

Abstract
We sought to determine incidence, etiologies, and yield of genetic testing in infantile onset developmental and epileptic encephalopathies (DEEs) in a population isolate, with an intensive multistage approach. Infants born in Tasmania between 2011 and 2016, with seizure onset <2 years of age, epileptiform EEG, frequent seizures, and developmental impairment, were included. Following review of EEG databases, medical records, brain MRIs, and other investigations, clinical genetic testing was undertaken with subsequent research interrogation of whole exome sequencing (WES) in unsolved cases. The incidence of infantile DEEs was 0.44/1000 per year (95% confidence interval 0.25 to 0.71), with 16 cases ascertained. The etiology was structural in 5/16 cases. A genetic basis was identified in 6 of the remaining 11 cases (3 gene panel, 3 WES). In two further cases, WES identified novel variants with strong in silico data; however, paternal DNA was not available to support pathogenicity. The etiology was not determined in 3/16 (19%) cases, with a candidate gene identified in one of these. Pursuing clinical imaging and genetic testing followed by WES at an intensive research level can give a high diagnostic yield in the infantile DEEs, providing a solid base for prognostic and genetic counseling.
Keywords: developmental and epileptic encephalopathy, incidence, whole exome sequencing
1. INTRODUCTION
Infantile developmental and epileptic encephalopathies (DEEs) have a range of etiologies including >60 genetic causes, and in many patients, the cause remains unknown. The infantile DEEs comprise a group of epilepsy syndromes with infantile spasms (West syndrome) typically accounting for over half the cases.1, 2 The incidence of DEEs overall was estimated as 0.27/1000 births in the North London study1 and as 0.54/1000 per year in our recent Victorian study;2 the difference was probably related to methodology as each had different inclusion criteria and only those with a specific syndrome were counted in the North London estimate. Reliable epidemiological data for the infantile DEEs is required for health services planning and will inform the need for genetic testing in the diagnosis and management in these severe epilepsies.
Genetic investigations have revolutionized understanding of the causes of DEEs. This knowledge has begun to be implemented in the clinic, predominantly using gene panel testing, and diagnostic yields are growing as new discoveries are made.3 Incorporating whole exome sequencing (WES) into the diagnostic protocol of DEEs increases the yield and reduces overall costs associated with reaching a diagnosis.2
The Australian island state of Tasmania is a population isolate of ~515 000 residents served by only two EEG laboratories in its major cities, making it an ideal population for epidemiological studies. More than 94% of Tasmanians have a Caucasian ancestry. In this paper, we characterize the phenotypic, genetic, and epidemiologic features of infantile DEEs in a captured population over a six‐year period, utilizing next‐generation molecular investigations in a multistage approach from clinical gene panels to whole exome sequencing.
2. METHODS
Inclusion criteria were infants born in Tasmania between 2011 and 2016, with onset of seizures <2 years of age, epileptiform features on EEG, frequent seizures defined as >daily for a week or >weekly for a month, and evidence of developmental delay, plateauing, or regression. Infants with infantile spasms were included irrespective of seizure frequency. Patients with acute symptomatic seizures such as those associated with hypoxic‐ischemic encephalopathy were excluded. Patients were identified through contact with all Tasmanian pediatricians and pediatric neurologists and comprehensive review of EEG reports. EEG reports were reviewed from 2009 to 2016 to ensure full inclusion. All EEGs reported abnormal were reviewed by two neurologists (TLW and DLJ) to confirm the presence of definitive epileptiform discharges.
The ascertainment process is shown in Figure S1. We screened >1200 EEG records and identified 89 patients with epileptiform features. Seventy‐three cases were excluded; 40 due to age or date of onset; 24 had a mild or self‐limited epilepsy syndrome; 4 had normal developmental outcome; and 5 had acute symptomatic seizures (four with neonatal hypoxic‐ischemic encephalopathy and one with traumatic brain injury).
Clinical details obtained included developmental history, seizure semiology, and comorbidities from interview, review of medical records, and a validated seizure questionnaire.4 EEG findings, neuroimaging, and metabolic investigations were reviewed. Specific epilepsy syndromes were diagnosed where possible.
