0.07 | 0.79 ± 0.02 |
| S149A | Linker | – | 0.06 ± 0.01 | 0.12 ± 0.01 |
| I150A | Linker | – | 0.07 ± 0.04 | 0.09 ± 0.02 |
| S146N | Linker | – | 0.13 ± 0.03 | 0.53 ± 0.09 |
| S149N | Linker | + | 0.99 ± 0.09 | 0.46 ± 0.05 |
| S149D | Linker | – | 0.00 ± 0.00 | 0.09 ± 0.01 |
| S149K | Linker | – | 0.00 ± 0.00 | 0.31 ± 0.08 |
Virus growth properties are summarized based on the results of repeated infection experiments. −, no detectable virus growth in the test period (15 days); +, retarded virus growth relative to NL4-3; ++, positive-control NL4-3.
Input virus amounts were normalized by the virion-associated RT activity produced from 293T cells transfected with the indicated proviral clones. For the particle production assays, H9 cells were transfected with 2 μg of proviral DNA clones and cultured for 2 days in the presence of CXCR4 antagonist AMD3100.
Values relative to those for NL4-3 are shown. Data presented are means ± standard errors of three independent experiments.
As shown in Table 1, all control mutants (EE75/76AA, K158A, and Y164A) exhibited an undetectable growth ability in H9 cells, a drastic decrease in both infectivity (mean values: 0.07 to 0.08) and particle production (mean values: 0.04 to 0.12), as expected from previous reports (19, 21, 22). Concurrent with the replication defects, these mutants (EE75/76AA and K158A) have been shown to display an aberrant viral morphology including enlarged and irregularly shaped cores (19). Among five mutants having single alanine substitutions in the linker region, the S149A and I150A mutants exhibited a remarkably decreased viral infectivity and virus particle production and were resultantly found to be noninfectious for H9 cells. The S146A mutant grew more poorly than NL4-3 but the virus production level was similar to that of NL4-3, indicating its only defect at the early replication phase. The P147A and T148A mutants displayed a reduced viral replication potential in H9 cells, and both viral infectivity and virus particle production were moderately decreased, indicating relatively mild mutational effects at these sites. The above-described results for the linker mutants (S146A, T148A, S149A, and I150A) were in line with the electron microscopic data previously described (28). Upon transfection, viral populations generated by infectious S146A and T148A mutants contained viruses with conical cores at percentages similar to that observed for the wild-type clone, whereas noninfectious S149A and I150A mutants produced progenies with conical cores at a severely reduced ratio (28). Our results for the P147A mutant were different from those previously reported (28), for unknown reasons. While the P147A mutant was reported to be unable to produce virus particles as judged by RT assays, we readily detected RT activity in the supernatant fluids of transfected cells, and the supernatants were infectious for H9 cells, albeit poorly. The results described above indicate that the effects of single amino acid substitutions are dependent on the site in the linker region.
To address effects of amino acid types to be substituted, we constructed four additional mutants that have single substitutions specific to the Gag-CA protein of simian immunodeficiency virus (SIV) clone SIVmacMA239 (S146N and S149N) (Fig. 1B) or those reported previously (S149D and S149K) (28–32). Unlike the S146A mutant, the S146N mutant displayed a striking reduction in viral infectivity and growth ability with moderately decreased particle production (Table 1). Furthermore, unlike the S149A mutant, the S149N mutant was found to display a moderately impaired phenotype, retaining viral infectivity comparable to that of HIV-1NL4-3 (Table 1). Meanwhile, the S149D and S149K mutants showed a marked reduction in virus particle production and infectivity, as seen for the S149A mutant (Table 1). These results are consistent with previous reports on the phenotypes of the S149A, S149D, and S149K mutants (28–32) and indicate that the type of amino acid residues in the linker region is critical to determine HIV-1 replication phenotypes. In sum, our results demonstrate that single amino acid substitutions within the linker region of HIV-1 Gag-CA diminish virus particle production in a site- and property-dependent manner.
Amino acid sequence in the Gag-CA interdomain linker is highly conserved among HIV-1 subtype B viruses except for the T148 site.
Amino acid variations in the Gag-CA protein of HIV-1 subtype B were examined using the public HIV Sequence Database (http://www.hiv.lanl.gov/content/sequence/HIV/mainpage.html) (n = 14,120) with Shannon entropy scores as the indicator of amino acid variations at individual positions (34–36). Scores obtained for individual sites ranged from 0.007 to 1.683 bits, with an average score of 0.16 bit, indicating that Gag-CA diversity in the subtype B population is relatively small, as seen in many viral enzymes (36–38). This is in contrast to virion surface proteins, for which entropy scores often exceeded 2.0 bits, reaching close to a maximum value of 4.4 bits in the antigenic sites (34, 39, 40).
The entropy scores for amino acid residues at positions 146, 147, 149, and 150 in the Gag-CA linker region were 0.041, 0.012, 0.106, and 0.012 bits, respectively (Fig. 2A). The scores were as low as those for residues at positions 75 (0.194), 76 (0.025), 158 (0.012), and 164 (0.011), which are critical for virion production (Table 1) (11, 19–22). The entropy analysis clearly demonstrated that the interdomain linker is highly conserved among HIV-1 subtype B strains and suggests the presence of strong constraints against changes in these amino acid residues during HIV-1 subtype B circulation and maintenance in nature. Notably, however, subtype B was found to accept variation at position 148, with an entropy score of 1.446 (Fig. 2A), and various residues, including T, V, A, G, I, L, P, and S, were detected at this position (Fig. 2B, upper row). It is worth mentioning here that T148 is important for optimal virus growth (Table 1), and the most dominant amino acid residues found at position 148 were different between subtype B alone and HIV-1 sequence populations, including group M (n = 25,222) (Fig. 2B, lower row). Our results may imply that this position is variable to optimize growth ability of viruses under the different CA backbone sequences. In total, amino acid residues in the linker region were conserved or functionally important in the HIV-1 subtype B population, suggesting a significant role for the interdomain linker in viral replication.
FIG 2.

Variation around the interdomain linker in HIV-1 CA. (A) Shannon entropy scores at individual amino acid positions. Scores were calculated (34–36) using a total of 14,120 full-length capsid amino acid sequences of HIV-1 subtype B from different geographic regions in the world (http://www.hiv.lanl.gov/content/sequence/HIV/mainpage.html). The majority consensus sequence around the linker region is shown on the x axis along with corresponding Shannon entropy scores on the y axis. An entropy score of 0 indicates absolute conservation, whereas a score of 4.4 indicates complete randomness. (B) Types and frequencies of amino acid residues detected in the linker region. Full-length capsid amino acid sequences of HIV-1 subtype B (n = 14,120) and group M (n = 25,222) from the HIV Sequence Database were used to analyze the types and frequencies of amino acid residues at indicated positions in the linker region.
Mutations in the linker region (S149A and I150A) impede accumulation of assembly intermediates formed by multimerized Gag proteins at the PM.
It has been reported that mutations in the amino acid residues affecting immature capsid assembly have little effect on Gag expression itself (19, 21, 22). We assumed that the reduction in virus particle production observed for the mutants of the Gag-CA linker region may be associated with their Gag assembly defects. For verification, we performed velocity sedimentation assays previously established (21, 22) to monitor the intracellular assembly process of mutant Gag proteins. With this assay, we can distinguish several assembly intermediates as 10S (sedimentation value), 80S, 150S, 500S, and 750S. The 10S intermediate contains newly synthesized Gag precursor proteins. The 80S intermediate is a complex of Gag oligomer and gRNA, which targets the PM. The 150S intermediate appears to be highly transient. Following binding of the 80S/150S intermediates to the PM, 500S and subsequent 750S intermediates are formed by further Gag multimerization there. When cells expressing Gag mutant proteins with some defects in the assembly process are examined by these sedimentation assays, intermediates that precede the step at which assembly is blocked are readily detectable. For example, mutants with defects in 500S intermediate formation (i.e., failure in Gag multimerization at PM) accumulate 10S to 150S intermediates (21, 22).
To analyze effects of the linker mutations on Gag assembly, we chose two alanine substitution mutants (S149A and I150A) that exhibit remarkably reduced virus particle production (Table 1). MHR K158A and Y164A mutants, which have been shown to be arrested at 80S intermediates in velocity sedimentation assays (22), were used as controls. To facilitate detection and quantification of the immature Gag precursor protein, we generated HIV-1NL4-3-derived constructs without Pol-protease and Env. Parental (NL4-3ΔPro/ΔEnv) and mutant (S149AΔPro/ΔEnv, I150AΔPro/ΔEnv, K158AΔPro/ΔEnv, and Y164AΔPro/ΔEnv) clones were transfected into HeLa cells, and at 24 h posttransfection, cells were lysed for velocity sedimentation assays (Fig. 3A). Steady-state expression levels of Gag proteins in total cell lysates were similar among test samples. Parental NL4-3ΔPro/ΔEnv showed a small amount of intermediates at early Gag assembly steps (10S to 80S), and 500S/750S intermediates were readily detected. Consistent with a previous report (22), MHR mutants (K158AΔPro/ΔEnv and Y164AΔPro/ΔEnv) showed defects in Gag multimerization on PM. While 150S to 750S intermediates were undetectable, a much higher level of 10S to 80S intermediates accumulated for the MHR mutants than for NL4-3ΔPro/ΔEnv. The linker mutants (S149AΔPro/ΔEnv and I150AΔPro/ΔEnv) exhibited lower and higher accumulations of early 10S to 80S and late 500S/750S intermediates, respectively, than did the MHR mutants. The linker mutants, however, clearly displayed increased 10S to 80S intermediates, comparable 80S/150S intermediates, and decreased 500S/750S intermediates relative to those of NL4-3ΔPro/ΔEnv. These results strongly suggest that the mutations in the interdomain linker impede Gag multimerization after the binding of 80S intermediates to the PM.
FIG 3.
