

Hereditary hearing loss is one of the most common disabilities among newborns, affecting approximately 1 in 1000 live-born babies. Most forms of hereditary hearing loss are nonsyndromic; 80% of affected newborns have hearing loss that is inherited in an autosomal recessive pattern, and in the remaining 20%, inheritance shows a dominant pattern.1
Many forms of hereditary hearing loss are caused by mutations in genes that affect the formation and function of cochlear hair cells — highly specialized sensory cells that play an important role in the detection and processing of sound.1 The hair cell has bundles of hair-like projections, called stereocilia, on its apical surface (Fig. 1). The deflection of these bundles by sound results in the opening of mechanotransduction ion channels, which are located at the tips of the stereocilia, and consequently, in the depolarization of the hair-cell membrane. Mutations that affect the protein transmembrane channel-like 1 (TMC1), an integral component of the mechanotransduction complex, cause autosomal dominant and autosomal recessive forms of hearing loss.2 Correction of the dominant form of hearing loss in a mouse model of Tmc1 (termed “Beethoven”) was recently reported by Gao and colleagues.3
Figure 1. Role of TMC1 in Hair-Cell Mechanotransduction.
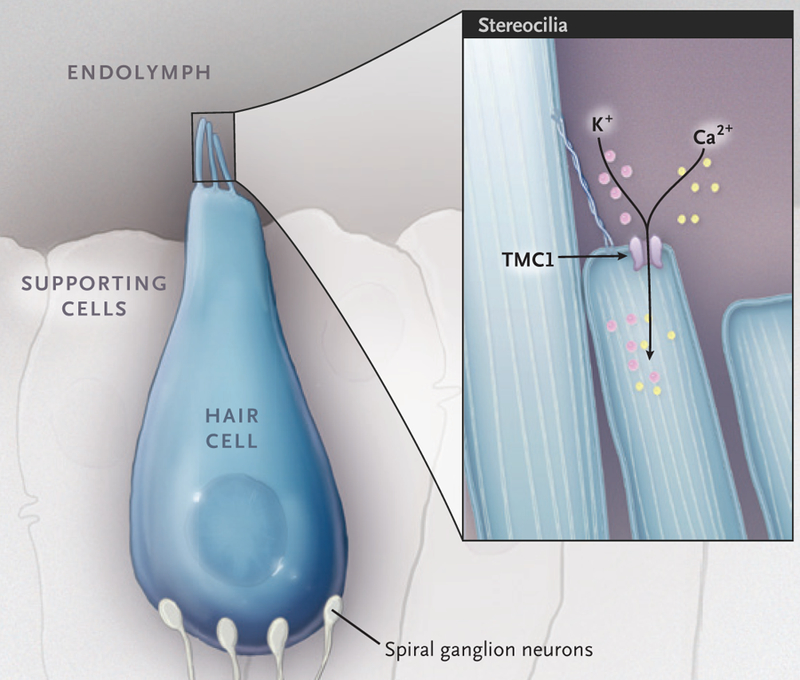
Shown is a cochlear inner hair cell with a stereocilia bundle on its apical surface. When the stereocilia bundle is deflected by sound, the mechanotransduction channels are opened, which leads to depolarization of the hair-cell membrane. The protein transmembrane channel-like 1 (TMC1) is an integral component of the mechanotransduction channel complex.