Research WES was conducted using Agilent SureSelect XT Human All Exon + UTR v5 (75 Mb) kit and 100 bp paired‐end sequencing on the HiSeq 2500 System. Trio WES analysis including both parents was performed to allow segregation of variants, where possible. WES read mapping, alignment processing, and variant calling were performed using GATK best practices.5 Initially, a panel of 423 candidate genes was interrogated from the WES for single nucleotide variants and, if this was negative, exome‐wide analysis was performed. Ultrarare variants predicted to result in functional changes and segregating with affected status were validated by Sanger sequencing. Plausible connections to epilepsy or neurodevelopmental conditions were investigated by review of the literature and performing functional studies (reported separately;6 Berecki et al, in submission). Pathogenicity of variants was assessed according to ACMG criteria.7 Data from unsolved cases were regularly re‐reviewed at 6‐ to 12‐month intervals. Ethical approval was obtained from the Human Research and Ethics Committee (Tasmania) Network (Reference H0013627).
3. RESULTS
Sixteen patients met the criteria for incident infantile DEEs in the six‐year period. All were of Anglo‐Australian background. All patients were identified from review of EEG records or by pediatric neurologists servicing Tasmania. Correspondence with pediatricians around the state did not identify any additional patients.
Of the 16 patients, 5 were male. Clinical and molecular findings are summarized in Table 1. Twelve had abnormal development prior to seizure onset or as newborn infants. Median seizure onset was 6 months (range 3 days to 20 months). Six patients had West syndrome with infantile spasms.
Table 1.
Clinical and etiology findings in epidemiological cohort of infantile DEEs
| Subject number/Gender age at last review |
Onset age of seizures Syndrome |
Clinical details | Etiology |
|---|---|---|---|
|
1/ F 2 y4 mo |
8 mo West syndrome |
Epileptic spasms Profound GDD EEG: Hypsarrhythmia MRI: Moderate lissencephaly, gradient: posterior more severe than anterior |
Lissencephaly Miller‐Dieker 17p13.3 microdeletion |
|
2/ M 6 y1 mo |
7 mo West syndrome |
Spasms, focal motor seizures Profound GDD EEG: Hypsarrhythmia MRI: Moderate lissencephaly, gradient: posterior more severe than anterior, pontine hypoplasia |
Lissencephaly 17p mosaic microduplication |
|
3/ M 2 y6 mo |
20 mo DEE |
Unifocal seizures Regression with seizures. Surgery curative. Mild language delay EEG: Left frontotemporal IEDs MRI: Segmental focal cortical dysplasia, subependymal nodules |
Focal cortical dysplasia Genetic testing not done |
|
4/ F 4 y4 mo |
2 wk DEE |
Focal tonic, FIAS Plateau with seizures. Mild language delay. EEG: Multifocal IEDs. MRI: Multifocal tubers. |
Tuberous sclerosis complex Genetic testing not done |
|
5/ F 4 y1 mo |
5 mo West syndrome |
Spasms, Focal motor seizures Hemiplegia, regression with spasms EEG: Hypsarrhythmia; unifocal centro‐temporal spike IEDs MRI: Antenatal venous infarction with multicystic encephalomalacia |
Antenatal clastic vascular Genetic testing not done |
|
6/ F 2 y3 mo |
3 d EIMFS |
Focal seizures, migrating focal seizures Mild GDD EEG: Ictal rhythms migrating between hemispheres; 6 mo & 13 mo normal MRI: Normal |
KCNQ2
c.637C>T p.Arg213Trpa de novo Pathogenicb |
|
7/ F 2 y1 mo |
2 mo West syndrome |
Focal tonic seizures, spasms, multifocal myoclonia Acquired microcephaly, dyskinesia, profound GDD EEG: Hypsarrhythmia; multifocal discharges MRI: Acquired moderate cerebral atrophy |
KCNQ2
c.593G>A p.Arg198Glna de novo Pathogenicb |
|
8/ M 19 mo |
4 wk DEE |
Tonic‐clonic seizures, focal tonic seizures Severe GDD EEG: Bilateral occipital IEDs MRI: Hypoplastic corpus callosum |
ARX
c.1449‐1 G>C p.Leu484*c Pathogenicb |
|
9/ F 5 y4 mo |
6 mo DEE |