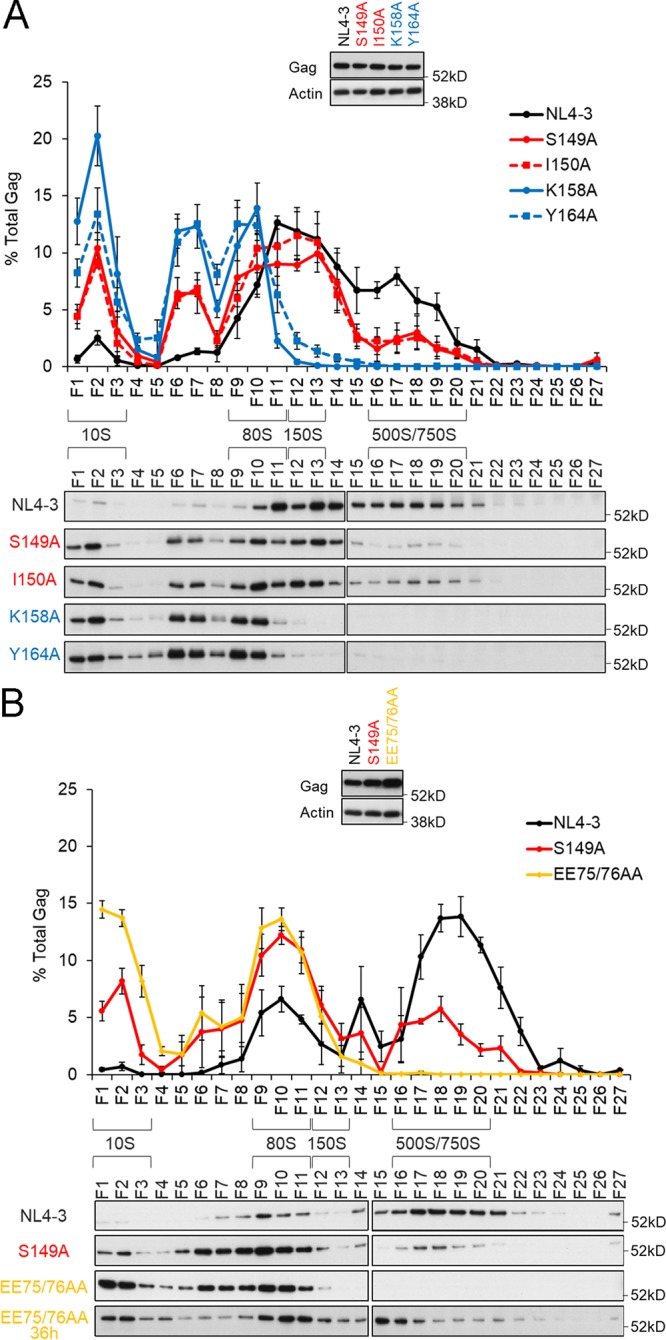
Effects of linker mutations on Gag assembly process as revealed by velocity sedimentation analysis. Cell lysates prepared from transfected HeLa cells were ultracentrifuged through sucrose gradients for sedimentation analysis (22), and fractions (F) were collected stepwise from centrifuge tubes (top to bottom) for Gag monitoring by the Western blotting method. Sedimentation coefficients (S values) were also determined. Results of three independent experiments with representative immunoblotting data are shown for comparison among the linker and MHR mutants (A) and for comparison between the linker and NTD mutants (B). Immunoblotting data for Gag proteins in total cell lysates are presented at the top of each panel.
It has been shown that the assembly process of a CA-NTD mutant (EE75/76AA) is arrested at the 500S intermediates prior to 750S intermediates (21). Thus, in another series of experiments, the linker and CA-NTD mutants were comparatively analyzed for formation of 500S/750S intermediates to see if there is any substantial difference between the mutants. Velocity sedimentation assays were performed as described above using parental NL4-3ΔPro/ΔEnv and the mutants (S149AΔPro/ΔEnv and EE75/76AAΔPro/ΔEnv). Since the Gag expression level for the EE75/76AAΔPro/ΔEnv mutant in transfected cells was always higher than those for NL4-3ΔPro/ΔEnv and S149AΔPro/ΔEnv (Fig. 3B), sample amounts for the sedimentation assays were adjusted to contain equal Gag proteins. As shown in Fig. 3B, while Gag proteins of NL4-3ΔPro/ΔEnv at 24 h posttransfection were predominantly detected in the 500S/750S fractions, the 500S intermediates for the EE75/76AAΔPro/ΔEnv mutant were not observed (Fig. 3B). The result for the EE75/76AAΔPro/ΔEnv mutant appeared to be different from that previously described (21) in that 500S intermediates were undetectable under this condition. Robinson et al. (21) have reported that the EE75/76AA mutant displays a pattern of Gag assembly intermediates similar to that of the wild type and that it exhibits inefficient progression of Gag assembly following 500S intermediate formation. When samples prepared at 36 h posttransfection were examined, the 500S intermediates became detectable in our system (Fig. 3B) as reported previously. The EE75/76AAΔPro/ΔEnv mutant prepared at this timing exhibited accumulated 500S intermediates and substantially reduced 750S intermediates relative to those for NL4-3ΔPro/ΔEnv, suggesting its arrest prior to 750S intermediates. Consistent with the previous report (21), this result showed that the EE75/76AAΔPro/ΔEnv mutant is impaired at the final step of the Gag assembly process, just before virus budding. It should be noted that an apparent difference was also noted for the results for NL4-3ΔPro/ΔEnv (Fig. 3). The different appearance of data obtained could be attributable to variations in Gag assembly kinetics among experiments, possibly due to transfection efficiency, cell conditions, and so on. Nevertheless, in three independent experiments, amounts of the 10S/80S intermediates were constantly increased in the linker mutants relative to those for NL4-3ΔPro/ΔEnv, and those of the 500S/750S intermediates were always decreased in the linker mutants relative to the wild type, as shown by the representative results (Fig. 3). These results suggested that the S149AΔPro/ΔEnv mutant, unlike the EE75/76AAΔPro/ΔEnv mutant, is impeded in formation of 500S intermediates, i.e., Gag multimerization on the PM, compared with NL4-3ΔPro/ΔEnv. It is conceivable that the linker mutations affect the ability of Gag to assemble to a lesser extent than the MHR mutations, because a relatively high level of 500S/750S intermediates was seen for the S149AΔPro/ΔEnv and I150AΔPro/ΔEnv linker mutants than that for the K158AΔPro/ΔEnv and Y164AΔPro/ΔEnv MHR mutants.
Linker mutations as well as MHR mutations strongly affect Gag multimerization ability at the PM but only moderately reduce membrane targeting of Gag.
Of virus production-defective mutants reported so far, mutations (e.g., G2A) in Gag-MA, which is involved in anchoring Gag at the PM, have been shown to diminish markedly the Gag expression level at the PM (less than 10% relative to the wild type) (19, 21, 22). On the one hand, mutations of the residues which are located in the MHR and CA-CTD (e.g., K158A and WM184/185AA) affect only moderately the amount of Gag targeted to the PM (40 to 70% relative to the wild type), yet they drastically reduce virus production (19, 21, 22). These results imply that the process(es) leading to virus particle production from Gag targeting to the PM onward is severely attenuated for MHR and CA-CTD mutants. In previous studies (21, 22), MHR and CA-CTD mutants with only a modest reduction in PM-targeted Gag level were found to accumulate substantial amounts of 10S to 80S intermediates without the formation of 500S intermediates, suggesting their impaired ability to multimerize on the PM after reaching it. Our velocity sedimentation assays of the particle production-defective linker mutants (S149AΔPro/ΔEnv and I150AΔPro/ΔEnv) showed increased and decreased accumulations of 10S to 80S and 500S intermediates, respectively (Fig. 3). Thus, we predicted that the linker mutants would also diminish Gag multimerization on the PM without severe reduction in Gag amounts there. To examine mutational effects of the linker region on membrane targeting of Gag, we performed membrane flotation assays (22) that can quantify Gag accumulation at the PM. HeLa cells were transfected with NL4-3ΔPro/ΔEnv, S149AΔPro/ΔEnv, I150AΔPro/ΔEnv, K158AΔPro/ΔEnv, or Y164AΔPro/ΔEnv, and at 24 h posttransfection, postnuclear supernatants were prepared and mixed with a high concentration of sucrose solution. Samples were then placed at the tube bottoms and fractionated by ultracentrifugation through sucrose gradients to float the membranes (22). As shown in Fig. 4, most Gag proteins were located at membrane fractions (MF) rather than in non-MF for NL4-3ΔPro/ΔEnv. Mutant Gag proteins showed that a substantial portion of Gag proteins was present at MF, although their relative amounts in non-MF were increased for linker mutants (S149AΔPro/ΔEnv and I150AΔPro/ΔEnv) and MHR mutants (K158AΔPro/ΔEnv and Y164AΔPro/ΔEnv). Gag amounts in MF, relative to that for NL4-3ΔPro/ΔEnv, were estimated to be 80% for the S149AΔPro/ΔEnv mutant, 78% for the I150AΔPro/ΔEnv mutant, 65% for the K158AΔPro/ΔEnv mutant, and 61% for the Y164AΔPro/ΔEnv mutant. Although statistical difference was noted between the data for wild-type and MHR mutant clones, it can be summarized from the results in Fig. 4 that mutant (linker and MHR) Gag proteins on the PM were only moderately reduced.
FIG 4.
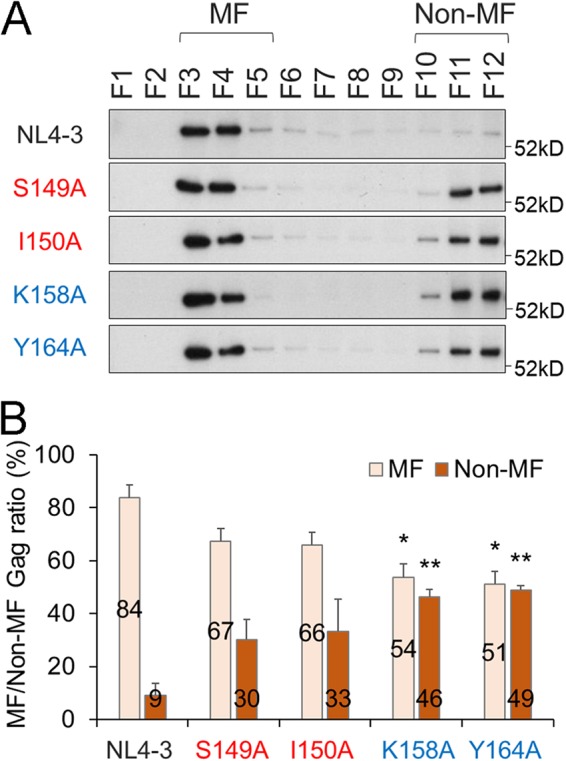
Effects of linker mutations on Gag targeting to cell membranes as judged by membrane flotation analysis. HeLa cells were transfected with the indicated clones and lysed to prepare the postnuclear supernatant for membrane flotation analysis (22). The sample in a high concentration of sucrose solution was placed at the bottom of a centrifugation tube and layered with lowered concentrations of sucrose from the bottom to the top. After ultracentrifugation, fractions (F) were collected from centrifugation tubes (top to bottom) for examination of Gag by the Western blotting method. Gag proteins in membrane (MF) and in nonmembrane (non-MF) fractions are indicated. Representative immunoblotting data from three independent experiments are shown in panel A. In panel B, the ratios of MF and non-MF Gag proteins for various clones are presented (n = 3; mean ± SE). Significance relative to control NL4-3 was determined by Welch’s t test. **, P < 0.01; *, P < 0.05.