The Beethoven mouse model has a point mutation (1235T→A) in Tmc1 that causes progressive hair-cell loss and hearing loss in heterozygous mice by 30 days after birth. To prevent hair-cell loss and improve hearing, Gao et al. used a type of genome-editing tool called CRISPR-Cas9 (clustered regularly interspaced short palindromic repeats associated with Cas9 endonuclease) to knock down the expression of the dominant mutant allele in the Beethoven mouse model. The CRISPR-Cas9 complex consists of a guide RNA that can be engineered to target a specific DNA sequence in the genome, as well as an endonuclease enzyme (Cas9) that makes a double-stranded cut in the genome (Fig. 2). Once the double-stranded cut is made, random insertions and deletions can be incorporated into the gap by the cells’ own DNA-repair mechanism, thereby effectively inactivating the target allele. To initiate the process of gene editing, Gao et al. engineered several guide RNAs and tested the ability of each to recognize the mutant Tmc1 allele in vitro. The authors identified a guide RNA that could efficiently target the mutant Tmc1 allele without affecting the wild-type allele. The guide RNA was then injected into the cochlea of neonatal (0 to 2 days old) Beethoven heterozygous mutant mice in vivo, along with the Cas9 endonuclease, with the use of a lipid-mediated delivery method. Gao et al. found that the CRISPR-Cas9 complex was able to target the mutant Tmc1 allele in vivo and to generate insertions and deletions around the mutation locus, thereby knocking down the expression of the mutant allele. The wild-type allele remained intact to support normal hair-cell mechanotransduction. The injected Beethoven mutant mice showed greater hair-cell survival and better hearing than the noninjected control mice 30 days after birth.
Figure 2. Use of CRISPR-Cas9 in Knockdown of Mutant Tmc1 Allele to Restore Hair-Cell Function.

Shown is the application of genome editing to the Beethoven mouse model of hereditary hearing loss. A guide RNA is designed to target the mutant Tmc1 allele and recruits Cas9 endonuclease to make a double-stranded cut. Random insertions and deletions are incorporated into the cut, and the mutant Tmc1 allele is inactivated.
Although several studies have shown that gene therapy (in which the effect of a mutated gene is compensated through supplementation with a wild-type version) can be used to improve hearing in mouse models of hereditary hearing loss, Gao et al. showed that genome editing can achieve the same goal. What remains to be determined is the longevity of hair-cell survival and the duration of effect. In a recent study4 in which RNA interference was used to knock down the mutant allele in the same Beethoven mouse model as that used by Gao et al., initial hearing improvement was found to gradually dissipate in most of the treated mice. Would we observe a more durable improvement with genome editing, since we expect changes to the genome to be permanent? One might imagine so, but supplementary data from Gao et al. suggest that hearing in the treated mice deteriorated over time. Would genome editing improve hair-cell survival and hearing in the Beethoven mouse model if it were delivered after the neonatal period? The authors showed that genome editing is possible in the adult mouse cochlea, but they did not report results from treatment in the adult Beethoven mouse. This question is particularly salient because the cochlea of the neonatal mouse is not fully developed, whereas the human cochlea is fully developed at birth.
That being said, because the Tmc1 mutation of the Beethoven mouse model has also been described in humans,5 the translation of this work could potentially improve hearing in patients who have the same Tmc1 mutation as the mouse model as well as in patients with other autosomal dominant mutations. More generally, the study by Gao et al. provides proof of concept that genome editing can be used to treat hereditary hearing loss.
Footnotes
Disclosure forms provided by the author are available with the full text of this article at NEJM.org.
References
- 1.Chien WW, Monzack EL, McDougald DS, Cunningham LL. Gene therapy for sensorineural hearing loss. Ear Hear 2015;36: 1–7. [DOI] [PubMed] [Google Scholar]
- 2.Pan B, Géléoc GS, Asai Y, et al. TMC1 and TMC2 are components of the mechanotransduction channel in hair cells of the mammalian inner ear. Neuron 2013;79:504–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gao X, Tao Y, Lamas V, et al. Treatment of autosomal dominant hearing loss by in vivo delivery of genome editing agents. Nature 2018;553:217–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shibata SB, Ranum PT, Moteki H, et al. RNA interference prevents autosomal-dominant hearing loss. Am J Hum Genet 2016;98:1101–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhao Y, Wang D, Zong L, et al. A novel DFNA36 mutation in TMC1 orthologous to the Beethoven (Bth) mouse associated with autosomal dominant hearing loss in a Chinese family. PLoS One 2014;9(5):e97064. [DOI] [PMC free article] [PubMed] [Google Scholar]