Tonic‐clonic seizures, FBTC Language delay EEG: 11 mo‐Ictal rhythm midline to frontocentral regions; 2 y1 mo‐GSW, PSW MRI: Normal |
SCN8A
c. 5009T>G p.Met1670Argc Uncertain significanceb (de novo status unproven) |
|
10/ F 5 y10 mo |
6 mo DEE |
Absence with eyelid myoclonia, absence, eyelid myoclonia, myoclonic jerks, tonic‐clonic seizures, NCSE Profound GDD, visual impairment EEG: Marked photosensitivity, 3‐4Hz GSW, PSW, myoclonic‐atonic seizure MRI: Normal |
DHDDS
c.632G>A p.Arg211Glna de novo Pathogenicb |
| 11/ M deceased 17 d |
5 d EME |
Myoclonic jerks Decreased activity, poor feeding, jitteriness EEG: Burst suppression MRI: Normal |
GABRB2
c.851C>A p.Thr284Lysc de novo Likely pathogenicb |
|
12/ F 8 y3 mo |
7 mo DEE |
Febrile seizures, vibratory tonic seizures, tonic‐clonic seizures, absence Profound GDD, hypotonia, truncal ataxia, ambulating with walker at 4 y EEG: GSW, multifocal discharges, PSW MRI: Normal, no cerebellar atrophy |
CACNA1G
c.2727G>C p.Leu909Phec Likely pathogenic (gain of function in vitro [unpublished]; de novo status unproven)b |
|
13/ F 4 y7 mo |
5 mo West syndrome |
Spasms Remission at 8 mo, severe ID and GDD, seizure free without medication EEG: Hypsarrhythmia MRI: Normal |
SNAP25
c. 526C>T p.Arg176Cysc Uncertain significanceb (de novo status unproven) |
|
14/ F 4 y10 mo |
13 mo DEE |
Myoclonic jerks; Focal motor seizures at 2 y Hypotonia, delayed visual maturation, severe ID and GDD EEG: Multifocal discharges, normal at 23 mo MRI: Normal |
Unknown Candidate gene: FAT1 c.8626G>C p.Asp2876Hisc c.7655A>G p.Glu2552Glyc Uncertain significanceb |
|
15/ M 4 y6 mo |
9 mo West syndrome |
Spasms Developmental plateau with spasms EEG: Hypsarrhythmia MRI: Normal |
Unknown |
|
16/ F 6 y9 mo |
10 mo DEE |
Focal tonic seizures, tonic‐clonic seizures, FIAS Specific learning difficulties EEG: 15 mo‐normal; 21 mo‐occipital ictal rhythm; 22 mo‐occipital IEDs; 4 y‐normal MRI: Normal |
Unknown |
Abbreviations: BS, burst suppression; DEE, developmental and epileptic encephalopathy; EIMFS, epilepsy of infancy with migrating focal seizures; EME, early myoclonic encephalopathy; FBTC, focal to bilateral tonic‐clonic seizure; FIAS, focal impaired awareness seizure; GDD, global developmental delay; GSW, generalized spike‐wave; ID, intellectual disability; IEDs, interictal epileptiform discharges; MFDs, multifocal discharges; NCSE, nonconvulsive status epilepticus; PSW, polyspike‐wave.
previously published variant.
ACMG classification.
Novel variant.
There were 36 408 births in Tasmania in 2011‐2016 with net migration in this period being negligible.8 The incidence of infantile DEEs and infantile spasms in Tasmania was 0.44/1000 (95% confidence interval 0.25 to 0.71) per year and 0.17/1000 (95% confidence interval 0.06 to 0.36) per year, respectively.
At ascertainment, five infants had an established etiology based on history and neuroimaging (Table 1). Cases 1 and 2 had lissencephaly including one with a causative copy number variant (17p13.3 Miller‐Dieker microdeletion) and the second with a mosaic microduplication at 17p regarded as likely pathogenic. One patient had focal cortical dysplasia, and one had clinically confirmed tuberous sclerosis (genetic testing not performed). Case 5 had extensive unilateral cystic encephalomalacia, consistent with a large perinatal anterior circulation infarct. As this patient presented with infantile spasms, she met our inclusion criteria despite the acquired cause. There were no metabolic etiologies identified in our cohort.
The etiology was unknown at ascertainment in the remaining 11 patients (cases 6‐16); however, three subsequently had positive clinical genetic testing. Two had de novo KCNQ2 variants, and one had an intronic change in ARX resulting in retention of intron 4 and predicted early termination of the ARX protein;6 the variant segregated with autism spectrum disorder in his mildly affected mother and brother.