To confirm biochemical results shown in Fig. 4, we examined intracellular Gag distribution by confocal microscopy. A construct that expresses wild-type Gag-enhanced green fluorescent protein (EGFP) fusion protein (NL4-3Gag-EGFPΔEnv) and its derivative mutants (S149AGag-EGFPΔEnv, I150AGag-EGFPΔEnv, K158AGag-EGFPΔEnv, and Y164AGag-EGFPΔEnv) were transfected into HeLa cells. At 10 h posttransfection, cells were fixed for confocal microscopic analyses. Expression patterns of Gag-EGFP observed in transfected cells were grouped as follows: (i) widely diffuses in cytoplasm (Diff), (ii) accumulates at the PM (aPM), or (iii) accumulates intracellularly and presents dot-like structures in cytoplasm (Int) (Fig. 5). Based on these subcellular localization patterns of Gag-EGFP, cells were divided into two categories: a PM distribution group (aPM+Int+Diff and aPM+Diff) and an internal distribution group (Diff+Int) (Fig. 5). As shown in Fig. 5, EGFP-positive cells transfected with parental NL4-3Gag-EGFPΔEnv were classified predominantly in the PM distribution group, and a minor population belonged to the internal distribution group. These results were in good agreement with those obtained by membrane flotation assays (Fig. 4). For the linker and MHR mutants, EGFP-expressing cells in the internal distribution and PM distribution groups were significantly increased and decreased, respectively, relative to NL4-3Gag-EGFPΔEnv. These results for mutant Gag proteins were also consistent in essence with those obtained by membrane flotation assays. Apparent differences in the MF/non-MF ratio observed for mutants (Fig. 4 and 5) could be due to experimental conditions (timing for sample preparations posttransfection, etc.) and/or the used method itself. Overall, results from two very different analyses show that a significant amount of mutant (linker and MHR) Gag proteins accumulated at the PM (61 to 80% relative to the wild type and 47 to 52% relative to the wild type as shown in Fig. 4 and 5, respectively), consistent with previous reports (21, 22). Meanwhile, these mutants (S149A, I150A, K158A, and Y164A) exhibited marked reduction in virus particle production (4 to 12% relative to the wild type [Table 1]). These results suggest that Gag-Gag interactions after PM binding are important determinants for the attenuation in Gag assembly of the mutants.
FIG 5.
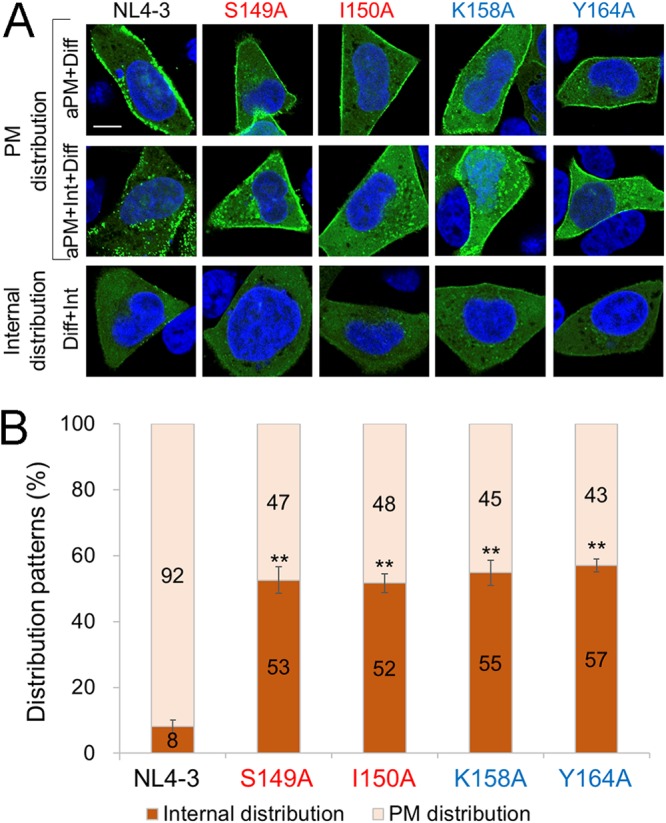
Effects of linker mutations on Gag subcellular localization as revealed by confocal microscopy analysis. HeLa cells were transfected with pNL4-3Gag-EGFPΔEnv or derivative Gag-CA mutant clones by Lipofectamine 2000, fixed with paraformaldehyde at 10 h posttransfection, and processed for confocal microscopy as previously described (63, 66). At least 100 EGFP-positive cells were examined for each sample to determine the subcellular localization pattern of Gag-EGFP. Typical distribution patterns of Gag (EGFP) in cells are shown in panel A. aPM, plasma membrane Gag accumulation; Diff, diffused Gag localization; Int, intracellular Gag accumulation. Bar = 10 μm. In panel B, Gag distribution patterns are compared among clones. Patterns aPM+Diff and aPM+Int+Diff are considered plasma membrane distribution, whereas pattern Diff+Int is considered internal distribution, as indicated in panel A. Results of three independent experiments are shown (means ± SE). Significance relative to the positive control, NL4-3, was determined by Welch’s t test. **, P < 0.01.
Following membrane targeting, Gag forms high-order oligomers at the PM (41). We directly measured Gag-Gag interactions in living cells using nano-bioluminescence resonance energy transfer (NanoBRET) technology (42, 43). We have previously demonstrated that the NanoBRET system can detect specific intracellular Gag-Gag interactions of HIV-1NL4-3 (43). For this NanoBRET analysis, we selected some linker mutants that show normal (S146A), moderately impaired (S146N and S149N), and impaired (S149A, S149D, and I150A) phenotypes with respect to virus particle production (Table 1). The K158A and Y164A MHR mutants were used for controls as virus particle production-defective mutants. As shown in Fig. 6, while mutants with a normal or moderately impaired phenotype (S146A, S146N, and S149N) exhibited signals comparable to those of wild-type NL4-3, those with an impaired phenotype (S149A, S149D, and I150A) produced signals at significantly reduced levels. MHR mutants showed further reduction in signal production. Thus, the ability of mutants to mediate intracellular Gag-Gag interactions was well correlated with their ability to produce virus particles. However, relative reduction values obtained for the two markers of the mutants (virus production levels in Table 1 and NanoBRET signals in Fig. 6) were different. While virus particle production of the S149A, S149D, and I150A mutants decreased to around 10% of that of NL4-3, their NanoBRET signals were approximately 25 to 50% of that of NL4-3. Similarly, the attenuation levels of the K158A and Y164A mutants in the two assays were not the same. The differences in experimental values may be accounted for by differences in transfection efficiency and/or protein expression level, which could result from the use of distinct cells (H9 versus HEK293T) and constructs (ΔPro/ΔEnv proviral clones versus codon-optimized expression vectors). It is thus possible that NanoBRET assays gave relatively overestimated values for the Gag-Gag interaction ability of the mutants. A greater reduction in the Gag-Gag interaction signals observed for MHR mutants relative to the linker mutants (Fig. 6) was also noteworthy. Considering that the linker mutants but not the MHR mutants could form 500S intermediates albeit slightly (Fig. 3), it is conceivable that the mutations in the interdomain linker may affect Gag assembly more weakly than those in MHR. In sum, while somewhat impaired in the membrane binding and PM localization (Fig. 4 and 5), the mutants showed quantitatively more significant effects on the Gag-Gag interactions as revealed by NanoBRET assays (Fig. 6). Despite the presence of substantial amounts of Gag proteins, the reduced interaction ability of the mutants would severely impede Gag multimerization on the PM in the HIV-1 assembly process. Overall, our results suggest that the decrease in virus particle production observed for the linker and MHR mutants could result mainly from their reduced capacities to drive Gag-Gag interactions.
FIG 6.

Detection of Gag-Gag interactions by NanoBRET. HEK293T cells were cotransfected with expression vectors encoding Gag-HaloTag and its corresponding Gag-NanoLuc (43). At 48 h posttransfection, NanoBRET signals were measured, and values relative to that for control NL4-3 are presented (n = 4; means ± SE). NanoBRET data were normalized by subtracting the signal value for a Gag CA linker/CTD deletion mutant (deletion of CA amino acids 146 to 231). Significance relative to control NL4-3 was determined by Welch’s t test. **, P < 0.01; *, P < 0.05. A black bar, red bars, and blue bars indicate NL4-3, linker mutants, and MHR mutants, respectively.
Two types of hydrophobic patches for molecular interactions are elucidated on the surface of HIV-1 immature CA protein.
The above-described results strongly suggested that the linker mutations except for S146A influence the structural property of CA interaction surfaces primarily for Gag assembly. To gain structural insights into the effects of linker mutations on Gag assembly, we conducted in silico analyses. Because hydrophobic interplay is a major contributor for macromolecule interactions, including protein oligomerization, we examined three-dimensional (3D) distributions of hydrophobic patches (a minimal patch area of 50 Å2) on the surface of HIV-1NL4-3 CA protein. A near-full-length immature CA model of HIV-1NL4-3 was constructed from the reported structure of HIV-1 immature capsid lattice in the virus particles (25) and searched for hydrophobic patches for molecular interactions (44–47).
The analysis disclosed two types of hydrophobic patches (Fig. 7): those along the CA-CA interfaces in an immature capsid lattice (25) (Fig. 7A, pink areas) and those located outside the CA-CA interfaces (Fig. 7B, light green areas). The former included the reported regions for direct CA-CA interactions in an immature CA lattice, such as H1, H2, and H9 (25). The latter included reported regions important for formation of Gag assembly intermediates, such as H4, H6, H8, H10, and H11 (21). Although its importance in the Gag assembly has not formally been described, H7 forms a hydrophobic patch along the CA-CA interface (Fig. 7A) and was recently shown by us to play a critical role in virus particle production (48). The significance of the H3 patch (Fig. 7B) in the late stage of HIV-1 replication has not been reported. H5 did not participate in formation of the hydrophobic patch (Fig. 7A and B). Hydrophobic patches were predominantly formed along the helices, and the amino acid residues contributed to form the patches were generally highly conserved among the global HIV-1 subtype B strains (Fig. 7C). Together with the functional and structural data on the immature CA protein so far reported (21, 25), the results suggest a critical role of the hydrophobic patches in the late stage of HIV-1 replication. In sum, the CA helices seem to play important roles in folding, stabilization, and interactions of the HIV-1 immature CA protein as generally believed.