Three patients had pathogenic or likely pathogenic variants identified on research genetic testing in DHDDS, GABRB2 (reported in9), and CACNA1G. For case 12 with a variant in CACNA1G, in vitro electrophysiological evaluation showed a pathogenic gain of function (Berecki et al, in submission). Cases 9 and 13, with heterozygous variants in SCN8A and SNAP25, respectively, were regarded as likely solved, as their clinical patterns were consistent with the literature, and there was strong in silico data. However, as both were novel variants and paternal DNA was unavailable for segregation, the ACMG classification for these two cases remained “uncertain significance.”
Three cases were regarded as currently unsolved. Case 14 had compound heterozygous variants in FAT1; segregation analysis confirmed one variant was inherited from each parent. Both variants were regarded as damaging on in silico analysis, but the gene is not established as a DEE gene, so the variants were regarded as of uncertain significance.
Etiology for two patients remains unknown despite detailed review of exome‐wide variants. For one, trio data are available, but for the other parental DNA was not available. Further clinical descriptions and variant details are given in the supporting information.
4. DISCUSSION
The incidence of the infantile DEEs in Tasmania (0.44/1000 per year) is consistent with estimates from North London, UK, and Victoria, Australia.1, 2 Our incidence of infantile spasms (0.17/1000 per year) was at the lower end of previous estimates, which ranged from 0.2‐0.45/1000 per year).1, 2, 10, 11 The Victorian study also included patients with acquired brain injuries (12% of the cohort), whereas these patients were excluded from our study with the exception of one who had infantile spasms (an automatic inclusion).
The etiology of the epilepsy was definitively identified in 11 of our 16 patients (69%). Five had a major structural abnormality. Genetics was important in this group with a defined genetic etiology in the two lissencephaly cases and a presumed but unstudied genetic abnormality in the tuberous sclerosis case.
Of the nonstructural cases, 6/11 had a definite genetic etiology. If we include cases 9 and 13, where the identified genes were highly plausible in terms of pathogenicity (SCN8A, SNAP25), but the absence of proof of a de novo etiology precluded strict ACMG classification as pathogenic, then the diagnostic success rate for the whole cohort climbs to 81% (Figure 1). Although our sample size was small, this high success rate in identifying the etiology can be attributed in part to the intensive scrutiny of WES data. This included trio testing where possible, consideration of various modes of inheritance including mosaicism and repeated interrogation incorporating newly published data into our analysis after standard clinical testing. Our rigorous testing identified genetic etiologies in two cases that were negative on initial exome‐wide analysis, as has also been shown to be valuable in other studies of unsolved DEE cases.12, 13
Figure 1.
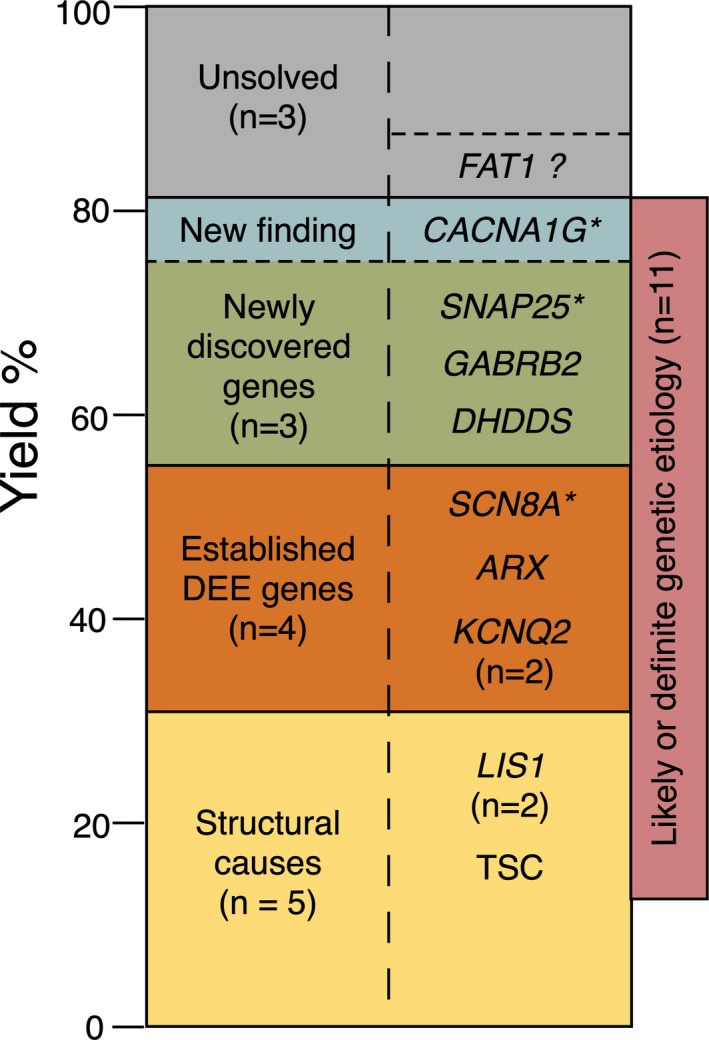
Yield of etiological diagnosis in 16 epidemiologically ascertained cases of Developmental and Epileptic Encephalopathy. Three of the “structural” cases had a definite or likely genetic etiology. *De novo status not confirmed
The reported diagnostic yield of genetic testing in infantile DEEs depends on the methods used, the inclusion criteria of the sample population, and whether “solved” patients with prior testing have been excluded. Recent studies generally find that 25%‐50% of cases are solved,2, 14, 15, 16, 17 although one study of a selected group of 14 cases studied by trio whole genome sequencing claimed diagnostic findings in all.18 Our findings provide a “real‐world” estimate reflecting that parental samples are not always obtainable from an epidemiologically ascertained cohort.