FIG 7.

Elucidation of the hydrophobic patches for molecular interactions of HIV-1NL4-3 immature CA protein. An HIV-1NL4-3 CA protein model was constructed by the homology modeling method from the reported immature CA structure (PDB code 4USN) (25). Distribution of the hydrophobic-interaction-prone patches (a minimal patch area of 50 Å2) on the surface of the CA model was assessed and visualized using the Protein Patch Analyzer tool in MOE (44–47). (A) Hydrophobic patches on the CA-CA interface. Patches that participate in the direct CA-CA interactions in an immature CA lattice (25) are shown in pink. (B) Hydrophobic patches outside the CA-CA interface. Patches that are not involved in the direct CA-CA interactions in an immature CA lattice (25) are shown in green. (C) Amino acid residues involved in forming the hydrophobic patches and their variations. Residues involved in the formation of hydrophobic patches are marked with colored circles: pink circles, residues for patches involved in direct CA-CA interactions; green circles, residues for patches that are not involved in direct CA-CA interactions. Shannon entropy scores calculated with the CA amino acid sequences of HIV-1 subtype B (n = 14,120) are shown above the corresponding residues.
MD simulations predict that the linker and MHR mutations remotely affect structural fluctuations of the immature CA interaction surfaces.
Because the direct CA interaction surfaces for Gag assembly have been suggested to be located in the CA-CTD and CA-NTD rather than in the linker region (21, 25, 49) (Fig. 7A), the linker mutations were predicted to influence the structures away from the mutation sites. To address this issue, we performed molecular dynamics (MD) simulations of the HIV-1NL4-3 near-full-length CA protein with or without a single linker mutation, S146A, S149A, or I150A. Among the linker mutants, the S149A and I150A mutants, but not the S146A mutant, exhibited attenuation in viral particle production (Table 1) along with attenuations in Gag-Gag interactions (Fig. 6). We also performed MD simulations of an MHR mutant, the Y164A mutant, whose assembly was more severely impaired than the linker mutants (Fig. 3 to 6), and its mutation is located outside the direct CA-CA contact site (Fig. 7A and C). Because structural fluctuations in solution play key roles in molecular interactions (50–53), we analyzed the root mean square fluctuation (RMSF) of the Cα atoms of the main chains (54) using 15,000 snapshots in the equilibrium states during MD simulations.
Notably, all mutations tested induced changes in fluctuations at various regions away from the mutation sites, as indicated by changes in the RMSF (Fig. 8). These remote effects of single mutations on the fluctuation profile of immature CA protein were reproducible with repeated MD simulations, suggesting that the effects are physicochemically inevitable. The changes were greatest with Y164A, and the fluctuations were augmented throughout the CA protein containing various CA-CA interfaces (Fig. 8, upper portion). The results suggest a critical role for MHR in maintaining proper CA dynamics for Gag assembly. Although less extensive, S149A mutation also induced overall augmentation of the CA fluctuations (Fig. 8, upper middle portion), suggesting that the linker region has a similar function with MHR in controlling CA surface dynamics. In contrast, effects of I150A and S146A mutations were limited in particular regions (Fig. 8, lower middle and bottom portions). I150A with marked attenuation in both virus production and viral infectivity (Table 1) induced changes in H6 and its downstream loop, H7 and its downstream linker region, and H10 and its downstream loop (asterisks at the lower middle portion of Fig. 8). These regions are all involved in the formation of the hydrophobic patches in an immature CA lattice (Fig. 7). Among them, H7 region is involved in the direct interactions between CA and CA in the immature CA lattice (Fig. 7A). Meanwhile, S146A mutation with no detectable attenuation in virion production but with attenuation in viral infectivity (Table 1) induced a pattern of changes distinct from that of I150A: the changes were seen in the H4/H5 loop (cyclophilin A [CypA] binding loop), H6 and its downstream loop, H8 and its downstream loop, and H10 and its downstream loop (asterisks at the bottom portion of Fig. 8). Although the last three regions participate in the formation of hydrophobic patches (Fig. 7), no changes in the regions for the CA-CA direct interactions were detected. These results show that the effects of the linker mutations on the CA dynamics are unique in individual mutations and suggest that the change in the H7 fluctuation may be a causative factor for the I150A phenotype (Table 1). Alternatively, the multiple changes on the CA surfaces rather than the change at the specific site may generate the I150A phenotype.
FIG 8.

Effects of the linker and MHR mutations on the structural dynamics of HIV-1NL4-3 immature CA protein. Gag-CA protein models of HIV-1NL4-3 and its mutants were constructed from the reported structure of HIV-1 immature capsid (PDB code 4USN) (25). MD simulations of Gag-CA proteins of HIV-1NL4-3 and its mutants were carried out at 1 atm, at 310 K, and in 150 mM NaCl for 200 ns using the Amber 16 program package (54). RMSF values, which indicate the atomic fluctuations of the main chains of individual amino acids during MD simulations, were calculated using 15,000 snapshots in the equilibrium states during MD simulations. Distributions of RMSF in the Gag-CA proteins and its mutants are shown. Numbers on the horizontal axes indicate amino acid positions in the HIV-1NL4-3 Gag-CA protein. Arrows indicate the positions of mutated residues. Blue asterisks for I150A and S146A indicate sites where the fluctuation profiles differ between the wild type and the mutants.
In summary, our results suggest that the linker and MHR mutations could remotely control the physical properties of regions outside the mutation sites. The MHR mutation Y164A and the linker mutations S149A and I150A, which caused attenuation in the Gag assembly and virus particle production, were predicted to induce distinct fluctuation changes in the CA-CA contact site(s), whereas linker mutation S146A, which did not cause significant effects on the virus particle production, did not cause such changes.
Distribution of the hydrophobic patches is distinct between mature and immature CA proteins of HIV-1.
HIV-1 CA protein changes its structure upon virion maturation (23, 25, 55). To gain structural insights into the effects of linker mutations on the early stages of viral replication, we conducted hydrophobic patch analysis and MD simulations of the mature CA. A full-length mature CA model of HIV-1NL4-3 was constructed from the reported structure of the HIV-1 mature capsid from a tubular HIV-1 CA assembly (55) and was searched for hydrophobic patches for molecular interactions as described above (44–47).
The analysis showed marked differences in the 3D distribution of the hydrophobic patches between the immature and the mature CA proteins, in parallel with the structural changes (Fig. 7 and 9). The patches for the CA-CA direct interactions on the mature CA lost the ordered parallelism along the CA lattice in the immature CA, being positioned irregularly to allow more compact CA-CA interactions in the CTD (Fig. 7A and 9A). Meanwhile, the patches outside the CA-CA contact sites on the mature CA were primarily positioned in the NTD more widely than those in the immature CA (Fig. 7B and 9B). Notably, however, the amino acid residues responsible for the formation of the patches in the mature CA were generally highly conserved, as was seen for those in the immature CA (Fig. 7C and 9C). These results strongly suggest that the hydrophobic-interaction-prone patches play distinct but conserved roles in HIV-1 Gag assembly and disassembly.
FIG 9.

Elucidation of the hydrophobic patches for molecular interactions of HIV-1NL4-3 mature CA protein. An HIV-1NL4-3 mature CA protein model was constructed from the reported structure of HIV-1 mature capsid (PDB code 3J34) (55). Hydrophobic patches were assessed and visualized as described for Fig. 7. Hydrophobic patches on the CA-CA interfaces in the mature CA (A) and those outside the CA-CA interfaces (B) are shown in pink and green, respectively. In panel C, amino acid residues involved in the formation of hydrophobic patches and their variations are indicated. Pink circles, residues for patches involved in the direct CA-CA interactions; green circles, residues for patches that are not involved in the direct CA-CA interactions. Shannon entropy scores calculated with the CA amino acid sequences of HIV-1 subtype B (n = 14,120) are shown above the corresponding residues.
MD simulations predict that the linker and MHR mutations remotely affect structural fluctuations of the mature CA interaction surfaces.
Influences of the linker mutations on the fluctuations of the mature CA were examined using a panel of CA mutants (Y164A, S146A, and S149N). The Y164A mutant exhibited marked attenuation in both virus production and viral infectivity (Table 1). The S146A mutant showed no detectable attenuation in virus production and moderate attenuation in the viral infectivity, while the S149N mutant exhibited no detectable attenuation in the viral infectivity and moderate attenuation in virus production (Table 1). Molecular models of the mature CAs of these HIV-1NL4-3 mutants were constructed, and MD simulations were performed as described above for the immature CA mutant models.
The mature CA of HIV-1NL4-3 tended to fluctuate more greatly in the NTD than the immature CA, especially at the CypA binding loop (Fig. 10A). This may increase the probability of interactions with trans-acting factor(s) in the NTD during Gag disassembly. Notably, all mutations tested again induced changes in fluctuations at regions apart from the mutation sites (Fig. 10B). Y164A mutation caused augmentation of the fluctuations primarily in the CTD, including a region near the critical CTD-CTD interface on H9 for stabilizing mature core (WM184/185) (55) (asterisk in the upper portion of Fig. 10B). The S146A mutation also induced similar changes near the WM184/185 region (asterisk in the middle portion of Fig. 10B). These changes may decrease viral infectivity via destabilization of the CA core. Consistently, such changes at the WM184/185 region were not detected for S149N (asterisk in the bottom portion of Fig. 10B). Meanwhile, all the mutations induced changes at the CypA binding loop (“#” in Fig. 10B) and around the linker and MHR (Fig. 10B). These remote effects of single mutations on the structural dynamics of mature CA protein were reproducible with repeated MD simulations. Although it is unclear at present how the changes can influence viral infectivity, multiple changes on the CA surfaces in addition to the changes at the specific sites may be important to realize distinct viral phenotypes. In sum, these results suggest that unoptimized structural fluctuation around the WM184/185 region may be a factor to decrease viral infectivity and highlight the role of the interdomain linker in controlling fluctuations of HIV-1 CA interaction surfaces away from the mutation sites.
FIG 10.

Effects of the linker and MHR mutations on the structural dynamics of HIV-1NL4-3 mature CA protein. Gag-CA protein models of HIV-1NL4-3 and its mutants were constructed from the reported structure of HIV-1 mature capsid (PDB code 3J34) (55). MD simulations were carried out under the same conditions as described for Fig. 8. RMSF values were calculated using 15,000 snapshots in the equilibrium states during MD simulations. Distribution of RMSF in the Gag-CA immature and mature proteins of NL4-3 (A) and those in the CA mature proteins of NL4-3 and mutants (B) are shown. Numbers on the horizontal axes indicate amino acid positions in the HIV-1NL4-3 Gag-CA protein. Arrows indicate the positions of mutated residues. #, positions of CypA binding loop; orange asterisks, sites upstream of WM184/185 residues in H9.
DISCUSSION
In this study, by a systemic mutational analysis, we aimed to elucidate how the Gag-CA interdomain linker works for HIV-1 replication, with a special interest in the late phase. Virus particle production by some linker mutants (S149A, S149D, S149K, and I150A) was markedly reduced, and that by the other mutants (S146N, P147A, T148A, and S149N) was moderately reduced (Table 1). Only the S146A mutant produced viral particle at a wild-type level. Most mutants in Table 1 displayed a considerable or drastic reduction in viral infectivity in TZM-bl cells. The S149N mutant is defective for viral particle production only but not for its early infectivity. These results strongly suggest that the interdomain linker functions in virus particle production. Biochemical and microscopic analyses of Gag-Gag assembly, which drives virus particle production, revealed that the linker mutations (S149A and I150A) impede the progression of Gag multimerization on the PM in spite of the accumulation of certain amounts of membrane-targeted Gag proteins as previously reported for MHR mutations (K158A and Y164A) (Fig. 3 to 5) (19, 21, 22). NanoBRET assays directly showed that these four mutations (S149A, I150A, K158A, and Y164A) significantly diminish the ability of Gag to drive Gag-Gag interactions (Fig. 6). In sum, we conclude that the decreased ability of the linker mutants for Gag-Gag interactions/multimerization on the PM rather than their reduced ability to target the PM leads to the decrease in virus particle production (Table 1). MD simulations showed that the linker mutations can remotely affect the structural fluctuations of interaction surfaces on the immature Gag-CA (Fig. 8). Taken together, the results lead us to propose that the interdomain linker facilitates Gag assembly by supporting the interactions of the CA-NTD and CA-CTD.
Although several mutational analyses on the linker region have been reported (28–30, 33), it remained unclear whether the linker acts on virus particle production. In this study, we clearly showed that all mutations except for one in the linker region have negative effects on virus particle production (Table 1). This may be due to the experimental systems used in this study. Cells used for transfections (lymphocyte line H9) and details of the transfection experiments (DNA amount, incubation time after transfection, and/or transfection methods) may have allowed us to obtain a positive readout on the changes in virus particle production.
Our MD simulations suggest that linker mutations would cause changes in fluctuations of amino acid residues away from the linker region (Fig. 8 and 10). These results were reproducible with repeated MD simulations, suggesting that the phenomena are inherent in the mutations. In this regard, a recent study has revealed that a disordered element of protein plays key roles in regulating overall structural dynamics and thus activity of the protein (52). The dynamic aspect of the disordered region theoretically increases entropy associated with the structure. Increasing evidence indicates that the structural dynamics in solution is critical to executing its physiological function involving recognition and release of substrates (50–53). Therefore, it is possible that the disordered linker region in the CA protein has similar regulatory functions for the overall structural dynamics and molecular interactions of CA protein. It remains unsolved, however, how such a remote control of the dynamics of CA protein could be attained. A possible explanation is that there exist networks of hydrophobic and hydrophilic interactions on the protein surface to remotely influence the mobility of amino acid residues apart from mutation sites. Alternatively, structural fluctuations may have resonant features, as often seen in the physical phenomena, and proper synchronization of fluctuations may be modulated by mutations. Whatever the physicochemical mechanisms are, what is clear in this study is that the linker region is a key site to regulate remotely structural dynamics and interaction potency of the CA domain.
Changes in fluctuations by mutations were primarily seen in the potential interaction surfaces, including helices and loops (Fig. 7 to 10). The mutations that caused changes in the fluctuations of the CA-CA contact sites induced attenuation in the Gag assembly and virus particle production, whereas S146A, which did not cause changes in the CA-CA contact sites, did not induce significant effects on the viral phenotypes. Thus, proper fluctuations in the CA-CA contact sites seem to be critical to optimize Gag assembly and virus particle production, as expected from protein science (50–53). Y164A and S149A, which induced augmentation of amino acid fluctuations throughout the protein (Fig. 8), caused greater attenuations in the Gag assembly and virus particle production. The results suggest that overall amino acid fluctuations of CA protein are optimized in the wild type for Gag assembly and virus particle production. Meanwhile, the sites with changes in fluctuations were differently distributed among the mutants. The results may imply that overall structural dynamics in addition to the specific changes in the CA-CA contact sites are also important for optimizing Gag assembly and virus particle production.
On the one hand, virus particle production was differently affected by amino acid residues altered and their locations within the linker region (e.g., S146A versus S149A and S149N versus S149A/D) (Table 1). Underlying molecular mechanisms for the observations remain unsolved. In this regard, changes in the size and/or electrostatic characteristics of a side chain in the linker region and MHR could modify differently the already optimized fluctuation profiles of HIV-1NL4-3 wild-type CA. Such differences in the fluctuation profiles with distinct mutation types may be a factor to cause distinct effects on Gag-Gag interactions and thus viral replication phenotypes. Results for the MHR mutants may imply that greater deviations of the CA fluctuation profiles from that of the wild type can cause greater effects on the viral replication phenotypes. In addition, changes in other structural factors for molecular interactions, such as those in the secondary structure on the interaction sites and the 3D distributions of the interaction-prone patches, may be involved in shaping the interaction efficiencies. Further study is necessary to address each of these issues.
Our in silico analyses provide structural insights into the experimental observations on the linker and MHR mutants of HIV-1 Gag-CA. They revealed that the linker and MHR mutations could induce changes in fluctuations at the regions for Gag assembly and disassembly, most of which were located away from the mutation sites (Fig. 8 and 10). Since structural fluctuations play key roles in molecular interactions (50–53), physical changes induced by the linker mutations would affect interaction efficiencies between CA contact sites and/or between CA and the trans-acting molecule(s) for Gag assembly and disassembly. This, in turn, could lead to the observed defects in Gag assembly, virus production process, and viral infectivity (Table 1 and Fig. 3 to 6). In this regard, structural influences caused by mutations were much greater for MHR, covering many important interaction sites, than those for the linker region (Fig. 7 to 10). The results are consistent with experimental data in that Gag assembly was more severely impaired by MHR mutations than by linker mutations. Thus, the present experimental and in silico findings strongly suggest that the HIV-1 Gag-CA linker region and MHR are both cis-acting protein elements governing structural properties of various Gag-CA sites for Gag assembly and disassembly. Taken together, our results have demonstrated that Gag interdomain linker has a role for HIV-1 replication in the late phase in addition to that in the early phase by influencing Gag-Gag interactions.
Our data thus illustrate a hitherto-unknown linker function, an allosteric regulation of CA structure by a disordered protein element, for HIV-1 replication. Although nearly half of human proteins contain intrinsically disordered peptide segments (56), their functions largely remain unknown. It is possible that the segments function to tune the protein structure and activity. In fact, the unstructured segment has been reported recently to be important in the regulation of structural dynamics and activity of human UDP-α-d-glucose-6-dehydrogenase (52). In the case of proteins of highly mutable pathogens, such tuning mechanisms may be necessary to maintain their replication abilities under constantly changing protein backbone sequences. In this regard, Shannon entropy analysis has shown that the linker region of HIV-1 subtype B allows practically no amino acid variations except for at position 148 (Fig. 2). The results strongly suggest the presence of potent selective constraints that restrict structural changes in the linker region during subtype B circulation and evolution in the field. Such constraints may be generated by strict requirement for the linker size and chemical property to deliver proper structural flexibility to CA during Gag assembly. Increasing evidence suggests that Gag assembly is a highly ordered CA-mediated process involving CTD-CTD interactions, interhexameric CTD-CTD interactions, intrahexameric CTD-CTD interactions, and interhexameric NTD-NTD interactions (21, 49). Thus, it is reasonable to postulate that the maintenance of appropriate physicochemical properties of the linker region is critical to successfully accomplish the assembly in order. Consistently, experimental data showed that a single mutation in the linker region often leads to reduction in particle production along with Gag assembly and infectivity defects (Table 1 and Fig. 3 to 6). Interestingly, however, position 148, in the center of the linker region, was found to accept amino acid changes (Fig. 2). This finding may imply that the interdomain linker of HIV-1 CA has a certain genetic space for maintaining the tuning function.
In conclusion, we demonstrated that the Gag-CA interdomain linker of HIV-1 plays a role in virus particle production via effects on Gag-Gag interactions/assembly. Since immature capsid assemblies would be a potential target for antiretroviral therapy (24, 57), it is necessary to comprehensively understand this process through clarification of the critical viral and cellular factors which are involved in virus particle production, as well as of the structural characteristics of the whole Gag precursor protein.
MATERIALS AND METHODS
Cells.
Monolayer cell lines HEK293T (ATCC CRL-1573), HeLa (ATCC CCL-2), and HeLa-derived reporter TZM-bl (58) were cultured and maintained in Eagle’s minimal essential medium containing 10% heat-inactivated fetal bovine serum as previously described (59). Lymphocyte line H9 (ATCC HTB-176) was cultured and maintained in RPMI 1640 medium containing 10% heat-inactivated fetal bovine serum as previously described (59).
Plasmid DNAs.
Proviral full-length clone pNL4-3 has been previously described (60). Its site-specific Gag-CA mutant clones (Table 1) were generated by PCR-based standard cloning procedures. Parental clones in this study, designated pNL4-3ΔPro/ΔEnv (Fig. 3 and 4) and pNL4-3Gag-EGFPΔEnv (Fig. 5), were constructed from pNL4-3. Briefly, two mutations that abolish functional expression of viral protease (NL-Hc) (61) and Env (NL-Kp) (62) were introduced into pNL4-3 to generate pNL4-3ΔPro/ΔEnv, and the Env mutation was inserted into a protease-deficient pNL4-3Gag-EGFP (63) to construct pNL4-3Gag-EGFPΔEnv. The test clones designated HaloTag-fused Gag and NanoLuc-fused Gag (Fig. 6) have been previously described (43). Finally, to perform the experiments whose results are shown in Fig. 3 to 6, Gag-CA mutant clones derived from the four parental test clones (pNL4-3ΔPro/ΔEnv, pNL4-3Gag-EGFPΔEnv, HaloTag-fused Gag, and NanoLuc-fused Gag) were constructed by standard recombinant DNA and PCR-based cloning techniques.
Assays for virus replication.
Input virus samples for infections (Table 1) were prepared from HEK293T cells transfected with various full-length clones by the calcium phosphate precipitation method as previously described (60, 64). Virus amounts were determined by virion-associated reverse transcriptase (RT) assays as previously described (64, 65). To monitor multicycle virus growth property, 1 × 105 RT units of virus samples were inoculated into 1 × 105 of H9 cells, and culture supernatants were collected every 3 days for RT assays. To evaluate viral single-cycle infectivity, 1 × 104 RT units of virus samples were inoculated into 4 × 103 TZM-bl cells, and on day 1 postinfection, cell lysates were prepared for luciferase assays (Promega) as previously described (59). To determine single-cycle virion productivity, 1 × 106 H9 cells were transfected with 2 μg of proviral clones by the Amaxa Cell Line Nucleofector kit V (Lonza) using Nucleofector II (Lonza) with program X-005. To prevent reinfection of progeny virions, CXCR4 antagonist AMD3100 (1 μM) was added to culture medium. Amounts of Gag-p24 in culture supernatants at 48 h posttransfection were measured by the HIV-1 p24 antigen enzyme-linked immunosorbent assay (ELISA) kit (ZeptoMetrix Corporation) according to the manufacturer’s protocol.
Analysis for Gag velocity sedimentation.
HeLa cells were transfected with pNL4-3ΔPro/ΔEnv or its derivative Gag-CA mutant clones by the polyethylenimine method (22), and at 24 h posttransfection, cells were harvested for analysis for Gag velocity sedimentation as previously described (22). Prepared cell lysates were then centrifuged through a sucrose gradient (22) using a P40ST rotor (Hitachi, Tokyo, Japan) at 36,000 rpm (162,700× g) for 160 min at 4°C. After centrifugation, gradients were fractionated from top to bottom. Sedimentation coefficients were calculated with a computer program, himac ASSIST (Hitachi), based on their actual Brix values. To detect Gag precursor protein, Western blot analysis using mouse anti-HIV-1 Gag p24 monoclonal antibody (NIH AIDS Reagent Program; catalog no. 6457) was performed as previously described (59). Gag bands were quantified by the Quantity One imaging program (Bio-Rad).
Membrane flotation assays.
HeLa cells were transfected with various pNL4-3ΔPro/ΔEnv clones as described above. On day 1 posttransfection, cells were harvested and lysates were prepared for the flotation assay by centrifugation through a sucrose gradient as previously described (22). Briefly, transfected cells were lysed with hypotonic lysis buffer using a Dounce homogenizer, and nuclei were removed by centrifugation. Postnuclear supernatants were mixed with the solution with a high sucrose concentration (final sucrose concentration was 75%). Samples were then placed at the bottom of an ultracentrifugation tube and overlaid with 65% to 10% sucrose solution. After centrifugation using a P40ST rotor (Hitachi) at 28,000 rpm (98,400 × g) for 20 h at 4°C, fractions (from top to bottom) were examined for Gag precursor by Western blotting as previously described (22).
Confocal microscopy analysis.
HeLa cells were transfected with pNL4-3Gag-EGFPΔEnv or derivative Gag-CA mutant clones by Lipofectamine 2000 (Thermo Fisher Scientific), and at 10 h posttransfection, cells were fixed with paraformaldehyde for confocal microscopy as previously described (63, 66). Nuclei were stained with 4′,6-diamidino-2-phenylindole (DAPI; Dojindo), and confocal images were obtained with an LSM 700 confocal microscope (Carl Zeiss). More than 100 EGFP-positive cells were examined for each sample to determine the subcellular localization pattern of Gag-EGFP.
NanoBRET analysis.
HEK293T cells in 96-well plates were transfected with vectors encoding HaloTag-fused Gag (100 ng) and NanoLuc-fused Gag (1 ng) by the Effectene transfection reagent (Qiagen). At 48 h posttransfection, NanoBRET activity was measured by the NanoBRET Nano-Glo detection system (Promega) in accordance with the manufacturer’s protocol. To remove minor effects of CA-independent Gag-Gag interactions, NanoBRET data were normalized by subtracting the signal value for a Gag CA linker/CTD deletion mutant (deletion of 146 to 231 amino acids in CA).
Shannon entropy analysis.
Amino acid variation at each position of Gag-CA was analyzed with Shannon entropy as described previously (34–36). Full-length capsid amino acid sequences of HIV-1 subtype B (n = 14,120) and group M (n = 25,222) were obtained from the HIV Sequence Database (http://www.hiv.lanl.gov/content/sequence/HIV/mainpage.html). Shannon entropy was calculated on the basis of Shannon’s equation (67):
where H(i), p(xi), and i indicate the amino acid entropy score of individual position, the probability of occurrence of a given amino acid at the position, and the number of the position, respectively. An H(i) score of zero indicates absolute conservation, whereas a score of 4.4 bits indicates complete randomness.
Molecular modeling of HIV-1 capsid protein.
HIV-1 capsid monomer structures with various linker and MHR mutations were constructed by the homology modeling method with Molecular Operating Environment (MOE; Chemical Computing Group Inc., Montreal, QC, Canada) as described previously for modeling of various HIV-1 capsid mutants (64, 68–70). The reported structures of HIV-1 immature capsid (PDB code 4USN) (25) and mature capsid (PDB code 3J34) (55) were used as templates for modeling the immature and mature CA proteins, respectively, of HIV-1NL4-3 and its derivatives.
Protein patch analysis.
Assessment and visualization of hydrophobic patches on the CA models were carried out using the Protein Patch Analyzer tool in MOE (44–47). This program calculates hydrophobic patches with minimum area of 50 Å2 to show critical regions for protein-protein interactions (71, 72). The hydrophobic potential is calculated using the Wildman and Crippen octanol-water partition coefficients f = log P (73).
MD simulation of HIV-1 capsid monomer.
HIV-1 capsid models were subjected to MD simulations essentially as described previously for viral structural proteins and enzymes (36, 74–76). Briefly, the simulations were done by the pmemd module in the Amber 16 program package (54) with the ff14SB force field (77) and the TIP3P water model for simulations of aqueous solutions (78). A nonbonded cutoff of 10 Å was used. Bond lengths involving hydrogen were constrained with SHAKE, a constraint algorithm to satisfy a Newtonian motion (79), and the time for all MD simulations was set to 2 fs. After heating calculations for 20 ps to 310 K using the NVT ensemble, simulations were executed using the NPT ensemble at 1 atm, at 310 K, and in 150 mM NaCl for a total of 200 ns.
Calculation of RMSF.
RMSFs were calculated as described previously (74–76) to quantify structural dynamics of molecules in the MD simulations. RMSFs of the Cα atoms were calculated to obtain information on atomic fluctuations of individual amino acid residues during MD simulations. The 15,000 snapshots in the equilibrium states during 130 and 200 ns of MD simulations were used to calculate RMSF. The average structures were used as reference structures for RMSF calculation. RMSF, which quantifies the differences between the average values and those obtained at given times of MD simulations, was calculated using the ptraj module in Amber, a trajectory analysis tool (54).
ACKNOWLEDGMENTS
This study was supported in part by grants to M.N., K.M., and H.S. from the Japan Agency for Medical Research and Development (AMED; Research Program on HIV/AIDS; fund identifier, JP18fk0410004).
The numerical calculations for the MD simulations were carried out on the TSUBAME3.0 supercomputer in the Tokyo Institute of Technology. We are indebted to the NIH AIDS Research and Reference Reagent Program for Gag-p24 antibody. We appreciate the Support Center for Advanced Medical Sciences, Tokushima University Graduate School of Biomedical Sciences, for experimental facilities and technical assistance. We thank Kazuko Yoshida (Tokushima University, Tokushima, Japan) and Hiromi Nakamura (National Institute of Infectious Diseases, Tokyo, Japan) for assistance with editorial matters and Shannon entropy analyses, respectively. We also thank Yuko Morikawa (Kitasato University, Tokyo, Japan) for the generous gift of pNL4-3Gag-EGFP.
We declare no competing interests.
REFERENCES
- 1.Campbell EM, Hope TJ. 2015. HIV-1 capsid: the multifaceted key player in HIV-1 infection. Nat Rev Microbiol 13:471–483. doi: 10.1038/nrmicro3503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Freed EO, Martin MA. 2013. Human immunodeficiency viruses: replication, p 1502−1560. In Knipe DM, Howley PM, Cohen JI, Griffin DE, Lamb RA, Martin MA, Racaniello VR, Roizman B (ed), Fields virology, 6th ed, vol 2 Lippincott Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- 3.Bell NM, Lever AM. 2013. HIV Gag polyprotein: processing and early viral particle assembly. Trends Microbiol 21:136–144. doi: 10.1016/j.tim.2012.11.006. [DOI] [PubMed] [Google Scholar]
- 4.Briggs JA, Kräusslich HG. 2011. The molecular architecture of HIV. J Mol Biol 410:491–500. doi: 10.1016/j.jmb.2011.04.021. [DOI] [PubMed] [Google Scholar]
- 5.Freed EO. 2015. HIV-1 assembly, release and maturation. Nat Rev Microbiol 13:484–496. doi: 10.1038/nrmicro3490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lingappa JR, Reed JC, Tanaka M, Chutiraka K, Robinson BA. 2014. How HIV-1 Gag assembles in cells: putting together pieces of the puzzle. Virus Res 193:89–107. doi: 10.1016/j.virusres.2014.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sundquist WI, Kräusslich HG. 2012. HIV-1 assembly, budding, and maturation. Cold Spring Harb Perspect Med 2:a006924. doi: 10.1101/cshperspect.a006924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gres AT, Kirby KA, KewalRamani VN, Tanner JJ, Pornillos O, Sarafianos SG. 2015. X-ray crystal structures of native HIV-1 capsid protein reveal conformational variability. Science 349:99–103. doi: 10.1126/science.aaa5936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Worthylake DK, Wang H, Yoo S, Sundquist WI, Hill CP. 1999. Structures of the HIV-1 capsid protein dimerization domain at 2.6 A resolution. Acta Crystallogr D Biol Crystallogr 55:85–92. doi: 10.1107/S0907444998007689. [DOI] [PubMed] [Google Scholar]
- 10.Craven RC, Leure-duPree AE, Weldon RA Jr, Wills JW. 1995. Genetic analysis of the major homology region of the Rous sarcoma virus Gag protein. J Virol 69:4213–4227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mammano F, Ohagen A, Höglund S, Göttlinger HG. 1994. Role of the major homology region of human immunodeficiency virus type 1 in virion morphogenesis. J Virol 68:4927–4936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Strambio-de-Castillia C, Hunter E. 1992. Mutational analysis of the major homology region of Mason-Pfizer monkey virus by use of saturation mutagenesis. J Virol 66:7021–7032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.von Schwedler UK, Stemmler TL, Klishko VY, Li S, Albertine KH, Davis DR, Sundquist WI. 1998. Proteolytic refolding of the HIV-1 capsid protein amino-terminus facilitates viral core assembly. EMBO J 17:1555–1568. doi: 10.1093/emboj/17.6.1555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dorfman T, Bukovsky A, Ohagen A, Höglund S, Göttlinger HG. 1994. Functional domains of the capsid protein of human immunodeficiency virus type 1. J Virol 68:8180–8187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fitzon T, Leschonsky B, Bieler K, Paulus C, Schröder J, Wolf H, Wagner R. 2000. Proline residues in the HIV-1 NH2-terminal capsid domain: structure determinants for proper core assembly and subsequent steps of early replication. Virology 268:294–307. doi: 10.1006/viro.1999.0178. [DOI] [PubMed] [Google Scholar]
- 16.Reicin AS, Ohagen A, Yin L, Hoglund S, Goff SP. 1996. The role of Gag in human immunodeficiency virus type 1 virion morphogenesis and early steps of the viral life cycle. J Virol 70:8645–8652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Reicin AS, Paik S, Berkowitz RD, Luban J, Lowy I, Goff SP. 1995. Linker insertion mutations in the human immunodeficiency virus type 1 gag gene: effects on virion particle assembly, release, and infectivity. J Virol 69:642–650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Srinivasakumar N, Hammarskjöld ML, Rekosh D. 1995. Characterization of deletion mutations in the capsid region of human immunodeficiency virus type 1 that affect particle formation and Gag-Pol precursor incorporation. J Virol 69:6106–6114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.von Schwedler UK, Stray KM, Garrus JE, Sundquist WI. 2003. Functional surfaces of the human immunodeficiency virus type 1 capsid protein. J Virol 77:5439–5450. doi: 10.1128/jvi.77.9.5439-5450.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chang YF, Wang SM, Huang KJ, Wang CT. 2007. Mutations in capsid major homology region affect assembly and membrane affinity of HIV-1 Gag. J Mol Biol 370:585–597. doi: 10.1016/j.jmb.2007.05.020. [DOI] [PubMed] [Google Scholar]
- 21.Robinson BA, Reed JC, Geary CD, Swain JV, Lingappa JR. 2014. A temporospatial map that defines specific steps at which critical surfaces in the Gag MA and CA domains act during immature HIV-1 capsid assembly in cells. J Virol 88:5718–5741. doi: 10.1128/JVI.03609-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tanaka M, Robinson BA, Chutiraka K, Geary CD, Reed JC, Lingappa JR. 2016. Mutations of conserved residues in the major homology region arrest assembling HIV-1 Gag as a membrane-targeted intermediate containing genomic RNA and cellular proteins. J Virol 90:1944–1963. doi: 10.1128/JVI.02698-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bharat TA, Castillo Menendez LR, Hagen WJ, Lux V, Igonet S, Schorb M, Schur FK, Kräusslich HG, Briggs JA. 2014. Cryo-electron microscopy of tubular arrays of HIV-1 Gag resolves structures essential for immature virus assembly. Proc Natl Acad Sci U S A 111:8233–8238. doi: 10.1073/pnas.1401455111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bharat TA, Davey NE, Ulbrich P, Riches JD, de Marco A, Rumlova M, Sachse C, Ruml T, Briggs JA. 2012. Structure of the immature retroviral capsid at 8 Å resolution by cryo-electron microscopy. Nature 487:385–389. doi: 10.1038/nature11169. [DOI] [PubMed] [Google Scholar]
- 25.Schur FK, Hagen WJ, Rumlová M, Ruml T, Müller B, Kräusslich HG, Briggs JA. 2015. Structure of the immature HIV-1 capsid in intact virus particles at 8.8 Å resolution. Nature 517:505–508. doi: 10.1038/nature13838. [DOI] [PubMed] [Google Scholar]
- 26.Pornillos O, Ganser-Pornillos BK, Yeager M. 2011. Atomic-level modelling of the HIV capsid. Nature 469:424–427. doi: 10.1038/nature09640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shin R, Tzou YM, Krishna NR. 2011. Structure of a monomeric mutant of the HIV-1 capsid protein. Biochemistry 50:9457–9467. doi: 10.1021/bi2011493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jiang J, Ablan SD, Derebail S, Hercík K, Soheilian F, Thomas JA, Tang S, Hewlett I, Nagashima K, Gorelick RJ, Freed EO, Levin JG. 2011. The interdomain linker region of HIV-1 capsid protein is a critical determinant of proper core assembly and stability. Virology 421:253–265. doi: 10.1016/j.virol.2011.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Brun S, Solignat M, Gay B, Bernard E, Chaloin L, Fenard D, Devaux C, Chazal N, Briant L. 2008. VSV-G pseudotyping rescues HIV-1 CA mutations that impair core assembly or stability. Retrovirology 5:57. doi: 10.1186/1742-4690-5-57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cartier C, Sivard P, Tranchat C, Decimo D, Desgranges C, Boyer V. 1999. Identification of three major phosphorylation sites within HIV-1 capsid. Role of phosphorylation during the early steps of infection. J Biol Chem 274:19434–19440. doi: 10.1074/jbc.274.27.19434. [DOI] [PubMed] [Google Scholar]
- 31.Takeuchi H, Saito H, Noda T, Miyamoto T, Yoshinaga T, Terahara K, Ishii H, Tsunetsugu-Yokota Y, Yamaoka S. 2017. Phosphorylation of the HIV-1 capsid by MELK triggers uncoating to promote viral cDNA synthesis. PLoS Pathog 13:e1006441. doi: 10.1371/journal.ppat.1006441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wacharapornin P, Lauhakirti D, Auewarakul P. 2007. The effect of capsid mutations on HIV-1 uncoating. Virology 358:48–54. doi: 10.1016/j.virol.2006.08.031. [DOI] [PubMed] [Google Scholar]
- 33.Arvidson B, Seeds J, Webb M, Finlay L, Barklis E. 2003. Analysis of the retrovirus capsid interdomain linker region. Virology 308:166–177. doi: 10.1016/S0042-6822(02)00142-3. [DOI] [PubMed] [Google Scholar]
- 34.Motomura K, Oka T, Yokoyama M, Nakamura H, Mori H, Ode H, Hansman GS, Katayama K, Kanda T, Tanaka T, Takeda N, Sato H, Norovirus Surveillance Group of Japan. 2008. Identification of monomorphic and divergent haplotypes in the 2006-2007 norovirus GII/4 epidemic population by genomewide tracing of evolutionary history. J Virol 82:11247–11262. doi: 10.1128/JVI.00897-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sato H, Yokoyama M, Nakamura H, Oka T, Katayama K, Takeda N, Noda M, Tanaka T, Motomura K. 2017. Evolutionary constraints on the norovirus pandemic variant GII.4_2006b over the five-year persistence in Japan. Front Microbiol 8:410. doi: 10.3389/fmicb.2017.00410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yokoyama M, Oka T, Takagi H, Kojima H, Okabe T, Nagano T, Tohya Y, Sato H. 2017. A proposal for a structural model of the feline calicivirus protease bound to the substrate peptide under physiological conditions. Front Microbiol 8:1383. doi: 10.3389/fmicb.2017.01383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Takahata T, Takeda E, Tobiume M, Tokunaga K, Yokoyama M, Huang YL, Hasegawa A, Shioda T, Sato H, Kannagi M, Masuda T. 2016. Critical contribution of Tyr15 in the HIV-1 integrase (IN) in facilitating IN assembly and nonenzymatic function through the IN precursor form with reverse transcriptase. J Virol 91:e02003-16. doi: 10.1128/JVI.02003-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yokoyama M, Oka T, Kojima H, Nagano T, Okabe T, Katayama K, Wakita T, Kanda T, Sato H. 2012. Structural basis for specific recognition of substrates by sapovirus protease. Front Microbiol 3:312. doi: 10.3389/fmicb.2012.00312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kuroda M, Niwa S, Sekizuka T, Tsukagoshi H, Yokoyama M, Ryo A, Sato H, Kiyota N, Noda M, Kozawa K, Shirabe K, Kusaka T, Shimojo N, Hasegawa S, Sugai K, Obuchi M, Tashiro M, Oishi K, Ishii H, Kimura H. 2015. Molecular evolution of the VP1, VP2, and VP3 genes in human rhinovirus species C. Sci Rep 5:8185. doi: 10.1038/srep08185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yokoyama M, Naganawa S, Yoshimura K, Matsushita S, Sato H. 2012. Structural dynamics of HIV-1 envelope Gp120 outer domain with V3 loop. PLoS One 7:e37530. doi: 10.1371/journal.pone.0037530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kutluay SB, Bieniasz PD. 2010. Analysis of the initiating events in HIV-1 particle assembly and genome packaging. PLoS Pathog 6:e1001200. doi: 10.1371/journal.ppat.1001200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Machleidt T, Woodroofe CC, Schwinn MK, Méndez J, Robers MB, Zimmerman K, Otto P, Daniels DL, Kirkland TA, Wood KV. 2015. NanoBRET—a novel BRET platform for the analysis of protein-protein interactions. ACS Chem Biol 10:1797–1804. doi: 10.1021/acschembio.5b00143. [DOI] [PubMed] [Google Scholar]
- 43.Miyakawa K, Nishi M, Matsunaga S, Okayama A, Anraku M, Kudoh A, Hirano H, Kimura H, Morikawa Y, Yamamoto N, Ono A, Ryo A. 2017. The tumour suppressor APC promotes HIV-1 assembly via interaction with Gag precursor protein. Nat Commun 8:14259. doi: 10.1038/ncomms14259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Capponi S, Geuens T, Geroldi A, Origone P, Verdiani S, Cichero E, Adriaenssens E, De Winter V, Bandettini di Poggio M, Barberis M, Chiò A, Fossa P, Mandich P, Bellone E, Timmerman V. 2016. Molecular chaperones in the pathogenesis of amyotrophic lateral sclerosis: the role of HSPB1. Hum Mutat 37:1202–1208. doi: 10.1002/humu.23062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Dindo M, Montioli R, Busato M, Giorgetti A, Cellini B, Borri Voltattorni C. 2016. Effects of interface mutations on the dimerization of alanine glyoxylate aminotransferase and implications in the mistargeting of the pathogenic variants F152I and I244T. Biochimie 131:137–148. doi: 10.1016/j.biochi.2016.10.001. [DOI] [PubMed] [Google Scholar]
- 46.Jetha A, Thorsteinson N, Jmeian Y, Jeganathan A, Giblin P, Fransson J. 2018. Homology modeling and structure-based design improve hydrophobic interaction chromatography behavior of integrin binding antibodies. MAbs 10:890–900. doi: 10.1080/19420862.2018.1475871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Shah M, Anwar MA, Park S, Jafri SS, Choi S. 2015. In silico mechanistic analysis of IRF3 inactivation and high-risk HPV E6 species-dependent drug response. Sci Rep 5:13446. doi: 10.1038/srep13446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Nakanishi S, Watanabe S, Doi N, Koma T, Adachi A, Nomaguchi M. 2018. Virological characterization of HIV-1 CA-NTD mutants constructed in a virus-lineage reflected manner. J Med Invest 65:110–115. doi: 10.2152/jmi.65.110. [DOI] [PubMed] [Google Scholar]
- 49.Schur FK, Obr M, Hagen WJ, Wan W, Jakobi AJ, Kirkpatrick JM, Sachse C, Kräusslich HG, Briggs JA. 2016. An atomic model of HIV-1 capsid-SP1 reveals structures regulating assembly and maturation. Science 353:506–508. doi: 10.1126/science.aaf9620. [DOI] [PubMed] [Google Scholar]
- 50.Dodson GG, Lane DP, Verma CS. 2008. Molecular simulations of protein dynamics: new windows on mechanisms in biology. EMBO Rep 9:144–150. doi: 10.1038/sj.embor.7401160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Karplus M, Kuriyan J. 2005. Molecular dynamics and protein function. Proc Natl Acad Sci U S A 102:6679–6685. doi: 10.1073/pnas.0408930102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Keul ND, Oruganty K, Schaper Bergman ET, Beattie NR, McDonald WE, Kadirvelraj R, Gross ML, Phillips RS, Harvey SC, Wood ZA. 2018. The entropic force generated by intrinsically disordered segments tunes protein function. Nature 563:584–588. doi: 10.1038/s41586-018-0699-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bai H, Kath JE, Zörgiebel FM, Sun M, Ghosh P, Hatfull GF, Grindley ND, Marko JF. 2012. Remote control of DNA-acting enzymes by varying the Brownian dynamics of a distant DNA end. Proc Natl Acad Sci U S A 109:16546–16551. doi: 10.1073/pnas.1203118109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Case DA, Betz RM, Cerutti DS, Cheatham TE III, Darden TA, Duke RE, Giese TJ, Gohlke H, Goetz AW, Homeyer N, Izadi S, Janowski P, Kaus J, Kovalenko A, Lee TS, LeGrand S, Li P, Lin C, Luchko T, Luo R, Madej B, Mermelstein D, Merz KM, Monard G, Nguyen H, Nguyen HT, Omelyan I, Onufriev A, Roe DR, Roitberg A, Sagui C, Simmerling CL, Botello-Smith WM, Swails J, Walker RC, Wang J, Wolf RM, Wu X, Xiao L, Kollman PA. 2016. AMBER 16. University of California, San Francisco, San Francisco, CA. [Google Scholar]
- 55.Zhao G, Perilla JR, Yufenyuy EL, Meng X, Chen B, Ning J, Ahn J, Gronenborn AM, Schulten K, Aiken C, Zhang P. 2013. Mature HIV-1 capsid structure by cryo-electron microscopy and all-atom molecular dynamics. Nature 497:643–646. doi: 10.1038/nature12162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Oates ME, Romero P, Ishida T, Ghalwash M, Mizianty MJ, Xue B, Dosztányi Z, Uversky VN, Obradovic Z, Kurgan L, Dunker AK, Gough J. 2012. D2P2: database of disordered protein predictions. Nucleic Acids Res 41:D508–D516. doi: 10.1093/nar/gks1226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Waheed AA, Freed EO. 2012. HIV type 1 Gag as a target for antiviral therapy. AIDS Res Hum Retroviruses 28:54–75. doi: 10.1089/aid.2011.0230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Platt EJ, Bilska M, Kozak SL, Kabat D, Montefiori DC. 2009. Evidence that ecotropic murine leukemia virus contamination in TZM-bl cells does not affect the outcome of neutralizing antibody assays with human immunodeficiency virus type 1. J Virol 83:8289–8292. doi: 10.1128/JVI.00709-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Nomaguchi M, Doi N, Sakai Y, Ode H, Iwatani Y, Ueno T, Matsumoto Y, Miyazaki Y, Masuda T, Adachi A. 2016. Natural single-nucleotide variations in the HIV-1 genomic SA1prox region can alter viral replication ability by regulating Vif expression levels. J Virol 90:4563–4578. doi: 10.1128/JVI.02939-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Adachi A, Gendelman HE, Koenig S, Folks T, Willey R, Rabson A, Martin MA. 1986. Production of acquired immunodeficiency syndrome-associated retrovirus in human and nonhuman cells transfected with an infectious molecular clone. J Virol 59:284–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kawamura M, Shimano R, Inubushi R, Amano K, Ogasawara T, Akari H, Adachi A. 1997. Cleavage of Gag precursor is required for early replication phase of HIV-1. FEBS Lett 415:227–230. doi: 10.1016/S0014-5793(97)01131-9. [DOI] [PubMed] [Google Scholar]
- 62.Sakai H, Shibata R, Miura T, Hayami M, Ogawa K, Kiyomasu T, Ishimoto A, Adachi A. 1990. Complementation of the rev gene mutation among human and simian lentiviruses. J Virol 64:2202–2207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Haraguchi H, Sudo S, Noda T, Momose F, Kawaoka Y, Morikawa Y. 2010. Intracellular localization of human immunodeficiency virus type 1 Gag and GagPol products and virus particle release: relationship with the Gag-to-GagPol ratio. Microbiol Immunol 54:734–746. doi: 10.1111/j.1348-0421.2010.00276.x. [DOI] [PubMed] [Google Scholar]
- 64.Nomaguchi M, Yokoyama M, Kono K, Nakayama EE, Shioda T, Doi N, Fujiwara S, Saito A, Akari H, Miyakawa K, Ryo A, Ode H, Iwatani Y, Miura T, Igarashi T, Sato H, Adachi A. 2013. Generation of rhesus macaque-tropic HIV-1 clones that are resistant to major anti-HIV-1 restriction factors. J Virol 87:11447–11461. doi: 10.1128/JVI.01549-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Willey RL, Smith DH, Lasky LA, Theodore TS, Earl PL, Moss B, Capon DJ, Martin MA. 1988. In vitro mutagenesis identifies a region within the envelope gene of the human immunodeficiency virus that is critical for infectivity. J Virol 62:139–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Nomaguchi M, Doi N, Fujiwara S, Fujita M, Adachi A. 2010. Site-directed mutagenesis of HIV-1 vpu gene demonstrates two clusters of replication-defective mutants with distinct ability to down-modulate cell surface CD4 and tetherin. Front Microbiol 1:116. doi: 10.3389/fmicb.2010.00116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Shannon CE. 1997. The mathematical theory of communication. 1963. MD Comput 14:306–317. [PubMed] [Google Scholar]
- 68.Nomaguchi M, Nakayama EE, Yokoyama M, Doi N, Igarashi T, Shioda T, Sato H, Adachi A. 2014. Distinct combinations of amino acid substitutions in N-terminal domain of Gag-capsid afford HIV-1 resistance to rhesus TRIM5α. Microbes Infect 16:936–944. doi: 10.1016/j.micinf.2014.08.017. [DOI] [PubMed] [Google Scholar]
- 69.Nomaguchi M, Yokoyama M, Kono K, Nakayama EE, Shioda T, Saito A, Akari H, Yasutomi Y, Matano T, Sato H, Adachi A. 2013. Gag-CA Q110D mutation elicits TRIM5-independent enhancement of HIV-1mt replication in macaque cells. Microbes Infect 15:56–65. doi: 10.1016/j.micinf.2012.10.013. [DOI] [PubMed] [Google Scholar]
- 70.Sultana T, Nakayama EE, Tobita S, Yokoyama M, Seki Y, Saito A, Nomaguchi M, Adachi A, Akari H, Sato H, Shioda T. 2016. Novel mutant human immunodeficiency virus type 1 strains with high degree of resistance to cynomolgus macaque TRIMCyp generated by random mutagenesis. J Gen Virol 97:963–976. doi: 10.1099/jgv.0.000408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Agrawal NJ, Kumar S, Wang X, Helk B, Singh SK, Trout BL. 2011. Aggregation in protein-based biotherapeutics: computational studies and tools to identify aggregation-prone regions. J Pharm Sci 100:5081–5095. doi: 10.1002/jps.22705. [DOI] [PubMed] [Google Scholar]
- 72.Chennamsetty N, Voynov V, Kayser V, Helk B, Trout BL. 2009. Design of therapeutic proteins with enhanced stability. Proc Natl Acad Sci U S A 106:11937–11942. doi: 10.1073/pnas.0904191106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Wildman SA, Crippen GM. 1999. Prediction of physicochemical parameters by atomic contributions. J Chem Inf Comput Sci 39:868–873. doi: 10.1021/ci990307l. [DOI] [Google Scholar]
- 74.Hikichi Y, Yokoyama M, Takemura T, Fujino M, Kumakura S, Maeda Y, Yamamoto N, Sato H, Matano T, Murakami T. 2016. Increased HIV-1 sensitivity to neutralizing antibodies by mutations in the Env V3-coding region for resistance to CXCR4 antagonists. J Gen Virol 97:2427–2440. doi: 10.1099/jgv.0.000536. [DOI] [PubMed] [Google Scholar]
- 75.Yokoyama M, Fujisaki S, Shirakura M, Watanabe S, Odagiri T, Ito K, Sato H. 2017. Molecular dynamics simulation of the Influenza A(H3N2) hemagglutinin trimer reveals the structural basis for adaptive evolution of the recent epidemic clade 3C.2a. Front Microbiol 8:584. doi: 10.3389/fmicb.2017.00584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Yokoyama M, Nomaguchi M, Doi N, Kanda T, Adachi A, Sato H. 2016. In silico analysis of HIV-1 Env-gp120 reveals structural bases for viral adaptation in growth-restrictive cells. Front Microbiol 7:110. doi: 10.3389/fmicb.2016.00110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Maier JA, Martinez C, Kasavajhala K, Wickstrom L, Hauser KE, Simmerling C. 2015. ff14SB: improving the accuracy of protein side chain and backbone parameters from ff99SB. J Chem Theory Comput 11:3696–3713. doi: 10.1021/acs.jctc.5b00255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML. 1983. Comparison of simple potential functions for simulating liquid water. J Chem Phys 79:926–935. doi: 10.1063/1.445869. [DOI] [Google Scholar]
- 79.Ryckaert J-P, Ciccotti G, Berendsen H. 1977. Numerical integration of the cartesian equations of motion of a system with constraints: molecular dynamics of n-alkanes. J Comput Phys 23:327–341. doi: 10.1016/0021-9991(77)90098-5. [DOI] [Google Scholar]