Strengths of our study are that the cohort was ascertained by comprehensive review of all EEG recordings performed in Tasmania from 2009 to 2016 and contact with all pediatricians and neurologists caring for children. The data are thus likely to be complete, which is supported by our incidence estimates being in broad agreement with others. It is possible, but unlikely, that patients with infantile DEEs may have been missed if they had not come to the attention of the mainstream medical profession or are cared for solely by a general practitioner, especially in the remote areas of Tasmania. Also, our combined clinical and research genetic approach resulted in a very high yield.
A weakness of our study is the small sample size; thus, while our global estimates are robust, we cannot provide estimates of the frequency of individual syndromes or genes. Indeed, based on established estimates of the incidence of Dravet syndrome, one to two cases of Dravet syndrome might have been expected in our cohort.19 The lack of Dravet cases in our cohort is most likely because they will not have satisfied inclusion criteria. It is likely that their EEG is normal in the first 2 years of life, seizures may be infrequent in infancy and always associated with fever (therefore diagnosed as febrile seizures), and developmental decline may not yet be apparent.20 Whole genome sequencing studies may reveal pathogenic variants not identified by WES.
Our hypothesis‐free, intensive, multistage approach to genetic testing identified pathogenic or likely pathogenic variants in a number of DEE genes. Our findings directly informed diagnosis, treatment, and prognostic planning for these infants and enabled accurate reproductive counseling for their parents.
CONFLICTS OF INTEREST
None of the authors have any conflict of interest to disclose. We confirm that we have read the Journal's position on issues involved in ethical publication and affirm that this report is consistent with those guidelines.
Supporting information
ACKNOWLEDGMENTS
We thank the patients and their families for participating in this study and pediatric staff in Tasmania for their collaboration. The authors gratefully acknowledge support by The Epilepsy Society Tasmania and the Estate of Kathleen Beulah Grace; National Health and Medical Research Council (NHMRC) Program Grant 1091593 (SFB; IES); NHMRC Fellowship 1104831 (IES); NHMRC Program Grant 1054618 (MB); and NHMRC Fellowship 1102971 (MB). Support Program and the NHMRC Independent Research Institute Infrastructure Support Scheme (IRIISS). Kim Wilmot, Ranitha Nandkumar, and Dasha Nepustilova (EEG scientists), Olivia Henry, Amy Schneider, and Georgina Hollingsworth provided research support.
Ware TL, Huskins SR, Grinton BE, et al. Epidemiology and etiology of infantile developmental and epileptic encephalopathies in Tasmania. Epilepsia Open. 2019;4:504–510. 10.1002/epi4.12350
REFERENCES
- 1. Eltze CM, Chong WK, Cox T, Whitney A, Cortina‐Borja M, Chin RFM, et al. A population‐based study of newly diagnosed epilepsy in infants. Epilepsia. 2013;54:437–45. [DOI] [PubMed] [Google Scholar]
- 2. Howell KB, Eggers S, Dalziel K, Riseley J, Mandelstam S, Myers CT, et al. A population‐based cost‐effectiveness study of early genetic testing in severe epilepsies of infancy. Epilepsia. 2018;59(6):1177–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Møller RS, Larsen LHG, Johannesen KM, Talvik I, Talvik T, Vaher U, et al. Gene panel testing in epileptic encephalopathies and familial epilepsies. Mol Syndromol. 2016;7:210–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Reutens DC, Howell RA, Gebert KE, Berkovic SF. Validation of a questionnaire for clinical seizure diagnosis. Epilepsia. 1992;33:1065–71. [DOI] [PubMed] [Google Scholar]
- 5. Van derAuwera GA, Carneiro MO, Hartl C, Poplin R, DelAngel G, Levy‐Moonshine A, et al. FastQ data to high confidence variant calls: the Genome Analysis Toolkit best practices pipeline. Curr Protoc Bioinformatics. 2013;43:11–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Shoubridge C, Jackson M, Grinton B, Berkovic SF, Scheffer IE, Huskins S, et al. Splice variant in ARX leading to loss of C‐terminal region in a boy with intellectual disability and infantile onset developmental and epileptic encephalopathy. Am J Med Genet A. 2019. [Epub ahead of print]. 10.1002/ajmg.a.61216 [DOI] [PubMed] [Google Scholar]
- 7. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier‐Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Australian Bureau of Statistics . Population ‐ Australian Bureau of Statistics, 2016. Available at: http://www.abs.gov.au/Population. Accessed 14/03/2019.
- 9. Hamdan FF, Myers CT, Cossette P, Lemay P, Spiegelman D, Laporte AD, et al. High rate of recurrent de novo mutations in developmental and epileptic encephalopathies. Am J Hum Genet. 2017;101:664–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Trevathan E, Murphy CC, Yeargin‐Allsopp M. The descriptive epidemiology of infantile spasms among Atlanta children. Epilepsia. 1999;40:748–51. [DOI] [PubMed] [Google Scholar]
- 11. Jia JL, Chen S, Sivarajah V, Stephens D, Cortez MA. Latitudinal differences on the global epidemiology of infantile spasms: systematic review and meta‐analysis. Orphanet J Rare Dis. 2018;13:216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Li J, Gao K, Yan H, Xiangwei W, Liu N, Wang T, et al. Reanalysis of whole exome sequencing data in patients with epilepsy and intellectual disability/mental retardation. Gene. 2019;700:168–75. [DOI] [PubMed] [Google Scholar]
- 13. Epilepsy Genetics Initiative . The epilepsy genetics initiative: systematic reanalysis of diagnostic exomes increases yield. Epilepsia. 2019;60:797–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Helbig KL, Farwell Hagman KD, Shinde DN, Mroske C, Powis Z, Li S, et al. Diagnostic exome sequencing provides a molecular diagnosis for a significant proportion of patients with epilepsy. Genet Med. 2016;18:898–905. [DOI] [PubMed] [Google Scholar]
- 15. Mercimek‐Mahmutoglu S, Patel J, Cordeiro D, Hewson S, Callen D, Donner EJ, et al. Diagnostic yield of genetic testing in epileptic encephalopathy in childhood. Epilepsia. 2015;56:707–16. [DOI] [PubMed] [Google Scholar]
- 16. Palmer EE, Schofield D, Shrestha R, Kandula T, Macintosh R, Lawson JA, et al. Integrating exome sequencing into a diagnostic pathway for epileptic encephalopathy: Evidence of clinical utility and cost effectiveness. Mol Genet Genomic Med. 2018;6:186–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Snoeijen‐Schouwenaars FM, van Ool JS, Verhoeven JS, van Mierlo P, Braakman HMH, Smeets EE, et al. Diagnostic exome sequencing in 100 consecutive patients with both epilepsy and intellectual disability. Epilepsia. 2019;60:155–64. [DOI] [PubMed] [Google Scholar]
- 18. Ostrander BEP, Butterfield RJ, Pedersen BS, Farrell AJ, Layer RM, Ward A, et al. Whole‐genome analysis for effective clinical diagnosis and gene discovery in early infantile epileptic encephalopathy. NPJ Genom Med. 2018;3:22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Bayat A, Hjalgrim H, Moller RS. The incidence of SCN1A‐related Dravet syndrome in Denmark is 1:22,000: a population‐based study from 2004 to 2009. Epilepsia. 2015;56:e36–39. [DOI] [PubMed] [Google Scholar]
- 20. Brunklaus A, Ellis R, Reavey E, Forbes GH, Zuberi SM. Prognostic, clinical and demographic features in SCN1A mutation‐positive Dravet syndrome. Brain. 2012;135:2329–36. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials